

Abstract
A key component in the response of the nervous system to injury is the proliferation and switch to a “proinflammatory” phenotype by microglia (microgliosis). In situations where the blood–brain barrier is intact, microglial numbers increase via the proliferation and chemotaxis of resident microglia; however, there is limited knowledge regarding the factors mediating this response. After peripheral nerve injury, a dorsal horn microgliosis develops, which directly contributes to the development of neuropathic pain. Neuregulin-1 (NRG-1) is a growth and differentiation factor with a well characterized role in neural and cardiac development. Microglia express the NRG1 receptors erbB2, 3, and 4, and NRG1 signaling via the erbB2 receptor stimulated microglial proliferation, chemotaxis, and survival, as well as interleukin-1β release in vitro. Intrathecal treatment with NRG1 resulted in microglial proliferation within the dorsal horn, and these cells developed an activated morphology. This microglial response was associated with the development of both mechanical and cold pain-related hypersensitivity. Primary afferents express NRG1, and after spinal nerve ligation (SNL) we observed both an increase in NRG1 within the dorsal horn as well as activation of erbB2 specifically within microglia. Blockade of the erbB2 receptor or sequestration of endogenous NRG after SNL reduced the proliferation, the number of microglia with an activated morphology, and the expression of phospho-P38 by microglia. Furthermore, consequent to such changes, the mechanical pain-related hypersensitivity and cold allodynia were reduced. NRG1-erbB signaling therefore represents a novel pathway regulating the injury response of microglia.
Introduction
Microglia are the resident myeloid cells of the CNS and respond rapidly to injury of the nervous system. This injury response (or “microgliosis”) has a number of different components including: microglial cell proliferation (Ajami et al., 2007; Echeverry et al., 2008), migration to the site of injury (Yao et al., 1990; Peterson et al., 1997; Honda et al., 2001; Ifuku et al., 2007); phagocytosis of cellular debris (Neumann et al., 2009); antigen presentation (Fischer and Reichmann, 2001); adoption of an amoeboid morphology; and the release of a broad range of cytokines and chemokines (Milligan and Watkins, 2009). Microglia express receptors for and respond to a range of injury-related molecules including cytokines, chemokines, complement components, and purines. These molecules regulate different components of the injury response (a process that can be thought of as a continuum rather than an “all-or-nothing event”). The microglial response to peripheral nerve injury contributes to the generation of neuropathic pain, and these cells are also implicated in disease processes as diverse as traumatic brain injury, neurodegeneration, and demyelination (Hanisch and Kettenmann, 2007; Scholz and Woolf, 2007; Ransohoff and Perry, 2009). It has recently been demonstrated using chimeric animals obtained by parabiosis or bone marrow transplantation and selective irradiation that in situations where the blood–brain barrier is intact the principal means by which microglial numbers increase is through the proliferation of resident microglia (rather than recruitment of blood-borne, bone marrow-derived microglial progenitors) (Ajami et al., 2007; Mildner et al., 2007). In this study, we focus on the role of neuregulin-1 (NRG1) in modulating microglial proliferation chemotaxis and cytokine release.
NRG1 is one of a family of growth factors (NRG1–4). Nrg1 undergoes alternative splicing and differential promoter use to produce at least 15 isoforms, which include both secreted and transmembrane forms (Falls, 2003; Esper et al., 2006). NRG1 has a key role in neural and cardiac development (Lee et al., 1995); it can modulate synaptic plasticity (Mei and Xiong, 2008), and it can stimulate the proliferation, survival, and motility of a number of different cell types (Falls, 2003). All Nrg1 isoforms contain an epidermal growth factor (EGF)-like domain, which is critical for mediating biologic activity and binds to the tyrosine kinase receptors erbB3 and 4. These receptors subsequently heterodimerize with erbB2, which lacks a ligand-binding domain but is a key coreceptor in mediating signal transduction (Carraway and Cantley, 1994). ErbB3 lacks an active tyrosine kinase domain, and so its interaction with erbB2 is essential for signaling. ErbB4 can signal via homodimers but dimerizes preferentially with erbB2 (Graus-Porta et al., 1997). Microglia are reported to express erbB2, 3, and 4 receptors (Gerecke et al., 2001; Dimayuga et al., 2003); however, very little is known regarding the function of this signaling pathway within microglia. We therefore studied the effects of NRG1 on microglial survival, proliferation, chemotaxis, and cytokine release in vitro, and subsequently tested the hypothesis that this signaling pathway contributes to microgliosis and neuropathic pain after peripheral nerve injury in vivo.
Materials and Methods
Animals and surgery.
Adult male Wistar rats were used in accordance with UK Home Office regulations. In one group of animals, the left L5 spinal nerve was ligated and transected. Sham surgery animals were prepared in an identical manner but without injuring the nerve. To label dividing cells, rats were injected with 5-bromo-2′-deoxyuridine (BrdU; Sigma) dissolved in 0.007N NaOH/PBS (100 mg/kg body weight, i.p.) once daily for 3 consecutive days before perfusion and fixation.
Drugs and delivery.
PD 168393 (Calbiochem), an irreversible erbB inhibitor, was dissolved in 5% DMSO and delivered intrathecally at 1.25, 2.5, 5, or 10 μg/d by an Alzet osmotic pump (model 2002). Control animals were given the same vehicle solution, lacking the active compound. Anti-erbB2 antibodies [monoclonal antibody (mAb) 7.16.4 from Calbiochem and mAb 9 from Thermo Scientific, without azide] were delivered intrathecally via lumbar puncture 1 h before nerve injury (2.5, 5 or 10 μg diluted in 25 μl of sterile PBS). For perineural application of mAb 7.16.4 to the site of nerve injury, a 0.5 × 0.5 cm strip of oxidized regenerated cellulose (Surgicell, Ethicon) soaked in 10 μg of the antibody diluted in sterile PBS was applied directly over the site of spinal nerve ligation. Control animals received a nonimmune IgG2a antibody (Calbiochem) via the same method (intrathecally or perineurally). To sequester endogenous NRG, we used a fusion protein (HBD-S-H4) that was injected intrathecally every 3 d (3 μg in 20 μl of sterile saline per injection). The drug doses were selected on the basis of previous reports and our preliminary studies. Neuregulin-β1 EGF domain (rHRGβ1 aa177-244) was administered intrathecally via lumbar puncture (0.4 or 4 ng in a volume of 20 μl). To label dividing cells after NRG1 treatment, rats were injected with BrdU (Sigma) dissolved in 0.007N NaOH/PBS, 100 mg/kg body weight, i.p. 24 h before perfusion and fixation. Before surgery, animals were randomly allocated into experimental study groups (computer-generated randomization schedules). An independent investigator prepared the drugs individually and labeled them for each animal according to the randomization schedule. Operators and data analysts were blinded throughout the study.
Behavioral testing.
Mechanical withdrawal thresholds were tested using the Dynamic Plantar Aesthesiometer (Ugo Basile), which is an automated version of the von Frey hair assessment. A maximum cutoff of 50 g was used. The withdrawal threshold is calculated as the average of three consecutive tests with at least 10 min between each test. To measure cold allodynia, we applied a drop of acetone to the plantar hindpaw and measured the time that the animal spent licking, shaking, or lifting the paw during the following 2 min (Kontinen and Dickenson, 2000).
Histology.
After defined survival times, animals were killed by terminal anesthetization and transcardially perfused with 4% paraformaldehyde plus 1.5% picric acid in 0.1% phosphate buffer. The lumbar spinal cords and L5 dorsal root ganglia were excised, cryoprotected in 20% sucrose, cryostat cut (10 or 20 μm), and thaw-mounted onto glass slides. Spinal cord sections were incubated overnight with the primary antibody, rabbit anti-phospho-p38 mitogen-activated protein kinase (MAPK) (1:100; Cell Signaling Technology) or rabbit anti-phospho-erbB2 [p-Neu (Tyr 1248), 1:200; Santa Cruz Biotechnology], both of which were viewed by tyramide amplification (TSA Biotin System, PerkinElmer) or with anti-rabbit neuregulin-1 (H-210), Polyclonal Antibody (1:100; Santa Cruz Biotechnology). For colocalization studies, the slides were then incubated with rabbit anti-Iba1 (1:1000; WAKO), mouse anti-OX42 (1:200) (Serotec), rabbit anti-GFAP (1:1000; Dako), or mouse anti-NeuN (1:1000; Millipore Bioscience Research Reagents). DRGs sections were incubated overnight with the calcitonin gene-related peptide antibody (rabbit anti-CGRP, 1:2000; Sigma) and neurofilament 200 antibody (mouse anti-NF200, 1:1000; Millipore Bioscience Research Reagents) or with anti-rabbit Neuregulin-1 (H-210). After primary antibody incubation, sections were washed and incubated for 1.5 h with corresponding secondary antibody solution. Isolectin B4 (IB4) detection was performed using biotin-conjugated IB4 (1:50; (Sigma) and ExAvidin-AMCA (1:400; Vector Laboratories). Slides were washed, coverslipped with Vectashield mounting medium with or without DAPI (Vector Laboratories), and visualized under a Zeiss Axioplan 2 fluorescent microscope. The specificity of immunostaining was checked by the omission of the appropriate primary antibody or preincubation with the corresponding blocking peptide. Antibody detection of BrdU incorporated into DNA requires pretreatment of the tissue to expose the BrdU epitope. For this purpose, we used the antigen retrieval method described previously (Tang et al., 2007). The primary antibody solution contained mouse anti-BrdU (1:200; BD Biosciences) plus rabbit anti-Iba-1 (1:1000; Wako), and the secondary antibody solution contained corresponding IgG-conjugated FITC 1:200 plus IgG-conjugated Cy3 1:400 (both from Stratech).
