

Abstract
This paper reports a new strategy, Recursive Directional Ligation by Plasmid Reconstruction (PRe-RDL), to rapidly clone highly repetitive polypeptides of any sequence and specified length over a large range of molecular weights. In a single cycle of PRe-RDL, two halves of a parent plasmid, each containing a copy of an oligomer, are ligated together, thereby dimerizing the oligomer and reconstituting a functional plasmid. This process is carried out recursively to assemble an oligomeric gene with the desired number of repeats. PRe-RDL has several unique features that stem from the use of type IIs restriction endonucleases: first, PRe-RDL is a seamless cloning method that leaves no extraneous nucleotides at the ligation junction. Because it uses type IIs endonucleases to ligate the two halves of the plasmid, PRe-RDL also addresses the major limitation of RDL in that it abolishes any restriction on the gene sequence that can be oligomerized. The reconstitution of a functional plasmid only upon successful ligation in PRe-RDL also addresses two other limitations of RDL: the significant background from self-ligation of the vector observed in RDL, and the decreased efficiency of ligation due to nonproductive circularization of the insert. PRe-RDL can also be used to assemble genes that encode different sequences in a predetermined order to encode block copolymers or append leader and trailer peptide sequences to the oligomerized gene.
Introduction
The ease with which synthetic genes that encode protein-based biopolymers can be assembled has greatly improved over the past decade, due to advances in molecular biology for the synthesis and assembly of genes that encode multiple repeats of peptide “monomers”. These advances have been driven by the utility of these biopolymers for a variety of applications including biomaterials 1, 2, tissue engineering scaffolds 3, 4, as materials for surface modification 5 and as drug delivery vehicles 6–8. The driving force for the use of artificial repetitive polypeptides in these applications stems from the ability of genetically encoded synthesis to control the polypeptide sequence, architecture, molecular weight (MW), and polydispersity, with a precision that is, as yet, unmatched by chemical polymerization.
As a result, a number of strategies to rapidly assemble synthetic genes that encode repetitive polypeptides have been developed, which include PCR cloning 9, 10, concatemerization 11, and seamless cloning 12–14. We previously developed a method known as Recursive Directional Ligation (RDL) 15 that utilizes stepwise, recursive addition of an oligomer with itself, or with another compatible DNA sequence by ligation of an insert with a linearized parent vector containing a copy of the same or compatible insert (Figure 1). RDL has proven to be very useful for the deterministic synthesis of genes that encode large biopolymers with a precisely specified molecular weight. It is also a useful method for the synthesis of genes that encode block copolymers, when the orientation, sequence, and molecular weight of the two blocks are critical to their function. Although this method for the assembly of synthetic genes for repetitive polypeptides has proven to be extraordinarily useful, its limitations have also became apparent to us over the last decade of extensive use 6, 7, 15–26. These limitations are: first, it is not generalizable, a priori, to biopolymers of any arbitrary sequence. Although our implementation of RDL exploited codon degeneracy to select endonucleases that would enable seamless cloning of elastin-like polypeptides (ELPs), the endonuclease recognition sequence in RDL overlaps the coding region. The type II restriction enzymes used in RDL place restrictions on the codons (and therefore the biopolymer sequence) that can be used in the gene (Figure 2B). Second, there is a significant background in RDL –clones lacking the insert– due to self-ligation of a vector or due to incomplete digestion of the vector. Third, the insert itself can self-ligate and circularize, which reduces cloning efficiency at each oligomerization step. Fourth, RDL requires many time-consuming cloning steps. For example, our implementation of RDL was designed for a pUC vector that is not suitable for expression, so that the final product –the gene oligomer of interest– had to be cloned into an expression plasmid.
Figure 1.

Recursive Directional Ligation. The strategy to double the length of an oligomer in a parent vector involves: (1) digesting the parent vector with both RE1 and RE2 to isolate the ELP insert; (2) digesting the parent vector with only RE1 to linearize the vector, which retains the insert; and (3) dimerizing the two moieties to double the length of the desired gene. The bold areas on the vector represent the endonuclease recognition sequences, which are contained within the sequence that encodes the repetitive polypeptide. The black arrows indicate the location of the cleavage site.
Figure 2.

(A) Recursive directional ligation by plasmid reconstruction (PRe-RDL). One round in PRe-RDL involves: (1) purifying the ELP-containing DNA fragment from the parent vector that is digested with AcuI and BglI; and (2) purifying the ELP-containing fragment from the parent vector that is digested with BseRI and BglI; and then (3) ligating the two compatible halves to reconstitute the original vector, and thereby while doubling the length of the insert. (B) Original RDL vector design reported by Meyer 15, with a representative pentamer sequence. The identity of the capitalized base pairs are specified by the recognition site of the restriction enzyme listed above those nucleotides. Note that the sequence of the restriction endonucleases required for RDL are contained within the DNA sequence that is oligomerized. The vertical arrows indicate the endonuclease restriction sites. (C) PRe-RDL vector, which utilizes the type IIs restriction enzymes, BseRI and AcuI, to eliminate sequence dependence upon the recognition sites. The recognition sequence for BseRI has been designed directly into the Shine-Delgarno ribosomal binding sequence (RBS; underlined), AGGAGGAG, which is required to initiate translation. The BseRI cleavage site (‘CC’ in this vector) is 8-bases downstream of its recognition site. The recognition site for AcuI, CTGAAG, is 14-bases downstream of its degenerate cleavage site, which is ‘GG’ in this vector. The vertical arrows indicate the endonuclease cleavage site on the sense strand. (D) BseRI and AcuI have 2-bp overhangs, which reduces intra-sequence dependence within the repeating unit to a single amino acid. (E) Basic design for a leader sequence to be inserted into a vector restricted with NdeI and BseRI.
To address these limitations of RDL, we introduce, herein, a significantly improved cloning methodology –Recursive Directional Ligation by Plasmid Reconstruction (PRe-RDL) – to rapidly clone repetitive polypeptides of any sequence and length. PRe-RDL is a modified form of Recursive Directional Ligation (RDL) that has new features that overcome the limitations of RDL.15 In PRe-RDL, two halves of a parent plasmid, each containing the desired oligomer, are ligated together, thereby dimerizing the oligomer and reconstituting a functional plasmid. Unlike RDL, PRe-RDL is applicable to any arbitrary DNA sequence (and hence any peptide sequence) and produces peptide oligomers with no extraneous peptides at the junction between repeats, because it uses type IIs endonucleases (Figure 2C). Unlike most other classes of endonucleases, type IIs restriction enzymes cut at a defined number of nucleotides away from their recognition sequence. Because the cleavage site is degenerate, it allows the selection of any single amino acid to join two repeats. When this amino acid is selected as the leading or trailing peptide of the repeated segment, the junctions become seamless. This feature of PRe-RDL hence addresses the inability to oligomerize any arbitrary peptide sequence using type II restriction endonucleases, which was a serious limitation of RDL. The reconstitution of a functional plasmid only upon successful ligation in PRe-RDL also addresses two the other limitations of RDL: the significant background from self-ligation of the vector and the decreased efficiency due to circularization of the insert. The modular design of PRe-RDL is suitable for the rapid generation of any repetitive peptide sequence of a specified length, and can also be used to join blocks of different lengths and sequences in a predetermined order.