Microglia cells and macrophages were 4% PFA-fixed for immunohistochemistry. Microglia were double-stained with erbB receptors antibodies [erbB 2/Neu (C-18), erbB3 (C-17), and erbB4 (C-18) rabbit, 1:100; Santa Cruz Biotechnology) and isolectin B4 biotin-conjugated (to label microglia, 1:50; Sigma) and visualized with corresponding secondary antibodies [Cy3, 1:400, for erbB receptors (Stratech); extra-avidin FITC, 1:200, for IB4 (Sigma)]. For detecting class II major histocompatibility complex (MHC-2), we used the monoclonal mouse anti-rat RT1B (clone OX-6) antibody from Serotec (1:100). We used the ApopTag Fluorescein In Situ kit (Millipore Bioscience Research Reagents) to identify apoptotic nuclei by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL). The purity of macrophages and microglial cultures was assessed by staining cells with Iba1 and DAPI.
In situ hybridization. In situ hybridization (ISH) was performed after immunohistochemistry using 34-nucleotide-long probes as previously described (Michael et al., 1997). The sequence and specificity of the NRG1 probe has been described before (Fricker et al., 2009). Oligonucleotides were chemically synthesized (Sigma-Genosys) and radioactively end-labeled with 35S-dATP (PerkinElmer Life Sciences) using terminal deoxynucleotidyl transferase (Promega). After prehybridization treatments (acetylation, dehydration, and delipidation), the radioactive probe was added on the sections and hybridization was performed overnight at 37°C. The next day, slides were washed in standard saline citrate solutions with increasing stringencies, rapidly dehydrated through graded alcohols, air-dried, dipped in autoradiographic emulsion (LM1; GE Healthcare), and developed for 3–4 weeks.
Quantification and analysis.
Quantitative assessment was performed by determining the numbers of immunoreactive cells within four areas of 10,000 μm2 in the superficial dorsal horn on five to seven randomly selected L5 spinal sections from each animal. For BrdU staining, the whole dorsal horn was analyzed. Microglia in which process length was less than double the soma diameter were classified as presenting an activated (or effector) morphology. Microglia in which the process length was double the soma diameter were classified as resting (or surveying) cells (Stence et al., 2001) (see Fig. 7). Cells were sampled only if the nucleus was visible within the plane of section and if cell profiles exhibited distinctly delineated borders. For quantification of dorsal root ganglia ISH, cells exhibiting grain density twice that of background levels were counted as exhibiting a positive hybridization signal. All analyses were performed with the operator blinded to treatment groups.
Figure 7.
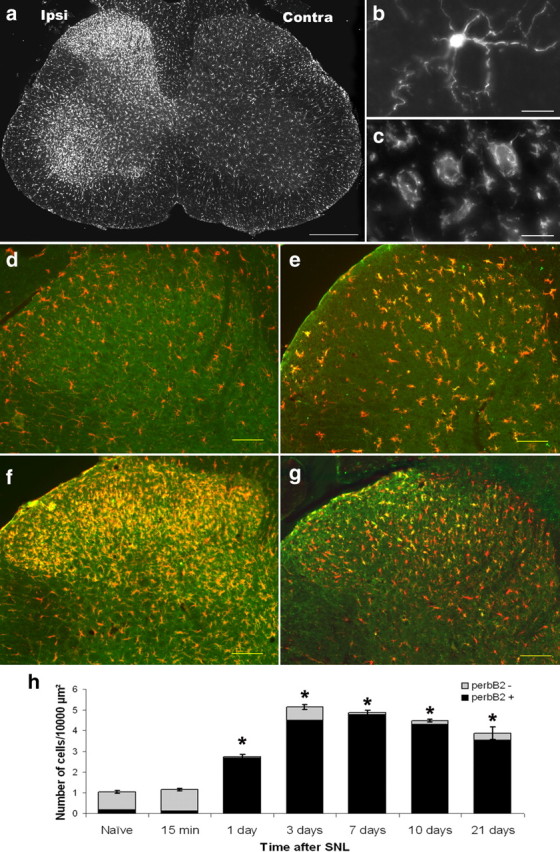
The time course of phospho-erbB2 expression is coincident with the development of microgliosis. The microglial response to nerve injury develops during the first week after injury (demonstrated by immunostaining with the microglial marker Iba1). a, An increase in microglial numbers is observed in the ipsilateral dorsal and ventral horn of the spinal cord of L5 SNL animals. b, c, A closer view shows that microglia change their morphology from having long processes and a small soma (b, naive animal, termed surveying microglia) to having retracted processes and a hypertrophic soma (c, 3 d after L5 SNL, termed an effector or activated morphology). Simultaneously with this microgliosis, we observed an increased expression of the p-erbB2 receptor (green) in the ipsilateral dorsal horn microglia (labeled with Iba1 in red). d–g, Naive (d), 1 d after SNL (e), 3 d after SNL (f), and 21 d after SNL (g). There was a significant increase in both total microglial number and the proportion that were p-erbB2-positive within the dorsal horn after nerve injury (quantification shown in h). *p < 0.05 (n = 3–4; one-way ANOVA on ranks, post hoc Student–Newman–Keuls method). Scale bars: a, 200 μm; b, c, 10 μm; d–g, 100 μm.
Detection of NRG1-β1 by ELISA.
Ipsilateral dorsal horns were homogenized in lysate buffer (20 mm Tris, pH 8, 137 mm NaCl, 1% NP-40, 1 mm sodium orthovanadate, and protease inhibitor mixture), and NRG1 was detected from lysates using a NRG1-β1-specific ELISA kit (detection range: 250–40,000 pg/ml; DuoSet ELISA DY377, R&D Systems). NRG1-β1 standards (250–40,000 pg/ml) and 100 μl of dorsal horn lysate (50 μg of protein) were run in duplicate following the manufacturer instructions. Samples were read at 450 nm with wavelength correction at 540 nm. Samples were considered NRG1-β1-positive if the signal was higher than background and within the range of the standard curve.
Primary microglia cell culture.
Mixed glial cultures were isolated from cortex of P3 Wistar rats according to the method of Giulian and Baker (1986). After mechanical and chemical dissociation, cells were seeded in DMEM with 10% fetal bovine serum (FBS) at a density of 500,000 cells/ml and cultured at 37°C in humidified 5% CO2/95% air. All reagents used were purchased from Invitrogen. Medium was replaced every 2–3 d, and confluency was achieved after 5 d in vitro. Confluent mixed glial cultures were manually shaken for 5 min, and the floating cells were pelleted and subcultured. After 15 min of plating, the medium was changed to discharge all nonadherent cells. This method resulted in 96–99% purity as assessed by Iba1 and DAPI staining. For proliferation assays, granulocyte monocyte colony stimulating factor (GM-CSF) (Cell Sciences, 1 nm) was used as a positive control (Giulian and Ingeman, 1988). Neuregulin-β1 EGF domain (rHRGβ1 aa177-244; Genentech) was used (0.5–10 nm). NRG1, GM-CSF (1 nm), and negative control (only medium) were applied in triplicate. For blocking the erbB2 receptor, we used PD168393 at 2.5, 5, or 10 μm, or mAb 7.16.4 at 1, 2, or 4 μg/ml. BrdU 10 μm was administered 15 h before fixation to label proliferating cells.
Peritoneal macrophage preparation.
Adult rats were killed with increasing CO2 concentrations and immediately after they were injected intraperitoneally with 20 ml of sterile HBSS. The elicited peritoneal exudate cells were harvested and resuspended in DMEM supplemented or not with 10% FBS as required. Cells were plated at a density of 300,000 cells/ml and cultured at 37°C in humidified 5% CO2/95% air. For chemotaxis assays, cells were used immediately after harvesting.
Chemotaxis assay.
Chemotaxis was assessed using the Boyden chamber (Neuroprobe). Polycarbonate filters (5 μm pore) were installed in the chamber, whose bottom wells were filled with serum-free DMEM or NRG1 at various concentrations. Freshly prepared microglia or macrophages were suspended in serum-free DMEM and were placed into the top wells (50,000 cells/well). The chamber was kept in a CO2 incubator at 37°C for 3 h. The filter was removed and stained with RapiDiffII (Biostain Ready Reagents). The cells on the top side of the filter were wiped off, and the number of cells that had migrated to the bottom side were counted.
Reverse transcriptase-PCR.
Reverse transcriptase (RT)-PCR was used for assessing mRNA expression of erbB receptors in microglial cultures. Total RNA was isolated using an RNeasy Mini Kit (Qiagen) and first-strand cDNA was reverse-transcribed. The sequences for primers used in this study were: erbB2, CCTGCCTCCACTTCAATCAT (forward), CAGGATCCCACTTCCGTAGA (reverse); erbB3 TGAAGATGTGTGAGCCTTGC (forward), GGTCCAGTGCGGGTATCTTA (reverse); and erbB4 ATGGCCTTCCAACATGACTC (forward), CACCTGCCATCACATTGTTC (reverse) (all 5′–3′). Amplification of templates was detected using SYBR Green 1 dye (Roche) on a Rotor-Gene thermal cycler (Corbett Life Science). Melting curve analysis and sequencing confirmed the specificity of the products. PCR products were visualized in a 2% agarose gel.
Detection of interleukin-1β by ELISA.
We used the Rat IL-1β/IL-1F2 Quantikine ELISA Kit (RLB00, R&D Systems), which mainly detects the mature or released form of interleukin-1β (IL-1β). IL-1β standards (31.2–2000 pg/ml) and 100 μl of microglia culture supernatants were run in duplicate following manufacturer instructions. Samples were read at 450 nm with wavelength correction at 540 nm. Samples were considered IL-1β-positive if the signal was higher than background and within the range of the standard curve. The supernatants of microglia were collected after the cells were primed with lipopolysaccharide (LPS) (1 μg/ml; Sigma for 3 h) and treated with ATP (1 mm; Sigma) or NRG1 (10 nm) for 30 min.
Western blots.
Microglial and macrophage cultures or spinal cord dorsal horn were homogenized in lysate buffer (20 mm Tris, pH 8, 137 mm NaCl, 1% NP-40, 1 mm sodium orthovanadate, and protease inhibitor mixture). The lysate was spun at 13,000 rpm at 4°C for 15 min, and the protein concentration of supernatant was determined using a BCA Protein Assay kit (Pierce). Proteins (50 μg/sample) were separated using 8% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were then blocked in 10% skimmed milk for 1 h at room temperature and washed in PBS-T three times for 5 min each. Membranes were incubated with primary antibody, rabbit erbB2 (1:200), rabbit erbB3 (1:100), and rabbit erbB4 (1:200; all from Santa Cruz Biotechnology) overnight at 4°C. After several PBS-T washes, as described before, membranes were incubated with donkey anti-rabbit HRP-conjugated secondary antibody (1:10,000; GE Healthcare) for 1 h at room temperature. As a loading control, we used mouse β-actin (1:10,000, Sigma), and the secondary antibody used was anti-mouse HRP-conjugated secondary antibody (1:10,000; GE Healthcare). Membranes were revealed using ECL-plus reagent for 5 min (GE Healthcare) for detection by autoradiography. Protein bands for all erbB receptors (185 kDa) and β-actin (42 kDa) were quantified using a model GS-700 imaging densitometer (Bio-Rad Laboratories). All quantifications were normalized against β-actin to ensure equal sample loading.
Statistical analysis.
Sample sizes for experiments were based on results from pilot studies. Datasets were tested for normality using the Kolmogorov–Smirnov test, and for homogeneity of variance using Levene's test. Parametric or nonparametric tests were used accordingly. Behavioral data were analyzed using repeated measures (RM) two-way ANOVA. When the assumptions of sphericity were violated (Mauchly's test; p < 0.05), the Greenhouse–Geisser correction was applied. p < 0.05 was considered as significant. Data are presented as the mean ± SEM.
Results
NRG1 promotes the survival, proliferation, and chemotaxis of microglia
Using RT-PCR analysis, we found that cultured rat microglia express erbB2, 3, and 4 receptor mRNA, and expression was confirmed by immunocytochemistry and Western blot analysis (Fig 1). When microglial cells are kept in serum-free conditions, apoptotic cell death is observed within 24 h (Koyama et al., 2000). We found that NRG1 significantly promoted the survival of microglia, which had been cultured in such conditions for 72 h (Fig. 2a–c). At NRG1 concentrations of 10 nm, the number of microglia was more than five times higher than seen in controls (p = 0.001, one-way ANOVA, four independent experiments). NRG1 application (10 nm) resulted in a threefold reduction in the number of apoptotic microglial nuclei after 24 h in serum-free conditions, as seen by TUNEL (p = 0.008, one-way ANOVA, three independent experiments) (Fig. 2d–f). This indicates that inhibition of apoptosis was responsible for the survival-promoting effects of NRG1. Because in many instances the erbB2 receptor is a key mediator of NRG1 signaling, we tested the consequences of erbB2 receptor blockade. We used either an irreversible specific erbB inhibitor, PD168393 (Bose et al., 2006), which blocks the ATP binding site of the receptor, or mAb 7.16.4, which specifically binds to the extracellular domain of erbB2 (sharing antigenic epitopes with Herceptin, Genentech) (Katsumata et al., 1995; Zhang et al., 1999). Both of these agents, which themselves had no effect on cell viability, could completely prevent the survival-promoting effects of NRG1 in a dose-dependent manner (there was no significant difference comparing treatment with NRG1 combined with PD168393 or mAb 7.16.4 and control, p = 1, one-way ANOVA, three independent experiments) (Fig. 2c,f; supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

ErbB receptor expression in primary cultured microglia. a, Using RT-PCR analysis, we found that primary cultured rat microglia express the erbB2, 3, and 4 receptors. The 2% agarose gels stained with ethidium bromide and photographed under ultraviolet light are shown for the PCR products of each of the genes amplified. Pos, Positive control (E16 embryonic rat brain cDNA); MG, microglia; Neg, negative control (water). b, ErbB receptors expression is shown in microglial cells by immunohistochemistry (IB4 labels microglia green, the erbB receptors are in red). Scale bar: 20 μm. c, Western blot analysis showing a 185 kDa band for erbB2, 3, and 4 in lysates from microglial cells.
Figure 2.

NRG1 stimulates microglial survival, proliferation, and chemotaxis in vitro. Survival of microglia in vitro was assessed by incubating cultured microglia for 3 d in serum-free medium, which was supplemented with increasing doses of NRG1 (0.5–10 nm) or GM-CSF (1 nm) (as a positive control). a, b, Representative wells are shown in which microglia are identified by Iba1 immunostaining after no treatment (a) or addition of 10 nm NRG1 (b). NRG1 promoted microglial survival, and erbB2 inhibition using PD168393 (INH, 10 μm) or mAb 7.16.4 (AB, 4 μg/ml) blocked this effect. c, Quantification of microglial numbers is shown. d–f, Cultured microglia were incubated for 1 day in serum-free medium with (e) or without (d) the addition of NRG1. DAPI staining (blue) demonstrated nuclei, and TUNEL (yellow) revealed those undergoing apoptosis. Treatment with NRG1 (10 nm) significantly decreased apoptosis, an effect that was prevented when microglia were treated with the erbB inhibitor PD168393 in combination with NRG1 (quantification in f). g–i, Proliferation was assessed by incubating microglia in medium supplemented with 5% FBS for 3 d and pulse-labeling with BrdU (yellow). NRG1 treatment significantly increased the proportion of BrdU-positive microglial nuclei, an effect that was erbB2-dependent. j–l, The effects of NRG1 on microglial migration were studied using a Boyden chamber. The addition of NRG1 to the lower well of the chamber (k) increased microglial migration to the inner membrane surface compared with control (j). Inhibition of erbB2 blocked this action. I, Quantification of migrated cells. Scale bars: 100 μm. Error bars represent ± SEM (three to five independent experiments). The statistical tests used were one-way ANOVA with Bonferroni post hoc analysis for all comparisons except for the proliferation experiment in which the data were not normally distributed and ANOVA on ranks with Dunn's post hoc test was used instead. *p < 0.05, **p < 0.005. CON, Control; INH, inhibitor (PD168393); AB, neutralizing antibody (7.16.4).
We subsequently investigated the effects of NRG1 on microglial proliferation. In serum-free conditions, we observed virtually no basal proliferation and very little response to either GM-CSF or NRG1 (data not shown). Microglia were therefore cultured in medium supplemented with 5% FBS, and cell proliferation in response to NRG1 was quantified using pulse labeling with BrdU. NRG1 treatment increased the proliferation of microglia in a dose-dependent manner (Fig. 2g,i) such that a dose of 10 nm increased the proportion of BrdU-labeled nuclei from 11 to 60% (p < 0.05, ANOVA on ranks, three to four independent experiments). This effect was again dependent on activation of the erbB2 receptor as it could be inhibited by PD168393 or mAb 7.16.4 in a dose-dependent manner (both were not significantly different from control, p = 0.215 and p = 0.516, respectively, for NRG1 plus PD168393 or mAb 7.16.4, t tests, three independent experiments) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The effect of NRG1 was comparable to that elicited by GM-CSF, a previously well described microglial mitogen (Giulian and Ingeman, 1988).
Microglia are motile cells and demonstrate directed migration toward regions of injury. Using a Boyden chamber, in which microglia migrate through pores in a polycarbonate filter across a concentration gradient, NRG1 was found to significantly increase microglial chemotaxis in a dose-dependent manner (Fig. 2j–l). A “checkerboard analysis” (Martinet et al., 1994) in which different concentrations of NRG1 were used in the upper and lower wells was performed. This demonstrated that, although there was migration when NRG1 was added to both upper and lower wells in equal concentrations, migration was greatest when the concentration gradient between the lower and upper well was maximal (Fig. 3). This is consistent with a true chemotactic response (i.e., directed migration along a concentration gradient) rather than a solely chemokinetic response (i.e., random migration) produced by NRG1. The chemotactic action of NRG1 was dose-dependently inhibited by erbB2 receptor blockade (NRG1 plus PD168393 or mAb 7.16.4, p = 1 and p = 0.36 vs control, respectively, t tests, three independent experiments) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Note that neither of these erbB2 receptor-blocking agents had any effect on basal migration (PD168393 vs control, p = 0.3; mAb 7.16.4 vs control, p = 0.6, Mann–Whitney rank sum test). Microglia are closely related to tissue macrophages, and indeed we found that peritoneal macrophages also express erbB 2, 3, and 4 receptors and demonstrate enhanced motility in response to NRG1 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Figure 3.