We demonstrate the utility of PRe-RDL by synthesis of genes that encode a class of artificial repetitive polypeptides – elastin-like polypeptides (ELPs) – that consist of VPGXG pentapeptide repeats derived from tropoelastin 27, 28. Furthermore, we illustrate the versatility of the system by incorporating additional –non-ELP– peptide sequences at the N- and C- terminus of the polypeptides. ELPs exhibit an inverse solubility phase transition as a function of solution temperature; below their inverse transition temperature (Tt), ELPs are soluble in aqueous solution, but when the solution temperature is raised above their Tt, ELPs become insoluble and form micron size aggregates that ultimately coalesce to form an ELP-rich coacervate phase 27. The phase transition of ELPs occurs over a narrow temperature range, and is dependent upon the guest residue composition (X), its molecular weight (MW), its concentration and the type and concentration of salts23. The guest residue (X) and the MW are two convenient and orthogonal structural variables that can be used to tune the Tt of an ELP, so that an ELP can be designed to have a desired Tt for a specific application. We chose ELPs to demonstrate the utility of PRe-RDL because the phase transition behavior of ELPs is precisely controlled by their sequence and MW, so that the successful synthesis of an ELP can be validated not only by physical characterization of the polypeptide, but also by its biophysical behavior. The ELPs were expressed in E. coli from their synthetic genes, and the molecular weights and phase transition behavior of the purified ELPs were characterized to verify that this methodology for gene assembly yields polypeptides with the expected size and biophysical behavior.
Experimental Details
Materials
Restriction enzymes and calf intestinal phosphatase (CIP) were purchased from New England Biolabs (Ipswich, MA). T4 DNA ligase was purchased from Invitrogen (Carlsbad, CA). The pET-24a+ cloning vector was obtained from Novagen Inc. (Madison, WI), and all custom oligonucleotides were synthesized by Integrated DNA Technologies Inc. (Coralville, IA). Top10™ cells were purchased from Invitrogen (Carlsbad, CA) and BL21™ E. coli cells were purchased from Novagen (Madison, WI). All E. coli cultures were grown in TBDry™ media purchased from MO BIO Laboratories, Inc (Carlsbad, CA). The DNA miniprep, gel purification, and PCR purification kits were purchased from Qiagen Inc. (Germantown, MD).
Modification of pET-24(+) for PRe-RDL
1.5 μg of the pET-24a(+) vector was digested with 20 U of XbaI and 20 U of BamHI in NEB buffer 3 for 4 h at 37 °C. The 5′ ends were dephosphorylated with 1 U CIP for 1 h at 37 °C, and the vector was then purified using the Qiagen PCR purification kit. The linearized vector was eluted in 30 μL of distilled, deionized water. Two oligonucleotides were designed: 5′- ctagaaataattttgtttaactttaagaagGAGGAGtacatatgggctactgataatgatCTTCAG -3′ and 5′- gatcCTGAAGatcattatcagtagcccatatgtaCTCCTCcttcttaaagttaaacaaaattattt– 3′. This DNA sequence was designed to encode two new endonuclease restriction sites for BseRI and AcuI (shown in capitalized letters), as well as a short leader sequence (Met) and trailer sequence (Tyr – Stop – Stop). The BseRI site was incorporated into the Shine-Delgarno ribosomal binding site adjacent to the start codon (underlined). The two oligonucleotides were annealed by heating 50 μL, at 2 μM concentration, of each oligonucleotide in T4 DNA ligase buffer at 95°C for 2 min, then slowly cooling the solution to room temperature over 3 h. This resulted in a double stranded (ds) DNA with XbaI and BamHI compatible sticky ends. Ligation of the modified cloning insert into the multiple cloning site within pET-24a(+) was carried out by incubating 20 pmol of the annealed dsDNA with 0.1 pmol of the linearized vector with 400 U of T4 DNA ligase at 20 °C for 1 h in T4 DNA ligase buffer. The product was transformed into Top10™ chemically competent cells, which were allowed to recover for 1 h at 37 °C in Luria broth. The cells were then plated on TBdry™ plates that were supplemented with 45 μg/mL of kanamycin. The sequence was then confirmed by DNA sequencing.
Monomer gene synthesis
A synthetic gene that encodes the (GVGVP)5 peptide sequence for ELP1 was synthesized as two 75 nt long single stranded oligonucleotides that encoded for the sense and antisense strands of the gene. The two oligonucleotides were annealed by heating 50 μL, at 2 μM concentration, of each oligonucleotide in T4 DNA ligase buffer at 95°C for 2 min, then slowly cooling the solution to room temperature over 3 h. This resulted in a double stranded (ds) DNA with nonpalindromic, 2 bp, 3′ overhangs. The same procedure was used to anneal the two 150 nt long, single stranded oligonucleotides that encoded the (GXGVP)10 peptide sequence for ELP2, where X alternates between the amino acids A and G in a 1:1 ratio.
Concatemerization
1.5 μg of the pET-24a(+) modified cloning vector was digested with 2 U of BseRI for 16 h at 37 °C. The 5′ ends were dephosphorylated with 1 U CIP for 1 h at 37 °C, and the vector was then purified using the Qiagen PCR purification kit. The linearized vector was eluted in 30 μL of distilled, deionized water. Ligation of concatemers was carried out by incubating 20 pmol of the annealed dsDNA with 0.1 pmol of the linearized vector with 400 U of T4 DNA ligase at 20 °C for 1 h in T4 DNA ligase buffer. The product was transformed into Top10™ chemically competent cells, which were allowed to recover for 1 h at 37 °C in Luria broth. The cells were then plated on TBdry™ plates that were supplemented with 45 μg/mL of kanamycin.