The migration of microglial cells in response to NRG1 is a true chemotactic response. In a Boyden chamber, microglial cells migrate from an upper to a lower well through a polycarbonate filter in response to a concentration gradient of the putative chemotactic agent. A checkerboard analysis of three independent experiments assessing the chemotactic response to NRG1 performed in triplicate and normalized to the unstimulated control is shown. Microglial cells were suspended in medium alone or with 0.1, 1, or 10 nm NRG1 and then allowed to migrate for 3 h at 37°C toward different concentrations of NRG1 in the lower compartments. Highlighted in gray boxes are the results achieved by using the same concentration of NRG1 in upper and lower wells. Note that in this circumstance NRG1 increases microglial migration, indicating chemokinesis. When there is an increasing concentration gradient from the upper to the lower well (values in gray), migration is clearly enhanced, indicating a true chemotactic response. *p < 0.05 for migration across a gradient vs migration when the NRG1 concentration is the same in both wells. Numbers represent the mean ± SEM.
In response to CNS injury, microglia adopt an effector (also termed “activated”) state, which is associated with development of an amoeboid morphology, proinflammatory cytokine expression, and expression of MHC-2 (Hanisch and Kettenmann, 2007). NRG1 had no effect on IL-1β release from naive microglia; however, it significantly enhanced IL-1β release from microglia that had been primed with LPS [a molecule known to activate microglia via the Toll-like receptor 4 (Lehnardt et al., 2003); p < 0.05 vs control, one-way ANOVA on ranks] (Fig. 4). The administration of LPS results in process retraction and increased OX6 (a marker of MHC-2 expression) immunoreactivity of microglia. NRG1, however, did not elicit such changes (the percentage of OX6 immunoreactivity in LPS-treated cells was five times higher than that in the control, p < 0.001; the NRG1-treated cells were no different from control, p = 1; one-way ANOVA, three independent experiments) (Fig. 4).
Figure 4.

NRG1 promotes the release of IL-1β from LPS-primed microglia but does not stimulate MHC-2 expression. a, An ELISA was used to quantify IL-1β released into the medium of cultured microglia. Neither ATP (1 mm) nor NRG1 (10 nm) alone produced IL-1β release from naive microglia; however, both significantly promoted IL-1β release from LPS (1 μg/ml)-primed microglia in a dose-dependent manner. Error bars: ± SEM. Statistical test: ANOVA on ranks. *p < 0.05, n = 3. b, Another aspect of the activation (or effector) response of microglia is that they increase expression of MHC-2. To investigate wherever NRG1 treatment leads to such a response in microglia, we incubated primary cultures in serum-free medium and treated them with LPS (1 μg/ml), NRG1 (10 nm), or medium alone (CON). Microglia were immunostained with OX-6 (which labels MHC-2), Iba1 (microglial marker), and DAPI (nuclear staining). LPS treatment led to a fivefold increase in OX-6 expression compared with control (p < 0.001), while NRG1 treatment did not elicit such a response (p = 1). Treatment with LPS also resulted in microglia adopting an amoeboid morphology, unlike NRG1. Scale bars: 100 μm. Error bars: ± SEM (three independent experiments) Statistical test: one-way ANOVA, Bonferroni post hoc analysis. **p < 0.001.
Intrathecal NRG1 produces dorsal horn microgliosis associated with mechanical and cold pain-related hypersensitivity
To investigate the effects of NRG1 on microglia in vivo, we administered this molecule intrathecally (0.4 or 4 ng administered daily for 3 d), after which we assessed both the response of dorsal horn microglia as well as pain-related behavior. Intrathecal NRG1 produced a dose-dependent increase in the number of microglia within the lumbar dorsal horn, and many of these cells were noted to develop an activated morphology with hypertrophy of the cell body and process retraction (Fig. 5a–c). Although a significant dorsal horn microgliosis was observed at day 1 (0.4 or 4 ng vs control, p = 0.02 and p = 0.002, respectively, one-way ANOVA, n = 4 per group), it was much more apparent at day 3 (Fig. 5a–c) (0.4 and 4 ng vs control, p < 0.05, ANOVA on ranks, n = 4). Furthermore, the number of microglia undergoing proliferation as assessed by pulse labeling with BrdU also significantly increased (Fig. 5d–l) (p < 0.05, 4 ng vs control, ANOVA on ranks, n = 4 per group). The microglial response within the dorsal horn after NRG1 treatment was mirrored by the development of a dose-dependent mechanical and cold pain-related hypersensitivity (Fig. 5m) (p = 0.009 and p < 0.001, respectively, at days 1 and 3 for 4 ng vs control; p = 0.04 at day 3 for 0.4 ng vs control) (Fig. 5n) (p < 0.001 at day 3 for 4 ng vs control, RM two-way ANOVA, n = 7–8 per group). As with the morphological changes in microglia, pain-related hypersensitivity was much more marked at day 3 than at day 1.
Figure 5.

Intrathecal administration of NRG1 results in dorsal horn microgliosis associated with mechanical and cold pain-related hypersensitivity. NRG1β was administered intrathecally (0.4 or 4 ng given daily for 3 d). a, b, Dorsal horn of animals treated with saline (control in a) or NRG1 (4 ng in b) 3 d after the first injection, immunostained with Iba1. Note the increase in the numbers of microglia with an activated morphology. c, Shows quantification of this response at 1 and 3 d after NRG1 injections. *p < 0.05, **p < 0.001 (comparing NRG1 doses vs control). d–l, We assessed proliferation (pulse labeling with BrdU) after NRG1 injections (Iba1 is shown in red, BrdU in green, DAPI in blue, and in the last panel merged images are shown). d–g, Dorsal horn microglia from a saline-treated animal (note that no BrdU is present). h–k, Dorsal horn microglia from a NRG1 (4 ng)-treated animal. l, Quantification of all BrdU-positive microglia in the dorsal horn of all groups at days 1 and 3 after injections were started. *p < 0.05 (comparing 4 ng vs control). Mechanical (m) and cold (n) pain-related hypersensitivity developed after NRG1 injections in a dose-dependent manner. *p < 0.05, **p < 0.001 for 4 ng vs control; #p < 0.05 for 0.4 ng versus control. Scale bars: (in b) a, b: 100 μm; (in k) d–k: 10 μm.
ErbB2 receptor inhibition or sequestration of endogenous NRG reduces microgliosis after peripheral nerve injury
We next explored the role of NRG1-erbB signaling in microglia in a nerve injury model [L5 spinal nerve ligation (Kim and Chung, 1992)], which results in a robust microgliosis. To visualize the location of erbB2 in its activated state, a phosphorylation state-specific antibody was used (Guertin et al., 2005). After L5 SNL, a significant increase in expression of the phosphorylated (i.e., activated) form of erbB2 receptor was seen in the ipsilateral dorsal horn of the spinal cord (Fig. 6a). No immunostaining was observed after preincubation of the antibody with the peptide used as the immunogen. Phospho-erbB2 expression was entirely restricted to microglia; phospho-erbB2 colocalized with the microglial markers Iba-1 (ionized calcium-binding adapter molecule 1) and OX-42 (which recognizes complement receptor type 3). Of the 518 phospo-erbB2-positive cells counted, 99% coexpressed Iba-1. There was no colocalization with either the neuronal marker neuronal-specific nuclear protein (NeuN) or the astrocyte marker glial fibrillary acidic protein (GFAP) (Fig. 6b-g). The time course of phosphorylated erbB2 (p-erbB2) expression, which was maximal at days 3–7, closely mirrored the temporal profile of the development of microgliosis (Fig. 7d–h). Using Western blot analysis, we saw a significant increase in erbB3 receptor expression in the dorsal horn after SNL. No change was observed in erbB2 and erbB4 expression (p = 0.02, naive vs day 3 after SNL for erbB3; p = 0.34 and p = 1, respectively, for erbB2 and erbB4; Mann–Whitney rank sum test, n = 4) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 6.

Peripheral nerve injury leads to increased phospho-erbB2 expression within dorsal horn microglia. a, Three days after L5 SNL, the expression of the phosphorylated form of the erbB2 receptor was increased in the ipsilateral dorsal horn. b–d, Phospho-erbB2 immunostaining (green) colocalized with Iba1 (microglial marker in red, b), but not with GFAP (astrocyte marker in red, c) or NeuN (neuronal marker in red, d). e, f, P-erbB2 (green, e) also colocalized with OX-42 (a marker of activated microglia; red, f). Scale bars: a, 200 μm; b–d, 50 μm; e–g, 10 μm.
The principal isoforms of NRG1 expressed within the nervous system are those with a β EGF domain (Meyer and Birchmeier, 1994; Shinoda et al., 1997). To identify cells expressing NRG1 in the adult, we used an oligonucleotide probe that recognizes a sequence common to all β isoforms (Fricker et al., 2009). In dorsal root ganglia, as previously noted, β EGF mRNA is expressed by both small-diameter (identified through the expression of the neuropeptide CGRP or binding of isolectin B4) and large-diameter [identified by the expression of phosphorylated neurofilament heavy chain (NF200)] DRG cells. Expression was highest in large-diameter DRG cells, which is consistent with the high level of expression of the type III β NRG isoform in these cells. In the naive situation, 67 ± 3.7% of DRG cells expressed β EGF mRNA, and at 3 d after SNL (when microgliosis is well established) 62 ± 1% of DRG cells expressed β EGF mRNA (Fig. 8a–f). This probe demonstrated neuronal expression within the spinal cord, particularly in motoneurons and occasionally in neurons in the deeper laminae of the dorsal horn (data not shown). We did not see expression in astrocytes either in the naive state or after injury. The most likely source of NRG within the dorsal horn is therefore likely to be primary afferent terminals.