Gene oligomerization by PRe-RDL
PRe-RDL was used to recursively double the ELP1 gene that encodes (GVGVP)30 and the ELP2 gene that encodes (GXGVP)20 where X = A and G in a 1:1 ratio, which were obtained after a single round of concatemerization. Using the dimerization of the 30 repeat fragment of ELP1 in the second round of PRe-RDL as an example, the designated ‘A’ fragment was obtained by digestion of 4 μg of ELP1-30 with 10 U AcuI and 40 U BglI for 3 hours at 37°C (see Figure 2A) in NEB Buffer 2 (New England Biolabs; Ipswich, MA). The ‘B’ fragment was obtained by digestion of 4 μg of ELP1-30 with 8 U BseRI and 40 U BglI for 3 hours at 37°C in NEB Buffer 2. Both DNA digests were run on a low melting point agarose gel. The ‘A’ digestion resulted in 3 bands: 1586 bp, 1821 bp, and a 2341 bp (1891 + ELP) fragment. The 2341 bp band was excised from the gel and purified with Qiagen’s gel purification kit. The ‘B’ digestion resulted in 2 bands: 1891 bp, and a 3857 (3407 + ELP) fragment, and the 3857 bp band was excised from the gel, purified, and eluted in 30 μL of distilled, deionized water. Equimolar amounts of the A and B fragments at a total DNA concentration of 5 ng/μL in a volume of 20 μL were ligated by incubation with T4 DNA ligase at 20 °C for 1 h. Top10™ chemically competent cells were transformed with the ligation product, as described previously. E. coli transformants were screened by colony PCR and by diagnostic restriction digests on an agarose gel. Each sequence was then confirmed by DNA sequencing.
Cloning of leader and trailer sequences
A plasmid containing a leader sequence and a plasmid containing a trailer sequence were also constructed. The gene segment encoding a (CGG)8WP trailer (T1) was created by annealing two chemically synthesized oligonucleotides that encode this peptide sequence, as described previously (Monomer gene synthesis). The annealed oligonucleotides were inserted into the linearized pET-24a(+) expression vector using the same preparation protocol and insert to vector ratios as provided in the Concatemerization section. The DNA segment encoding a MSKGPG leader (L1) was similarly synthesized by annealing two oligonucleotides, which provide the ‘TA’ and ‘CC’ overhangs that are compatible with NdeI and BseRI (Figure 2E). Because the NdeI restriction site is between the BseRI recognition sequence and BseRI cleavage site, 1.5 μg vector was first digested with 8 U BseRI for 2 hours at 37 °C in NEB buffer 2. 60 U NdeI was then added, and the sample was incubated at 37 °C for an additional hour. The linearized vector was purified using a Qiagen PCR purification kit, and eluted in 30 μL distilled, deionized water. Annealing of the oligonucleotides encoding L1 to yield a dsDNA cassette, the insertion of the cassette into the linearized vector, and transformation of the plasmid into E. coli, proceeded as described above. Colonies were screened by a diagnostic restriction endonuclease digestion using XbaI and BamHI and agarose gel electrophoresis, and the sequence of positive clones were confirmed by DNA sequencing.
The ligation of the plasmid encoding L1 to the ELP proceeded in a manner similar to a typical dimerization step in PRe-RDL. The construction of the ELP1-60 L1T1 oligomer is shown in Figure 3 to illustrate the modular process by which a leader or trailer gene sequence can be appended to the 5′- and 3′-end of the oligomerized gene that encode for peptide sequences that are distinct from the repetitive polypeptide. The plasmid containing the gene encoding L1 was subjected to an ‘A’ digestion (AcuI and BglI), resulting in 3 bands: 1586 bp, 1821 bp, and 1903 bp (1891 + leader), and the 1903 bp band was excised and purified, as previously described. The ELP1-60 construct was subjected to a ‘B’ digestion (BseRI and BglI), resulting in the 2 bands: 1891 bp and a 4307 bp (3407 + ELP1-60) fragment. These two fragments were ligated together under the previously described conditions, thereby joining the leader fragment to the N-terminus of ELP1-60 and reforming the parent plasmid. The colonies were then screened via diagnostic digest and sequenced. The plasmid containing the gene for T1 was then subjected to a ‘B’ digestion, resulting in 2 bands: 1891 bp and 3476 bp (3407 + trailer), and the 3476 band was excised and purified. The L1 ELP1-60 construct from the previous step was then subjected to an ‘A’ digestion, resulting in three bands: 1586 bp, 1821 bp, and 2803 bp (1891 + L1 ELP1-60), of which the 2803 band was excised and purified. The two purified fragments were ligated together, which ligated the T1 encoding DNA fragment at the 3′-end of the gene encoding L1 ELP1-60, forming L1 ELP1-60 T1. E. coli transformants were screened by colony PCR, followed by a diagnostic restriction digest, and positive clones were confirmed by DNA sequencing.
Figure 3.

PRe-RDL is a modular design that allows for the combination of multiple libraries. This figure demonstrates the stepwise ligation of a leader to an ELP, and then the ligation of a trailer to the Leader-ELP fragment. The ligation order shown here is not essential; leaders were appended to the ELPs before incorporating the trailers, though the order could be reversed if needed.
ELP Expression
100 ng of plasmid DNA was transformed into chemically competent E. coli BL21™ cells (Novagen; Carlsbad, CA), and used to inoculate a 250 mL flask containing 50 mL TBdry™ media supplemented with 45 μg/mL kanamycin. The cultures were incubated on a shaker at 200 rpm overnight at 37 °C. This starter culture was then used to inoculate 4 L flasks containing 1 L of TBdry media with 45 μg/mL kanamycin. The flasks were incubated on a shaker at 200 rpm for 24 hours at 37 °C. This “hyperexpression” expression protocol relies upon the leakiness of the T7 promoter to express the ELP29.
ELP Purification
After a 24 h incubation period, the cells were centrifuged at 3000 g for 15 min in 1 L bottles. The cell pellet was resuspended in 10 mL of PBS, and the cells were lysed by 54 cycles of sonication for 10 s separated by 20 s intervals (Sonicator 3000, Misonix, Farmingdale, NY) on ice. 0.7% w/v polyethyleneamine (PEI) was added to the cell lysate to precipitate nucleic acid contaminants. The ELP was then purified by inverse transition cycling (ITC) as follows: the cell lysate was centrifuged in a 30 mL round bottom tube at 11,000 g for 10 min at 4 °C to precipitate the insoluble fraction of cell lysate. The supernatant, containing soluble ELP, was then transferred to a new 30 mL round bottom tube and heated to 37 °C. For ELP2, which has a higher transition temperature, up to 3 M salt was added to trigger the phase transition at 37 °C. Once the solution became turbid, it was centrifuged at 11,000 g for 10 min at 37 °C to precipitate aggregated, insoluble ELP. The supernatant was decanted and the pellet was resuspended in 10 mL of cold, low ionic strength buffer (PBS). Typically, 3–5 rounds of ITC were enough to attain > 95% purity, as assessed by SDS-PAGE.
ELP Characterization
The purified ELP constructs were characterized with SDS-PAGE, matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) and UV-vis spectrophotometry. The ELP concentration was determined by UV-vis spectrophotometry (Nanodrop, Thermo Scientific, Waltham, MA) using the extinction coefficient of tyrosine at 280 nm (1285 M-1cm-1) or tryptophan at 280 nm (5630 M-1cm-1). The purity of the ELPs was determined by SDS-PAGE, using 4–20% Tris-HCl Ready Gels™ (Bio-Rad, Hercules, CA), which were stained with copper chloride. MALDI-MS was performed on a PE Biosystems Voyager-DE instrument equipped with a nitrogen laser (337 nm). The MALDI-MS samples were prepared in a 50% (v/v) aqueous – acetonitrile solution, containing 0.1% trifluoroacetic acid, using a sinapinic acid matrix. To characterize the inverse transition temperature (Tt) of the ELPs, the optical density at 350 nm (OD350) of ELP solutions were measured as a function of temperature on a UV-visible spectrophotometer equipped with a multicell thermoelectric temperature controller (Cary 300, Varian Instruments, Walnut Creek, CA) as follows: an ELP solution in PBS, at concentrations between 1–100 μM, was heated from 20 °C to 80 °C at a rate of 1 °C/min. The Tt was defined as the temperature that corresponds to the maximum of the first derivative of its turbidity profile with respect to temperature, which is indicative of the onset of the phase transition.
Discussion
PRe-RDL is a methodology in which two halves of a parent plasmid, each containing a copy of a DNA insert, are ligated together, thereby dimerizing the insert and reconstituting the full plasmid (Figure 2A). To implement PRe-RDL to oligomerize a gene directionally (i.e., head-to-tail) and with no extraneous nucleotides introduced at the ligation junctions, six criteria must be fulfilled in the selection of the restriction enzymes: (i) one restriction enzyme (RE) must be unique (RE1), i.e., only present once in the cloning plasmid, and is needed to linearize the vector in preparation for ligation of the insert. (ii) The second RE is preferably unique (RE2) to the cloning vector, but if it is present in the plasmid, it should be present only in Region II of the plasmid, which is defined as the region between RE2 and RE3, excluding the insert (see Figure 2A). (iii) RE1 and RE2 must have different recognition sequences so that their sites can be cut independently of each other. This last feature is essential to implement PRe-RDL, as it allows the isolation of the A and B fragments that together constitute an entire functional plasmid. (iv) The two enzymes must result in compatible overhangs, with at least a 2 bp overhang on their 3′-end so that the 3′-cohesive (“sticky”) end of the A fragment is compatible with the 5′-sticky end of the B fragment. (v) Palindromic overhangs at the ends of the repeat unit must be avoided to allow for directional and seamless ligation. (vi) The plasmid must also contain a unique restriction site (RE3) elsewhere on the plasmid, with overhangs that are incompatible with RE1 and RE2. This restriction site is the point at which the plasmid is cut into two halves, both of which must be unable to circularize. Although the exact location of RE3 is not critical, it would ideally be within the antibiotic resistance gene to further decrease viability of each half of the plasmid. We first examined all three classes of restriction endonucleases, particularly those that were asymmetric, to identify enzymes that are capable of fulfilling these requirements. Although the cleavage sites of type I restriction endonucleases are asymmetrically displaced from their recognition sequence, this distance is highly variable, reducing their usefulness for cloning. Type II enzymes cut symmetrically within a palindromic or interrupted palindromic sequence, either of which places local sequence constraints on the biopolymer. Type IIs and type III enzymes both cut asymmetrically at a defined distance from their recognition sequence, thereby eliminating any constraints on the sequence to be oligomerized. The only difference between the type IIs and type III endonucleases is structural, and either class of enzymes is suitable for PRe-RDL 30, 31.
Having identified type IIs or type III restriction endonucleases as the two classes of enzymes with the necessary attributes to implement PRe-RDL, we narrowed the choice of the enzymes by examining which of the many commercially available enzymes would be compatible with commercial vectors. We chose to directly carry out oligomerization directly in low-copy number expression vectors, as working with high-copy number cloning plasmids such as the pUC series – used for RDL – proved to be unnecessary, thereby eliminating one cloning step. Of the various expression vectors that are commercially available, we solely focused on the pET system (Novagen Inc.), given the high expression yield of diverse ELPs and their fusion proteins in this expression system 16, 22, 32.
These five requirements were met with BseRI, AcuI, and BglI as the three restriction endonucleases, and pET-24a(+) as the vector. BseRI is a type IIs restriction enzyme, whose recognition sequence is not present in the unmodified pET-24a(+), and AcuI is a type IIs enzyme, whose recognition sequence of CTGAAG only occurs once in the unmodified vector. BseRI cuts at a degenerate site 8 bps downstream of its recognition site (GAGGAG) on the coding strand, which allows any two amino acids to be encoded between the recognition site and the beginning of the oligomer. AcuI cuts 14 bps downstream of its recognition site, and permits any four amino acids to be encoded between its recognition and cleavage site. A dsDNA cassette was designed to incorporate both of these restriction endonucleases and include a short, default leader sequence (Met) and trailer (Tyr – Stop – Stop) that would encompass the oligomer until the desired length was obtained (Figure 2C). BseRI was chosen as RE1 for the 5′-end of the ELP because the minimal default leader sequence (Met) was shorter than the two amino acid limit imposed by the choice of this enzyme. In addition, the BseRI recognition site could be easily incorporated into the Shine-Delgarno ribosomal binding site proximal to the start codon. Similarly, AcuI was selected as RE2, because the default peptide trailer of Tyr-Stop-Stop was shorter than the four amino acid limit imposed by Acu I. This dsDNA cassette consisting of two chemically synthesized oligonucleotides with XbaI and BamHI compatible sticky ends was then inserted into the multiple cloning region of pET 24a(+) by cleaving pET 24(+) with XbaI and BamHI and ligation of this insert into the linearized plasmid. AcuI also only occurs once in pET 24a+ vector, within Region II (Figure 2A). Both AcuI and BseRI have different recognition sequences (Figure 2C), so that the first three requirements were met by this pair of REs.