Figure 8.

NRG1 expression after L5 spinal nerve ligation. a–f, In situ hybridization images using a probe directed against the βEGF domain of NRG1 in L5 DRG of naive animals (a–c) and 3 d after SNL (d–f). Expression is highest in large-diameter DRG cells positive for NF 200 (green) but is also observed in small DRG cells positive for either IB4 (blue) or CGRP (red). Arrows provide illustrative examples of double labeled cells. There was no significant change in the proportion of DRG cells expressing EGF mRNA at 3 d after SNL. g–k, IHC images. g, h, We used the H210 antibody, which is raised against the N-terminal extracellular domain of NRG1. To confirm specificity, we tested it in Nrg1 knock-out tissue (Nfh Cre Nrg1f1/f1 mice in which Nrg1 is ablated in sensory neurons in the late embryonic period). Note that normal NRG1 immunoreactivity is observed in DRG cells of Nrgf1/f1 mice (i.e., control), whereas no immunoreactivity is present DRG cells of Nfh Cre Nrg1f1/f1 mice. i–k, Immunohistochemistry confirmed that NRG1 was present in ∼60% of DRG cells (i, j) and within the dorsal horn of the spinal cord (especially within superficial laminae, k). l, NRG1 expression was also assessed using a NRG1β-specific ELISA. Lysate from dorsal horn spinal cord of naive, and 1 and 3 d after SNL animals were run (50 μg of protein for each sample). All sample values were within the range of the ELISA detection. A significant increase in NRG1 was detected at 3 d after SNL (p = 0.04, one-way ANOVA, Bonferroni post hoc test, n = 3). Scale bars: f, h, j, 50 μm; k, 200 μm.
The H 210 antibody that recognizes the N terminus of NRG1 was used to localize NRG1 immunoreactivity. To confirm antibody specificity in tissue from mice with conditional Nrg1, gene ablation in sensory neurons was used [mice expressing Cre under control elements of the neurofilament heavy chain gene Nfh Cre (Mallucci et al., 2002) were crossed with mice with loxp sites flanking the essential EGF domain of Nrg1 (Nrgf1/f1) (Yang et al., 2001)]. In Nfh Cre, Nrg1f1/f1 mice Nrg1 is ablated in sensory neurons in the late embryonic period, resulting in a severe dysmyelinating neuropathy and shortened survival (D.L.H. Bennett, unpublished observations). Note that normal NRG1 immunoreactivity is observed in DRG cells of Nrgf1/f1 (i.e., control) animals whereas no immunoreactivity is present DRG cells of Nfh Cre, Nrg1f1/f1 animals (Fig. 8g–h). NRG1 immunoreactivity was observed within DRG cells of all cell sizes (in naive situation: 61.9 ± 1.2% of all cells were positive; at 3 d after SNL: 58.4 ± 2.1% of all cells were positive) (Fig. 8i–j). NRG1 immunoreactivity was also seen within the dorsal horn of the spinal cord (especially within superficial laminae) (Fig. 8k), and this increased at 3 d after SNL (ipsilateral to lesion). An ELISA that recognizes NRG1 isoforms containing β EGF and IgG domains demonstrated a significant increase in the level of this protein within the dorsal horn at 3 d after SNL (Fig. 8l) (p = 0.04, one-way ANOVA Bonferroni post hoc test, n = 3 per group).
To test whether activation of the erbB2 receptor contributes to microgliosis in vivo, we inhibited it using PD168393, the erbB2 receptor-blocking antibodies mAb7.16.4 (discussed above), or mAb 9 (B10), which causes erbB2 homodimerization without activation of downstream signaling pathways and accelerates receptor endocytosis (Sawyer et al., 2002; Pugatsch et al., 2006). At day 3 after SNL, when the microglial response is well established, we found that blocking the erbB2 receptor could dose-dependently reduce the number of microglia within the ipsilateral dorsal horn with an activated morphology [using PD168393, 5 μg/d, i.t. infusion (p = 0.002); mAb 7.16.4, 5 μg, i.t. injection (p < 0.001); or mAb 9, 5 μg, i.t. injection (p < 0.001), compared with control, one-way ANOVA, n = 3–4 per group) (Fig. 9a–c) (supplemental Fig. 1d, available at www.jneurosci.org as supplemental material). The number of microglia within the dorsal horn that were phospho-p38 MAPK-positive was also significantly reduced after erbB2 inhibition (p < 0.001, after treatment with PD168393, 5 μg/d, i.t. infusion; mAb 7.16.4, 5 μg, i.t. injection; or mAb 9, 5 μg, i.t. injection, compared with control, one-way ANOVA, n = 3–4 per group) (Fig. 9f–j).
Figure 9.
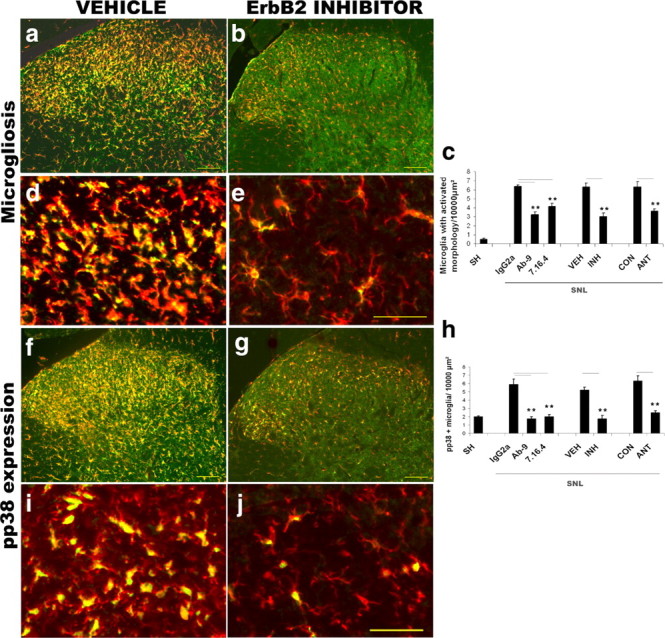
ErbB2 receptor blockade or sequestration of endogenous NRG inhibits the development of microgliosis after spinal nerve ligation. The effect of erbB2 receptor blockers (PD168393, 5 μg/d, i.t.; or mAb 9 or 7.16.4, 5 μg, i.t.) or a NRG-sequestering molecule (HBD-S-H4, 3 μg, i.t.) was determined at 3 d after SNL. a–e, ErbB2 receptor blockade (b and e, low- and high-power photomicrographs, respectively) resulted in a reduction in both p-erbB2 (green) expression and in the number of microglia in the dorsal horn (Iba1 immunostaining, red) compared with vehicle (a and d, low- and high-power photomicrographs, respectively). Quantification (c) demonstrates that SNL results in an increased number of microglia with activated morphology compared with sham surgery animals. This increase was significantly attenuated after treatment with erbB2 inhibitor, erbB2 receptor-blocking antibodies, or the NRG1 antagonist (HBD-S-H4) with respect to control. f–j, SNL results in increased numbers of microglia expressing phospho-p38. The number of microglia (red) expressing p-p38 MAPK (green) after SNL was significantly reduced after erbB2 receptor blockade or sequestration of NRG using HBD-S-H4. h, Quantification. Error bars represent ± SEM (n = 3–4 per group). SH, Sham; VH, vehicle; INH, inhibitor (PD168393); IgG2a, nonimmune IgG (control); ANT, antagonist (HBD-S-H4). Statistical test: t test (vehicle vs inhibitor and control vs antagonist) or one-way ANOVA, Bonferroni post hoc test (IgG2a vs erbB2 receptor-blocking antibodies).**p < 0.005. Scale bars: a, b, f, g, 100 μm; d, e, i, j, 50 μm.
To sequester endogenous NRG, we used a fusion protein (HBD-S-H4) consisting of the soluble ectodomain of erbB4, which has high affinity for NRG, and a heparin-binding domain, which helps target the molecule to the same heparan sulfate-rich cell surfaces that bind NRG. This has been shown to be highly effective in vitro and blocks the survival-promoting activity of NRG on Schwann cells in vivo (Ma et al., 2009). Intrathecal administration of HBD-S-H4 (3 μg, i.t. injection) significantly reduced both the number of microglia with an activated morphology within the dorsal horn and phospho-p38 expression after SNL (in both cases, p < 0.01, after treatment with HBD-S-H4 vs control, unpaired t test, n = 3–4 per group) (Fig. 9).
Pulse labeling with BrdU showed a marked increase in the rate of cell proliferation within the dorsal horn of the spinal cord after SNL and, as previously reported (Echeverry et al., 2008), the vast majority of these cells (94.6 ± 1.2%) were microglia. The number of proliferating microglia within the dorsal horn was reduced by ∼60–70% after blockade of erbB2 [p < 0.001, after treatment with PD168393 (5 μg/d, i.t. infusion), mAb 7.16.4 (5 μg/d, i.t. infusion), or mAb 9 (5 μg/d, i.t. infusion) compared with control, one-way ANOVA, n = 3–4 per group] (Fig. 10).