The fourth and fifth requirements were considered simultaneously. If the enzymes resulted in overhangs of 3 bp or less, the overhangs would be contained within the sequence of a single amino acid to maintain a seamless union. Because any repetitive gene can be designed to repeat seamlessly by partitioning the first amino acid into two complementary overhangs encompassing the monomer, RE’s yielding an overhang of 3 bp or less are generalizable to any polypeptide sequence (Figure 2D). In systems that are even more sensitive to overhang length, RE’s with only one bp overhangs can be selected, such as AlwI, BceAI and BspPI. The fifth requirement was met by the selection of BglI as RE3 because its recognition site occurs only once on pET-24a(+), on the opposite side of the plasmid as the site of gene oligomerization. In addition, BglI has incompatible overhangs with AcuI and BseRI.
In order to decrease the number of rounds in PRe-RDL, we exploited the fact that the inserts in PRe-RDL can be forced to concatamerize in the absence of a vector to create larger inserts that can serve as the starting point for PRe-RDL. The compatibility of Pre-RDL with concatemerization is especially useful to minimize the number of recursive steps required to arrive at genes with a high degree of oligomerization. We therefore concatemerized the monomer ELP1 genes that encoded to (GVGVP)5 to obtain a hexamer that encodes (GVGVP)30. Similarly, the ELP2 monomer (GXGVP)10 –a 10 pentamer ELP monomer where X alternates between A and G in a 1:1 ratio– was concatemerized to obtain the dimer, (GXGVP)20. One of the limitations of concatemerization is that it is difficult to reliably obtain a large number of higher order oligomers. While concatemerization of the ELP1 monomer provided a hexamer with no trouble, ELP2, despite several attempts only yielded a dimer.
With the genes for ELP1-30 and ELP2-20, the products of concatemerization in hand, we next proceeded to recursively dimerize these sequences by PRe-RDL. The gene for ELP1-30 was doubled in each round to create the -60, -120, and -240 mers (Figure 4A). The ELP2 library was similarly recursively oligomerized starting with the gene encoding (GXGVP)20 (ELP2-20) to obtain the -40, -80, -160, and -320 mers after each round of PRe-RDL (Figure 4C). After the ELP2 gene library was generated, an ‘A’ fragment of ELP2-60 and a ‘B’ fragment of ELP2-80 were then combined to generate ELP2-240 in order to demonstrate how PRe-RDL can be used to generate intermediate length ELPs (lane 7, Figure 4C). The same approach can also be used to join together two different sequences to create more complex block copolymers. Following the synthesis of the ELP2 gene library, the ELP2 constructs selected for expression (ELP2-80, -160, -240, and -320) were all modified with a leader sequence to demonstrate that PRe-RDL also enables attachment of a leader L1, or a trailer, in a single round of cloning in order to increase yield.
Figure 4.
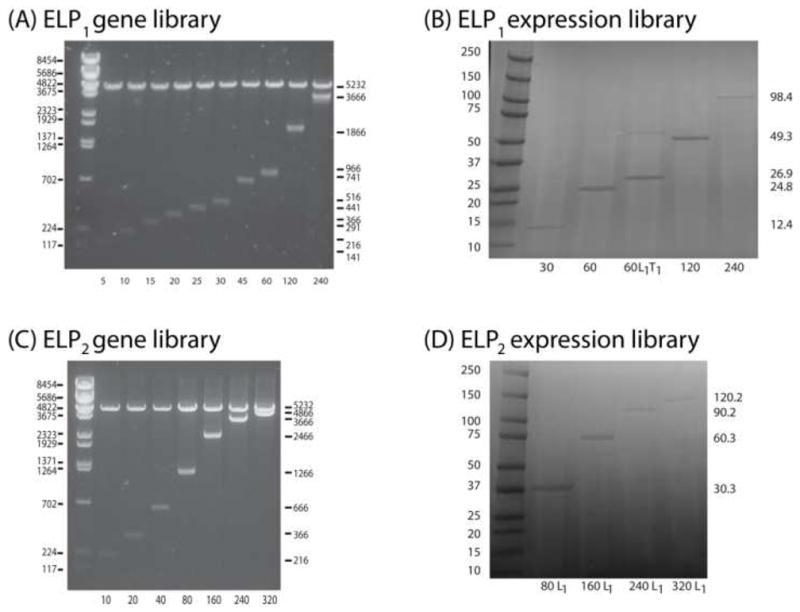
ELP1 and ELP2 libraries produced by PRe-RDL. (A) ELP1 gene library run on an agarose gel (1%). The left lane represents a size standard ladder, with sizes in base pairs shown on the left. Lanes 2–11 are diagnostic digests of each construct (restricted with XbaI and BamHI, which flank the ELP sequence with 66 bp), with the length shown on the right (in base pairs) and bottom (in pentapeptides). Lanes 2–8 were generated using concatemerization, while 9–11 were created using PRe-RDL. (B) ELP1 expression library run on an SDS-PAGE gel. The left lane is the Bio-Rad Kaleidoscope™ Ladder, with lengths in kDa on the left. Lanes 2–6 show the expressed ELPs with the lengths shown on the right (in kDa) and bottom (in pentapeptides). The dimer in lane 4 (ELP1-60 L1T1) is indicative of disulfide bonds formed between the cysteine residues present in T1. (C) ELP2 gene library run on an agarose gel. Lanes 2–3 were generated using concatemerization, whereas lanes 4–8 were formed via PRe-RDL. (D) ELP2 expression library run on an SDS-PAGE gel. To increase expression yields, the ELP2 library was modified with L1 (MSKGPG) on the amino terminus.
In the modified PRe-RDL cloning plasmid, the default DNA leader and trailer sequences are restricted in length by RE1 and RE2 to 2 and 3 amino acid residues, respectively. While this length of leader and trailer peptide may be adequate for some applications, in many instances having the flexibility to append other, longer leader or trailer peptides with any arbitrary sequence of interest is desirable, as they can provide peptide sequences for targeting, encode other purification tags (e.g. an oligohistidine tag) or provide unique reactive groups for site-specific conjugation of the polypeptide with drugs or imaging agents. To demonstrate the feasibility of appending an arbitrary leader and trailer peptide sequence (Figure 3), a leader plasmid (L1) consisting of MSKGPG and a trailer plasmid (T1) consisting of G(CGG)8WP were generated in separate cloning vectors, identical to the vector used in the ELP1 library. DNA encoding these leader and trailer peptides were added to ELP1-60 in a two-step process: 1) The ‘A’ fragment of L1 was ligated to the ‘B’ fragment of the ELP1-60, and 2) The ‘A’ fragment of the L1 ELP1-60 was ligated to the ‘B’ fragment of T1, generating L1ELP1-60T1. Once a desired length is reached, PRe-RDL can be used to add leaders and trailers of unrestricted length, although the addition of a leader or trailer prevents further seamless dimerization of the original monomer. However, this procedure enables the rapid generation of a library of leaders and a library of trailers that can be used to systematically modify the 5′-and 3′-ends of the gene encoding a repetitive polypeptide once it has been assembled. For example, the ELP2 constructs described in the following section (ELP2 - 80, -160, -240, and -320) were modified with L1 in a single round of cloning. While these modified constructs cannot be further dimerized with themselves in a seamless manner, each could be modified at its 3′-end, either with a library of trailers, or other compatible sequences lacking the 5′-modification.