Figure 10.

Microglial proliferation after spinal nerve ligation is significantly reduced after erbB2 receptor inhibition. a–h, Three days after L5 SNL, a proliferative response is seen in microglia within the dorsal horn, as shown here by labeling newly dividing cells with BrdU (yellow) and microglia with Iba1 (red). DAPI is shown in blue to delineate nuclei. The BrdU-labeled nuclei are almost exclusively within microglial cells (as seen in the merge image). Blocking the erbB2 receptor with PD168393 (5 μg/d, i.t.) or with the blocking antibodies (mAb 9 or 7.16.4, 5 μg, i.t.) resulted in a significant reduction in microglial proliferation (quantification in i). Scale bars: 100 μm. Error bars represent ± SEM (n = 3–4 per group). SH, Sham; VH, vehicle; INH, inhibitor (PD168393); IgG2a, nonimmune IgG (control). Statistical test: one-way ANOVA, Bonferroni post hoc test. **p < 0.005.
Inhibition of erbB2 significantly reduced the number of microglia with activated morphology, the expression of phospho-p38 MAPK, and microglial proliferation at a later time point after nerve injury, i.e., 7 d after SNL (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). In a further set of experiments, we assessed dorsal horn microgliosis at 7 d after pump emptying (i.e., 21 d after SNL). We found no significant difference in dorsal horn microgliosis at this time point comparing intrathecal treatment with PD168393 and control (p = 0.57, t test, n = 3–4) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Neuregulin is known to have a key role in signaling between axons and Schwann cells in peripheral nervous system (Chen et al., 2003; Nave and Salzer, 2006; Fricker et al., 2009). There is therefore the theoretical possibility that these changes could be secondary to changes in axoglial signaling in the periphery if the inhibitor or antibody were to spread outside of the intrathecal space. To exclude this possibility, we administered mAb 7.16.4 directly to the site of spinal nerve ligation and found that in contrast to intrathecal treatment this peripheral administration had no effect on dorsal horn microgliosis (p = 0.18, t test, n = 3–4) (supplemental Fig. 6, available at www.jneurosci.org as supplemental material).
A week after SNL, we observed a significant increase in astrocytes within the ipsilateral spinal cord (p = 0.004, t test, n = 4), which is in accordance with previous reports of a delayed astrocytosis after nerve injury (Colburn et al., 1999). However, unlike the microgliosis this was not reduced by erbB2 inhibition (p = 0.88, t test, n = 4) (supplemental Fig. 7, available at www.jneurosci.org as supplemental material).
ErbB2 receptor inhibition or sequestration of endogenous NRG reduces mechanical and cold pain-related hypersensitivity after nerve injury
There is increasing evidence that the microglial activation observed within the dorsal horn of the spinal cord after nerve injury contributes to the development of neuropathic pain (Tsuda et al., 2003; Milligan and Watkins, 2009). Intrathecal infusion of PD168393 over 7 d had no effect on mechanical withdrawal thresholds in naive animals (p = 0.25, RM two-way ANOVA, PD168393, 5 μg/d, vs vehicle; data not shown). We therefore monitored pain-related behavior over 14 d after L5 SNL in animals that were treated with the erbB2 inhibitor or vehicle delivered via continuous intrathecal infusion. In both groups of animals, a clear mechanical hypersensitivity was observed at day 1 after SNL; however, at subsequent time points erbB2 receptor inhibition resulted in a significant dose-dependent reduction in mechanical pain hypersensitivity on the side ipsilateral to the lesion (Fig. 11a) (p < 0.05, RM two-way ANOVA, PD168393, 10 or 5 μg/d, vs vehicle, n = 7–11/group) with no effect on the contralateral paw (data not shown). Once the pump had emptied, the mechanical withdrawal thresholds of the group receiving PD168393, 5 μg/d, returned to those of control (Fig. 11b). Delayed treatment with PD168393, 5 μg/d, from day 3 onward (by which time microgliosis is well established) was not effective at reversing mechanical pain-related hypersensitivity (Fig. 11c) (p = 0.75, RM two-way ANOVA, n = 7 per group). Cold allodynia (as assessed by nociceptive behavior in response to acetone application to the paw) also developed after SNL and was significantly attenuated in a dose-dependent fashion by erbB2 inhibition (Fig. 11d) (p < 0.05 PD168393, 10 or 5 μg/d, vs vehicle, RM two-way ANOVA, n = 7–11/group). Intrathecal injection of HBD-S-H4 (two 3 μg intrathecal injections administered on the day of surgery and on day 3 after surgery), which sequesters endogenous NRG, also significantly attenuated mechanical pain-related hypersensitivity (Fig. 11e) (p < 0.001 vs control, RM two-way ANOVA, n = 8 per group) and cold allodynia (Fig. 11f) (p < 0.05 vs control, RM two-way ANOVA, n = 8 per group).
Figure 11.

ErbB2 receptor inhibition or sequestration of endogenous NRG reduced mechanical pain hypersensitivity and cold allodynia after spinal nerve ligation. Animals underwent L5 SNL and received a continuous intrathecal infusion of the erbB2 inhibitor (PD168393, 1.25–10 μg/d) or vehicle for 14 d. a, Mechanical hypersensitivity developed at day 1 after SNL in both groups and after day 2 was significantly attenuated in animals receiving the erbB2 inhibitor in a dose-dependent fashion. *p < 0.05, **p < 0.001, for 10 μg/d vs vehicle; #p < 0.05 for 5 μg/d vs vehicle; n = 7–11/group. b, Once the pump had emptied, the withdrawal thresholds of the group receiving the erbB2 inhibitor (5 μg/d) returned to those of control. c, Delayed treatment with the erbB2 inhibitor (5 μg/d) from day 3 onward (by which time microgliosis is well established) was not effective at reversing mechanical pain-related hypersensitivity (p = 0.75) d, Cold allodynia was also significantly reduced in a dose-dependent fashion by inhibiting the erbB2 receptor. **p < 0.001 for 10 μg/d vs vehicle; #p < 0.05 for 5 μg/d vs vehicle, n = 7–11/group. e, f, Intrathecal administration of HBD-S-H4 (3 μg) at days 0 and 4 after SNL (shown by arrows) significantly reduced mechanical (p < 0.001) (e) and cold (p < 0.05) (f) pain-related hypersensitivity. Error bars represent ± SEM. Statistical tests: two-way ANOVA, Bonferroni test, or Fischer LSD post hoc analysis. When the assumptions of sphericity were violated (Mauchly's test; p < 0.05), the Greenhouse–Geisser correction was applied and independent two-tailed t tests were used to determine differences between groups. INH, Inhibitor (PD168393); NRG ANT, NRG antagonist (HBD-S-H4). The lines in a–d denote the period of pump infusion. The arrows in e and f denote the days of intrathecal injections.
Discussion
Microglia closely resemble tissue macrophages and are key sensors of injury to the CNS produced by many diverse pathologies. NRG1 has not previously been implicated in regulating microglial function. Here, we demonstrate that NRG1 is a survival, proliferative, and chemotactic factor for microglia in vitro, and that NRG1-erbB signaling is activated specifically within microglia after peripheral nerve injury, contributing to the development of microgliosis and consequently neuropathic pain.
Microglia express the NRG1 receptors erbB2, 3, and 4. This factor promotes the survival of microglia in vitro through inhibition of apoptosis and is a potent proliferative factor for these cells in an erbB2-dependent manner. In addition to proliferation, the other means by which microglia can accumulate at the site of injury is through directed migration in response to the release of chemotactic agents, which include the following: purines (Honda et al., 2001), complement components (Yao et al., 1990), bradykinin (Ifuku et al., 2007), and chemokines (Peterson et al., 1997). We have used the Boyden chamber chemotaxis assay and by manipulating the concentration gradient in the upper and lower wells have shown that NRG1 is a chemotactic agent for microglia acting in an erbB2-dependent manner. Neuregulin has previously been shown to increase the motility of a number of different cell types including malignant cell lines (Hijazi et al., 2000), keratinocytes (Schelfhout et al., 2002), and Schwann cells (Lyons et al., 2005). NRG1 could also enhance the release of interleukin-1β from LPS-primed microglia. Unlike LPS, however, NRG1 application did not promote the development of amoeboid morphology or MHC class II expression in microglia. We have found that macrophages express erbB2, 3, and 4 receptors. Therefore, in situations where there is major disruption of the blood–brain barrier (e.g., traumatic brain injury) resulting in macrophage infiltration into the CNS the function of macrophages may also be modulated by NRG1.
Because of the potent effects we observed of NRG1 on cultured microglia, we explored the relevance of this signaling pathway in vivo. Intrathecal administration of NRG1 resulted in a dorsal horn microgliosis and also produced mechanical and cold pain-related hypersensitivity [in agreement with a previous study by Lacroix-Fralish et al. (2008)]. Spinal nerve ligation provides a well characterized model of nerve injury associated with a robust microgliosis (Tsuda et al., 2003; Scholz and Woolf, 2007) and the development of neuropathic pain (Kim and Chung, 1992). We used an antibody that recognizes phosphorylated erbB2 as a measure of receptor activation (Guertin et al., 2005). In naive and sham surgery animals, there was only a low level of p-erbB2 expression in resting microglia. After SNL, p-erbB2 expression increased, and it was localized specifically within microglia, especially those with an activated morphology. The time course of p-erbB2 expression within the dorsal horn was coincident with the development of microgliosis being detectable at day1 after injury and peaking at day 3–7 after SNL.