The use of three restriction enzymes in PRe-RDL has the intrinsic benefit of reducing background by eliminating the possibility of self-ligation of the vector. This is possible because BglI (RE3) is incompatible with both BseRI and AcuI, so that each half of the vector can only ligate to its complement, rather than itself. PRe-RDL also increases cloning efficiency by eliminating the possibility of circularization of the insert. It has been our experience that RDL 15 has a high yield (up to 80%) when dimerizing small inserts < 1000 bp. However, the efficiency rapidly drops to 5–30% when working with inserts over 1000 bp. An added inconvenience of RDL is that the cloning is performed in a high copy number plasmid that is not designed for expression, so that the insert must be transferred to an expression vector once it reaches the desired length. Because this final length is commonly 1500–2400 bp, this step can be tremendously rate limiting. In contrast, this issue is eliminated in PRe-RDL, because cloning is carried out in an expression vector. PRe-RDL has shown efficiencies of 80–100% for dimerizing inserts less than 1500 bp, which gradually drops to 20–50% as the insert length approaches 2400 bp. However, even with the longest oligomer (ELP2-320, 4800 bp, 120 kDa), the final step of adding a leader was near 100%, suggesting that the reduced efficiency is limited to joining two large moieties.
Following validation of the genes by DNA sequencing, each of the higher molecular weight ELPs from each library was transformed into BL21™ cells and expressed in 4–6 L of media each. The ELP was then purified by 3–5 rounds of inverse transition cycling (ITC), followed by overnight dialysis into distilled, deionized water and subsequent lyophilization. Each of the ELPs produced between 30–70 mg purified protein/L media, which was gravimetrically quantified. The size and purity of each of the expressed ELP1 and ELP2 specimens was visualized by SDS-PAGE (Figure 4B, 4D). Each of the ELPs ran approximately 20% higher than expected, which matches previously published data 15, 33. To further verify the molecular weight of each of the constructs, ELPs < 240 pentapeptides in length were characterized by MALDI-MS (Table I). The MWs of all ELPs determined by MALDI-MS were close to their calculated MWs, indicating that the gene oligomerization in PRe-RDL proceeded as expected.
Table 1.
Theoretical molecular weight and the experimentally determined mass by MALDI-MS for the ELP1 and ELP2 libraries. Molecular weights could not be accurately determined (n.d.) for species larger than 160 pentapeptides. Theoretical masses were determined by inputting the theoretical amino acid composition of the peptide into the exPASy Proteomics Server 36, which then outputs the sum of the isotopically averaged mass of each individual amino acid.
| Mass (Da) | |||
|---|---|---|---|
| Pentamers | Theoretical | MALDI-MS | |
| ELP1 | 30 | 12465.7 | 12476.2 |
| 60 | 24750.3 | 23524.9 | |
| 60 L1T1 | 26914.7 | 26965.0 | |
| 120 | 49319.4 | 49309.3 | |
| 240 | 98457.7 | n.d. | |
| ELP2 | 80 L1 | 30561.1 | 30502.7 |
| 160 L1 | 60514.6 | 59539.6 | |
| 240 L1 | 90468.0 | n.d. | |
| 320 L1 | 120421.5 | n.d. | |
The thermal behavior of both ELP libraries was then assessed by monitoring the turbidity of a solution of each ELP as a function of temperature. The linear dependence of the Tt on the log of concentration can be seen for ELP1 in Figure 5A. The measured Tt’s for the ELP1 library were then compared with the fit generated for the same ELP sequence by Meyer and Chilkoti 23. This fit (Equation 1) describes the Tt dependence of ELP in terms of the critical Tt (Ttc; °C), the critical concentration (Cc; μM), the length (L; pentapeptides) of the sequence, the concentration of the ELP (C; μM), and k (°C), a proportionality constant.
Figure 5.

Thermal properties of the ELP1 and ELP2 series. (A) ELP1 Tt as a function of concentration and chain length. The dashed line represents the predicted Tt for each ELP1 length based on Meyer and Chilkoti’s model 23. (B) ELP2 Tt as a function of concentration and chain length in 1 M NaCl. The ELP2 Tt was > 80°C at these concentrations. 1 M NaCl was added to depress the Tt to demonstrate the presence of the thermal transition. (C) The ELP Tt as a function of chain length at a constant 25 μM ELP. ELP1 is in a PBS solution, whereas ELP2 is in 1 M NaCl in water. The dashed line represents the expected Tt for ELP1 based on the model developed by Meyer and Chilkoti 23.
| (1) |
Despite the small sequence variation at the C-terminus (the ELP1 library contains a Tyr trailer instead of the Trp-Pro trailer used in Meyer’s work) the model very accurately predicts the measured transition temperatures. The ELP2 constructs were also characterized in PBS, but their Tt’s were above 80°C, the highest measurable temperature on the temperature-controlled UV-vis spectrophotometer available for these studies. Their inverse transition behavior was hence analyzed in PBS + 1 M NaCl, which significantly depresses the Tt, allowing the thermally triggered transition behavior to be quantified in a temperature range that is experimentally accessible (Figure 5B). The Tt of ELP2 in 1M NaCl were not fit to Meyer’s model, because this model does not compensate for the depression in the Tt due to the addition of salt. Figure 5C also shows the Tt dependence on chain length at the given concentration of 25μM for both ELP1 in PBS, which also closely matches the data obtained by Meyer, and ELP2 in 1 M NaCl.