For NRG1 to act as a proliferative and chemotactic agent for microglia, it needs to be released at the site of injury. All NRG1 isoforms contain an EGF domain, which is required for biological activity and can be classified into subgroups according to the structure of their N termini. Type I and II isoforms have Ig-like domains and either can be directly secreted or alternatively transmembrane isoforms can undergo metalloprotease-mediated cleavage to be released as soluble proteins from the cell surface. These isoforms bind avidly to the extracellular matrix, which can act as a source of localized release (Loeb and Fischbach, 1995). Type III isoforms possess a cysteine-rich domain and undergo proteolytic cleavage by β-secretase enzyme (Willem et al., 2006); however, the EGF domain remains membrane tethered, and so this molecule signals in a juxtacrine fashion. DRG cells have been reported to principally express type I and III isoforms (Bermingham-McDonogh et al., 1997). We found clear expression of NRG1 by small- and large-diameter DRG cells using in situ hybridization and a probe that recognizes all β EGF isoforms (the nervous system principally expresses β rather than α isoforms). β EGF mRNA was expressed at low levels by occasional dorsal horn neurons, and so we propose that the most likely source of NRG1 within the dorsal horn is from primary afferent terminals. We did not observe a change in β EGF mRNA expression in DRG cells or the dorsal horn at 3 d after SNL, a time point when microgliosis is well established. We did find significantly increased β EGF containing NRG1 protein within the dorsal horn using an ELISA after SNL. Cultured neurons have been shown to release soluble NRG1 in an activity-dependent manner (Ozaki et al., 2004), and NRG1 activity has also been detected in human CSF (Pankonin et al., 2009). In culture systems, NRG1 activity has also been shown to be released from DRG cell axons (Esper and Loeb, 2004; Taveggia et al., 2005); this release is enhanced by BDNF [which is itself released by microglia after nerve injury (Coull et al., 2005)]. The rapid response of microglia after SNL suggests that increased NRG1 activity within the dorsal horn after nerve injury is due to the following: altered NRG1 protein trafficking or release from primary afferent terminals, or, possibly, release from the extracellular matrix. We suggest that such NRG1 release provides an injury-related signal from primary afferents triggering a microglial response within the dorsal horn.
We tested the hypothesis that inhibition of NRG-erbB signaling would reduce the microgliosis produced by SNL. We used two strategies for erbB2 receptor inhibition: intrathecal treatment with either an irreversible specific erbB inhibitor (PD168393), which blocks the ATP binding site of the receptor or monoclonal antibodies (mAb 7.16.4 and mAb 9). mAb 7.16.4 specifically binds to the extracellular domain of erbB2, downregulates surface expression of erbB2, and blocks activation of erbB2/3 and erbB2/4 heterodimers; this antibody has previously been shown to have efficacy in vivo (Katsumata et al., 1995; Yarden and Sliwkowski, 2001). Inhibition of the erbB2 receptor after nerve injury resulted in a significant reduction in the microgliosis within the dorsal horn as well as reducing the expression of phospho-p38 (a component of the MAPK signaling cascade), which is specifically expressed within activated microglia (Jin et al., 2003; Kobayashi et al., 2008). This effect on p38 signaling is likely to be indirect as NRG1 did not stimulate the p38 MAPK pathway in cultured microglia, although it does activate the extracellular signal-regulated kinase kinase–extracellular signal-regulated kinase (MEK–ERK) and phosphatidylinositol-3 kinase–Akt pathways (M. Calvo, N. Zhu, and D.L.H. Bennett, unpublished observations). Interestingly, ERK previously has been shown to be activated in microglia after peripheral nerve injury (Zhuang et al., 2005). We have also used a molecule (HBD-S-H4) to sequester endogenous NRG1. This is a fusion protein consisting of the soluble ectodomain of erbB4, which has high affinity for NRG1, and a heparin-binding domain, which helps target the molecule to the same heparan-sulfate rich cell surfaces that bind NRG1. This molecule could also effectively reduce dorsal horn microgliosis and pp38 expression after SNL.
Given the lack of major disruption to the blood–brain barrier after SNL (Abram et al., 2006; Lu et al., 2009), proliferation of resident microglia is likely to be the principal means by which microglial numbers increase after SNL. We found a clear increase in microglial cell proliferation within the dorsal horn in the first 3 d after peripheral nerve injury. ErbB2 receptor blockade significantly reduced this proliferative response. Inhibition of microglial proliferation provides one explanation for the reduction in microgliosis observed after erbB2 receptor inhibition, although impaired chemotaxis of local microglia may also play a role. ErbB2 inhibition specifically reduced microgliosis after nerve injury and did not prevent the development of astrocytosis.
There is now increasing evidence that glial cells (both microglia and astrocytes) play an important role in the generation of neuropathic pain. A number of molecules have been implicated in the recruitment and activation of microglia within the dorsal horn after nerve injury including cytokines, chemokines (such as CCL2 and Fractalkine), complement components, and purines (Jin et al., 2003; Tsuda et al., 2003; Clark et al., 2007; Scholz and Woolf, 2007; Zhang et al., 2007; Milligan and Watkins, 2009; Thacker et al., 2009). After activation, microglia release cytokines and BDNF, which enhance the excitability of dorsal horn neurons (Coull et al., 2005; Kawasaki et al., 2008). Inhibition of the microglial response to injury has been shown to effectively reduce the development of neuropathic pain (Tsuda et al., 2003; Coull et al., 2005; Ledeboer et al., 2005; Clark et al., 2007). ErbB2 receptor inhibition or sequestration of endogenous NRG1 could ameliorate (but not completely reverse) both mechanical and cold pain-related hypersensitivity after SNL.
After peripheral nerve injury, the proinflammatory microglial response is clearly deleterious as it contributes to neuropathic pain; however, in other contexts there is evidence that these cells may be neuroprotective (Hanisch and Kettenmann, 2007). NRG-erbB signaling represents a novel pathway stimulating microglial proliferation and chemotaxis. We have shown that this contributes to the development of neuropathic pain, and it will be of great interest in the future to determine the role of NRG-erbB signaling in mediating the microglial response to other forms of CNS injury.
Footnotes
D.B. is a Wellcome Trust Clinical Scientist and a member of the London Pain Consortium. M.C. is sponsored by the Chilean National Scholarship Program for Graduate Studies. C.T. is a student of the Wellcome Trust-funded London Pain Consortium. J.L. is supported by National Multiple Sclerosis Society Grant RG 3410B3. We thank Genentech for provision of NRG1. We thank M. Malcangio and S.B. McMahon for helpful advice. We thank G. Mallucci (MRC Toxicology Unit, Leicester University, Leicester, UK) for provision of Nfh Cre mice and C. Birchmeier (Max Delbrück Center for Molecular Medicine, Berlin, Germany) for provision of Nrg1f1/f1 mice. D.B. conceived of and coordinated this program of work. M.C. and N.Z. designed experiments, and performed animal surgery, microglial cultures, and histological analysis. J.G. and M.C. performed behavioral testing. C.T. performed ISH. Z.M. and J.A.L. developed and provided information on the use of the NRG antagonist. M.C. and D.B. performed data analysis and wrote the manuscript. All authors discussed the results.