PRe-RDL was motivated by the utility –and limitations– of RDL, and it also borrows from the work of Kempe et al. who used Type II palindromic endonucleases 34 to recursively clone up to 64 repeats of the 11-mer peptide substance P, and Lewis et al. 35 who also used Type II palindromic endonucleases to recursively synthesize genes consisting up to 32 repeats of spider silk protein 10. Both Kempe and Lewis used the similar strategy of compatible but nonregenerating restriction sites to ligate two halves of a parent vector, each containing the oligomer, to reconstitute the plasmid and thereby double the oligomer length. This method allowed for recursion because as the oligomers were ligated together, the restriction site internal to the oligomer was abolished. Although these methods displayed the low background inherent in PRe-RDL, each was also restricted to sequences compatible with the selected palindromic endonuclease restriction sites, making each not generalizable to any arbitrary peptide repeat. Lee et. al used the unique asymmetric cutting characteristics of type IIs endonucleases to eliminate sequence constraints to oligomerize a gene encoding the antimicrobial peptide, magainin, in an iterative strategy based upon multiple concatemerization reactions, although the use of 4 bp overhangs resulted in a 4 bp seam between each monomer 11. Ståhl et al. also capitalized on the asymmetry of type IIs endonucleases to build a polypeptide comprised of two different concatemerized sequences, each derived from malaria blood-stage antigen12. In a design similar to our own plasmid, they eliminate extraneous amino acid residues from the N and C terminus as well as the junction between inserts. Our work combines the advantages of these methodologies by using type IIs endonucleases to allow for directional head-to-tail oligomerization of the gene encoding any peptide of interest, with no extraneous peptides encoded at the junction between two repeats, while maintaining the ability to encode a biopolymer of a predetermined length. The plasmid reconstruction strategy also greatly increases cloning efficiency and significantly decreases background. and limits self-ligation of the insert, thereby increasing the efficiency of cloning by reducing the loss of the insert in an unproductive side-reaction.
Concatemerization, frequently used to create multimers, relies upon a high insert to linearized vector molar ratio to unidirectionally ligate multiple inserts into each vector. This is a very useful technique for rapidly generating a library of proteins with a distribution of molecular weights. Unfortunately, the statistical nature of concatemerization cannot guarantee a specific number of inserts. While concatemerization is more rapid than any RDL strategy, it sacrifices control over the order and number of inserted monomers, which can be critical for many applications. This was demonstrated in the concatemerization of a 5-pentapeptide repeating unit (75 bp) encoding an ELP with the guest residue Valine (i.e. ELP1). This step resulted in a library consisting of the 5, 10, 15, 20, 25, 30, and 45-pentamer length ELP1 (Lanes 2–8, Figure 4A). However, the 35 and 40-pentamer lengths were not isolated since concatamerization does not guarantee a product with a specific MW, and in cases where the length exceeds current DNA sequencing capabilities it also complicates the determination of the precise number and order of repeats. Our strategy uses concatemerization in an initial step to generate the largest length that can be fully sequenced, which minimizes the time spent cloning genes encoding short repeats are not of any intrinsic interest (typically less than 10 kDa) (Table 2). One of the products of concatemerization can then be used in PRe-RDL to generate the higher order oligomers of interest. In the construction of the ELP2 library, a 150 bp oligomer was used in the concatemerization protocol, which was found to be much less efficient than the 75 bp oligomer, as it only provided a dimer (20 pentapeptides), which was then used to clone the 40 pentapeptide length. These results also illustrate the statistical and unpredictable nature of concatemerization; in contrast to concatemerization of ELP1, which provided a hexamer as the largest product beginning with a gene that encoded a single (VPGVG)5 monomer, the concatemerization of the (VPGAGVPGGG)5 monomer in the ELP2 series only yielded a dimer.
Table 2.
The number of rounds of cloning before the polypeptide is ready for transformation into expression-ready cells. Using RDL, each pentapeptide requires an extra cloning step to transfer the insert from the pUC vector into a pET expression vector. PRe-RDL eliminates this step, and bypasses three to five rounds of time-consuming cloning by concatemerizing the monomer gene.
| # of cloning steps before expression | ||
|---|---|---|
| Length in pentapeptides | RDL [14] | PRe-RDL (ELP2) |
| 5 | 2 | 1 |
| 10 | 3 | 1 |
| 20 | 4 | 1 |
| 40 | 5 | 2 |
| 80 | 6 | 3 |
| 160 | 7 | 4 |
| 240 | 8 | 5 |
| 320 | 9 | 5 |
The use of type IIs enzymes permits the sequential gene amplification of any repeating peptide in this system. PRe-RDL is also compatible with concatemerization so that an initial concatemerization reaction can be used prior to PRe-RDL to significantly decrease the number of PRe-RDL cycles, and hence the time necessary to amplify the “monomer” gene to the desired oligomer. The strength of this system also lies in its flexibility. As each sequence can be combined with other members within the library, members of other sequence libraries, and leader and trailer modifications, a large number of biopolymers with specific sequences, molecular weights, and functionalities can be rapidly created.
Conclusion
This paper presents a straightforward and general method to rapidly produce a repetitive polypeptide of any desired sequence and length over a wide range of molecular weights. The key to this approach is to use type IIs restriction enzymes to produce seamless, head-to-tail repeats of any arbitrary DNA sequence by a process that requires reconstitution of the plasmid in every oligomerization step. This process was used to recursively oligomerize two different ELP genes up to a desired length, while avoiding the problems of poor ligation efficiency and high background that limit RDL. Carrying out PRe-RDL with an initial concatemerization step directly in an expression vector permitted the gene assembly of two ELP libraries that are up to 4800 bp in length with 3–4 fewer steps than required by conventional RDL. With a dimerization efficiency of 80–100%, PRe-RDL is also enormously efficient compared to RDL, thereby largely simplifying the screening process at each cloning step. This system is also flexible in that the genes for other, non-repetitive peptide sequences can be appended at the two ends of the gene, which allows leader and trailer peptide sequences with unique functional groups to be incorporated at the two ends of a repetitive polypeptide.
Acknowledgments
This work was supported by a grant from the National Institute of Health (R01 GM61232) to A.C. J.R.M acknowledges the financial support of a NIH Biotechnology predoctoral fellowship (T32 GM 8555).
References
- 1.Lazaris A, Arcidiacono S, Huang Y, Zhou JF, Duguay F, Chretien N, Welsh EA, Soares JW, Karatzas CN. Science. 2002;295(5554):472–476. doi: 10.1126/science.1065780. [DOI] [PubMed] [Google Scholar]
- 2.Zhou CC, Leng BX, Yao JR, Qian J, Chen X, Zhou P, Knight DP, Shao ZZ. Biomacromolecules. 2006;7(8):2415–2419. doi: 10.1021/bm060199t. [DOI] [PubMed] [Google Scholar]
- 3.Nettles DL, Elder SH, Gilbert JA. Tissue Engineering. 2002;8(6):1009–1016. doi: 10.1089/107632702320934100. [DOI] [PubMed] [Google Scholar]
- 4.Panitch A, Yamaoka T, Fournier MJ, Mason TL, Tirrell DA. Macromolecules. 1999;32(5):1701–1703. [Google Scholar]
- 5.Nath N, Chilkoti A. Advanced Materials. 2002;14(17):1243. [Google Scholar]
- 6.Chilkoti A, Dreher MR, Meyer DE, Raucher D. Advanced Drug Delivery Reviews. 2002;54(5):613–630. doi: 10.1016/s0169-409x(02)00041-8. [DOI] [PubMed] [Google Scholar]
- 7.Dreher MR, Raucher D, Balu N, Colvin OM, Ludeman SM, Chilkoti A. Journal of Controlled Release. 2003;91(1–2):31–43. doi: 10.1016/s0168-3659(03)00216-5. [DOI] [PubMed] [Google Scholar]
- 8.Kopecek J. European Journal of Pharmaceutical Sciences. 2003;20(1):1–16. doi: 10.1016/s0928-0987(03)00164-7. [DOI] [PubMed] [Google Scholar]
- 9.Kurihara H, Morita T, Shinkai M, Nagamune T. Biotechnology Letters. 2005;27(9):665–670. doi: 10.1007/s10529-005-4477-8. [DOI] [PubMed] [Google Scholar]
- 10.Mi LX. Biomacromolecules. 2006;7(7):2099–2107. doi: 10.1021/bm050158h. [DOI] [PubMed] [Google Scholar]
- 11.Lee JH, Skowron PM, Rutkowska SM, Hong SS, Kim SC. Genetic Analysis-Biomolecular Engineering. 1996;13(6):139–145. doi: 10.1016/s1050-3862(96)00164-7. [DOI] [PubMed] [Google Scholar]
- 12.Stahl S, Sjolander A, Hansson M, Nygren PA, Uhlen M. Gene. 1990;89(2):187–93. doi: 10.1016/0378-1119(90)90005-c. [DOI] [PubMed] [Google Scholar]
- 13.McMillan RA, Lee TAT, Conticello VP. Macromolecules. 1999;32(11):3643–3648. [Google Scholar]
- 14.Padgett KA, Sorge JA. Gene. 1996;168(1):31–35. doi: 10.1016/0378-1119(95)00731-8. [DOI] [PubMed] [Google Scholar]
- 15.Meyer DE, Chilkoti A. Biomacromolecules. 2002;3(2):357–367. doi: 10.1021/bm015630n. [DOI] [PubMed] [Google Scholar]
- 16.Chow DC, Dreher MR, Trabbic-Carlson K, Chilkoti A. Biotechnol Prog. 2006;22(3):638–46. doi: 10.1021/bp0503742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christensen T, Amiram M, Dagher S, Trabbic-Carlson K, Shamji MF, Setton LA, Chilkoti A. Protein Science. 2009;18(7):1377–1387. doi: 10.1002/pro.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christensen T, Trabbic-Carlson K, Liu WG, Chilkoti A. Analytical Biochemistry. 2007;360(1):166–168. doi: 10.1016/j.ab.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dreher MR, Liu WG, Michelich CR, Dewhirst MW, Chilkoti A. Cancer Research. 2007;67(9):4418–4424. doi: 10.1158/0008-5472.CAN-06-4444. [DOI] [PubMed] [Google Scholar]
- 20.Dreher MR, Liu WG, Michelich CR, Dewhirst MW, Yuan F, Chilkoti A. Journal of the National Cancer Institute. 2006;98(5):335–344. doi: 10.1093/jnci/djj070. [DOI] [PubMed] [Google Scholar]
- 21.Dreher MR, Simnick AJ, Fischer K, Smith RJ, Patel A, Schmidt M, Chilkoti A. Journal of the American Chemical Society. 2008;130(2):687–694. doi: 10.1021/ja0764862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer DE, Chilkoti A. Nat Biotechnol. 1999;17(11):1112–5. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 23.Meyer DE, Chilkoti A. Biomacromolecules. 2004;5(3):846–851. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 24.McHale MK, Setton LA, Chilkoti A. Tissue Engineering. 2005;11(11–12):1768–1779. doi: 10.1089/ten.2005.11.1768. [DOI] [PubMed] [Google Scholar]
- 25.Raucher D, Chilkoti A. Cancer Research. 2001;61(19):7163–7170. [PubMed] [Google Scholar]
- 26.Shamji MF, Betre H, Kraus VB, Chen J, Chilkoti A, Pichika R, Masuda K, Setton LA. Arthritis and Rheumatism. 2007;56(11):3650–3661. doi: 10.1002/art.22952. [DOI] [PubMed] [Google Scholar]
- 27.Urry DW. Journal of Physical Chemistry B. 1997;101(51):11007–11028. [Google Scholar]
- 28.Urry DW, Trapane TL, Prasad KU. Biopolymers. 1985;24(12):2345–2356. doi: 10.1002/bip.360241212. [DOI] [PubMed] [Google Scholar]
- 29.Guda C, Zhang X, McPherson DT, Xu J, Cherry JH, Urry DW, Daniell H. Biotechnology Letters. 1995;17(7):745–750. [Google Scholar]
- 30.Bickle TA, Kruger DH. Microbiological Reviews. 1993;57(2):434–450. doi: 10.1128/mr.57.2.434-450.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szybalski W, Kim SC, Hasan N, Podhajska AJ. Gene. 1991;100:13–26. doi: 10.1016/0378-1119(91)90345-c. [DOI] [PubMed] [Google Scholar]
- 32.Trabbic-Carlson K, Liu L, Kim B, Chilkoti A. Protein Sci. 2004;13(12):3274–84. doi: 10.1110/ps.04931604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McPherson DT, Xu J, Urry DW. Protein Expr Purif. 1996;7(1):51–7. doi: 10.1006/prep.1996.0008. [DOI] [PubMed] [Google Scholar]
- 34.Kempe T, Kent SBH, Chow F, Peterson SM, Sundquist WI, Litalien JJ, Harbrecht D, Plunkett D, Delorbe WJ. Gene. 1985;39(2–3):239–245. doi: 10.1016/0378-1119(85)90318-x. [DOI] [PubMed] [Google Scholar]
- 35.Lewis RV, Hinman M, Kothakota S, Fournier MJ. Protein Expression and Purification. 1996;7(4):400–406. doi: 10.1006/prep.1996.0060. [DOI] [PubMed] [Google Scholar]
- 36.Gasteiger EHC, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]