References
- Abram SE, Yi J, Fuchs A, Hogan QH. Permeability of injured and intact peripheral nerves and dorsal root ganglia. Anesthesiology. 2006;105:146–153. doi: 10.1097/00000542-200607000-00024. [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Bermingham-McDonogh O, Xu YT, Marchionni MA, Scherer SS. Neuregulin expression in PNS neurons: isoforms and regulation by target interactions. Mol Cell Neurosci. 1997;10:184–195. doi: 10.1006/mcne.1997.0654. [DOI] [PubMed] [Google Scholar]
- Bose R, Molina H, Patterson AS, Bitok JK, Periaswamy B, Bader JS, Pandey A, Cole PA. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc Natl Acad Sci U S A. 2006;103:9773–9778. doi: 10.1073/pnas.0603948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraway KL, 3rd, Cantley LC. A neu acquaintance for erbB3 and erbB4: a role for receptor heterodimerization in growth signaling. Cell. 1994;78:5–8. doi: 10.1016/0092-8674(94)90564-9. [DOI] [PubMed] [Google Scholar]
- Chen S, Rio C, Ji RR, Dikkes P, Coggeshall RE, Woolf CJ, Corfas G. Disruption of ErbB receptor signaling in adult non-myelinating Schwann cells causes progressive sensory loss. Nat Neurosci. 2003;6:1186–1193. doi: 10.1038/nn1139. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Dimayuga FO, Ding Q, Keller JN, Marchionni MA, Seroogy KB, Bruce-Keller AJ. The neuregulin GGF2 attenuates free radical release from activated microglial cells. J Neuroimmunol. 2003;136:67–74. doi: 10.1016/s0165-5728(03)00003-1. [DOI] [PubMed] [Google Scholar]
- Echeverry S, Shi XQ, Zhang J. Characterization of cell proliferation in rat spinal cord following peripheral nerve injury and the relationship with neuropathic pain. Pain. 2008;135:37–47. doi: 10.1016/j.pain.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Esper RM, Loeb JA. Rapid axoglial signaling mediated by neuregulin and neurotrophic factors. J Neurosci. 2004;24:6218–6227. doi: 10.1523/JNEUROSCI.1692-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esper RM, Pankonin MS, Loeb JA. Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Rev. 2006;51:161–175. doi: 10.1016/j.brainresrev.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Falls DL. Neuregulins: functions, forms, and signaling strategies. Exp Cell Res. 2003;284:14–30. doi: 10.1016/s0014-4827(02)00102-7. [DOI] [PubMed] [Google Scholar]
- Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol. 2001;166:2717–2726. doi: 10.4049/jimmunol.166.4.2717. [DOI] [PubMed] [Google Scholar]
- Fricker FR, Zhu N, Tsantoulas C, Abrahamsen B, Nassar MA, Thakur M, Garratt AN, Birchmeier C, McMahon SB, Wood JN, Bennett DL. Sensory axon-derived neuregulin-1 is required for axoglial signaling and normal sensory function but not for long-term axon maintenance. J Neurosci. 2009;29:7667–7678. doi: 10.1523/JNEUROSCI.6053-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerecke KM, Wyss JM, Karavanova I, Buonanno A, Carroll SL. ErbB transmembrane tyrosine kinase receptors are differentially expressed throughout the adult rat central nervous system. J Comp Neurol. 2001;433:86–100. doi: 10.1002/cne.1127. [DOI] [PubMed] [Google Scholar]
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Ingeman JE. Colony-stimulating factors as promoters of amoeboid microglia. J Neurosci. 1988;8:4707–4717. doi: 10.1523/JNEUROSCI.08-12-04707.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin AD, Zhang DP, Mak KS, Alberta JA, Kim HA. Microanatomy of axon/glial signaling during Wallerian degeneration. J Neurosci. 2005;25:3478–3487. doi: 10.1523/JNEUROSCI.3766-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Hijazi MM, Thompson EW, Tang C, Coopman P, Torri JA, Yang D, Mueller SC, Lupu R. Heregulin regulates the actin cytoskeleton and promotes invasive properties in breast cancer cell lines. Int J Oncol. 2000;17:629–641. doi: 10.3892/ijo.17.4.629. [DOI] [PubMed] [Google Scholar]
- Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K, Kohsaka S. Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci. 2001;21:1975–1982. doi: 10.1523/JNEUROSCI.21-06-01975.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifuku M, Färber K, Okuno Y, Yamakawa Y, Miyamoto T, Nolte C, Merrino VF, Kita S, Iwamoto T, Komuro I, Wang B, Cheung G, Ishikawa E, Ooboshi H, Bader M, Wada K, Kettenmann H, Noda M. Bradykinin-induced microglial migration mediated by B1-bradykinin receptors depends on Ca2+ influx via reverse-mode activity of the Na+/Ca2+ exchanger. J Neurosci. 2007;27:13065–13073. doi: 10.1523/JNEUROSCI.3467-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumata M, Okudaira T, Samanta A, Clark DP, Drebin JA, Jolicoeur P, Greene MI. Prevention of breast tumour development in vivo by downregulation of the p185neu receptor. Nat Med. 1995;1:644–648. doi: 10.1038/nm0795-644. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Yamanaka H, Fukuoka T, Dai Y, Obata K, Noguchi K. P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci. 2008;28:2892–2902. doi: 10.1523/JNEUROSCI.5589-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontinen VK, Dickenson AH. Effects of midazolam in the spinal nerve ligation model of neuropathic pain in rats. Pain. 2000;85:425–431. doi: 10.1016/S0304-3959(99)00298-5. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Kimura Y, Yoshioka Y, Wakamatsu D, Kozaki R, Hashimoto H, Matsuda T, Baba A. Serum-deprivation induces cell death of rat cultured microglia accompanied with expression of Bax protein. Jpn J Pharmacol. 2000;83:351–354. doi: 10.1254/jjp.83.351. [DOI] [PubMed] [Google Scholar]
- Lacroix-Fralish ML, Tawfik VL, Nutile-McMenemy N, Deleo JA. Neuregulin 1 is a pronociceptive cytokine that is regulated by progesterone in the spinal cord: implications for sex specific pain modulation. Eur J Pain. 2008;12:94–103. doi: 10.1016/j.ejpain.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995;378:394–398. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, Volpe JJ, Vartanian T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Fischbach GD. ARIA can be released from extracellular matrix through cleavage of a heparin-binding domain. J Cell Biol. 1995;130:127–135. doi: 10.1083/jcb.130.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Gonzales C, Chen Y, Adedoyin A, Hummel M, Kennedy JD, Whiteside GT. CNS penetration of small molecules following local inflammation, widespread systemic inflammation or direct injury to the nervous system. Life Sci. 2009;85:450–456. doi: 10.1016/j.lfs.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Lyons DA, Pogoda HM, Voas MG, Woods IG, Diamond B, Nix R, Arana N, Jacobs J, Talbot WS. erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr Biol. 2005;15:513–524. doi: 10.1016/j.cub.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Ma Z, Li Q, An H, Pankonin MS, Wang J, Loeb JA. Targeting human epidermal growth factor receptor signaling with neuregulin's heparin-binding domain. J Biol Chem. 2009;284:32108–32115. doi: 10.1074/jbc.M109.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci GR, Ratté S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002;21:202–210. doi: 10.1093/emboj/21.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet Y, Martinet N, Vignaud JM, Plenat F. Blood monocyte chemotaxis. J Immunol Methods. 1994;174:209–214. doi: 10.1016/0022-1759(94)90024-8. [DOI] [PubMed] [Google Scholar]
- Mei L, Xiong WC. Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci. 2008;9:437–452. doi: 10.1038/nrn2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C. Distinct isoforms of neuregulin are expressed in mesenchymal and neuronal cells during mouse development. Proc Natl Acad Sci U S A. 1994;91:1064–1068. doi: 10.1073/pnas.91.3.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, Salzer JL. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol. 2006;16:492–500. doi: 10.1016/j.conb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaki M, Itoh K, Miyakawa Y, Kishida H, Hashikawa T. Protein processing and releases of neuregulin-1 are regulated in an activity-dependent manner. J Neurochem. 2004;91:176–188. doi: 10.1111/j.1471-4159.2004.02719.x. [DOI] [PubMed] [Google Scholar]
- Pankonin MS, Sohi J, Kamholz J, Loeb JA. Differential distribution of neuregulin in human brain and spinal fluid. Brain Res. 2009;1258:1–11. doi: 10.1016/j.brainres.2008.12.047. [DOI] [PubMed] [Google Scholar]
- Peterson PK, Hu S, Salak-Johnson J, Molitor TW, Chao CC. Differential production of and migratory response to beta chemokines by human microglia and astrocytes. J Infect Dis. 1997;175:478–481. doi: 10.1093/infdis/175.2.478. [DOI] [PubMed] [Google Scholar]
- Pugatsch T, Abedat S, Lotan C, Beeri R. Anti-erbB2 treatment induces cardiotoxicity by interfering with cell survival pathways. Breast Cancer Res. 2006;8:R35. doi: 10.1186/bcr1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Sawyer DB, Zuppinger C, Miller TA, Eppenberger HM, Suter TM. Modulation of anthracycline-induced myofibrillar disarray in rat ventricular myocytes by neuregulin-1beta and anti-erbB2: potential mechanism for trastuzumab-induced cardiotoxicity. Circulation. 2002;105:1551–1554. doi: 10.1161/01.cir.0000013839.41224.1c. [DOI] [PubMed] [Google Scholar]
- Schelfhout VR, Coene ED, Delaey B, Waeytens AA, De Rycke L, Deleu M, De Potter CR. The role of heregulin-alpha as a motility factor and amphiregulin as a growth factor in wound healing. J Pathol. 2002;198:523–533. doi: 10.1002/path.1240. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Shinoda J, Nakao J, Iizuka Y, Toba Y, Yazaki T, Kawase T, Uyemura K. Multiple isoforms of neuregulin are expressed in developing rat dorsal root ganglia. J Neurosci Res. 1997;50:673–683. doi: 10.1002/(SICI)1097-4547(19971201)50:5<673::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Stence N, Waite M, Dailey ME. Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia. 2001;33:256–266. [PubMed] [Google Scholar]
- Tang X, Falls DL, Li X, Lane T, Luskin MB. Antigen-retrieval procedure for bromodeoxyuridine immunolabeling with concurrent labeling of nuclear DNA and antigens damaged by HCl pretreatment. J Neurosci. 2007;27:5837–5844. doi: 10.1523/JNEUROSCI.5048-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681–694. doi: 10.1016/j.neuron.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, Thompson SW, Marchand F, McMahon SB. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain. 2009;13:263–272. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 2001:399–410. doi: 10.1016/s0896-6273(01)00287-2. [DOI] [PubMed] [Google Scholar]
- Yao J, Harvath L, Gilbert DL, Colton CA. Chemotaxis by a CNS macrophage, the microglia. J Neurosci Res. 1990;27:36–42. doi: 10.1002/jnr.490270106. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Zhang H, Wang Q, Montone KT, Peavey JE, Drebin JA, Greene MI, Murali R. Shared antigenic epitopes and pathobiological functions of anti-p185(her2/neu) monoclonal antibodies. Exp Mol Pathol. 1999;67:15–25. doi: 10.1006/exmp.1999.2266. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]