

Abstract
Macrophages which play a central role in the injury, infection and sepsis, use glucose as their primary source of metabolic energy. Increased glucose uptake in inflammatory cells is well known to be one of the responsible processes that cause inflammatory response and cytotoxicity. We have shown recently that the inhibition of aldose reductase (AR) prevents bacterial endotoxin, lipopolysaccharide (LPS)-induced cytotoxicity and inflammatory response in macrophages. However, it is not known how AR inhibition prevents LPS-induced inflammation. Here in, we examined the effect of AR inhibition on LPS-induced glucose uptake and the expression of glucose transporter 3 (GLUT-3) in RAW264.7 murine macrophages. Stimulation of macrophages with LPS-increased glucose uptake as measured by using C14 labeled methyl-D-glucose and inhibition of AR prevented it. Similarly, ablation of AR by using AR–siRNA also prevented the LPS-induced glucose uptake in macrophages. Further, AR inhibition also prevented the LPS-induced upregulation of GLUT-3 expression, cyclic adenosine monophosphate (cAMP) accumulation and protein kinase A (PKA) activation in RAW264.7 cells. Moreover, LPS-induced down-regulation of cAMP response element modulator (CREM), phosphorylation of cAMP response element-binding protein (CREB) and DNA binding of CREB were also prevented by AR inhibition. Further, inhibition of AR or PKA also prevented the LPS-induced levels of GLUT-3 protein as well as mRNA in macrophages. These results indicate that AR mediates LPS-induced glucose uptake and expression of glucose transporter-3 via cAMP/PKA/CREB pathway and thus represents a novel mechanism by which AR regulates LPS-induced inflammation.
Keywords: cAMP, PKA, Aldose Reductase, CREB, LPS, Glucose transporters
1. Introduction
Bacterial endotoxin lipopolysaccharide (LPS) plays a central role in inflammation and in regulating the immune response (Miller et al., 2005, Palsson-Mc Dermott and O'Neill, 2004). LPS is released from the surface of replicating gram-negative bacteria into the circulation, where it is recognized by a variety of circulating cell types, triggering gene induction of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1), and biosynthesis of prostaglandins (PGE2) (Karin and Lin, 2002; Nathan, 2002). These and other cytokines act in an autocrine or paracrine manner to induce and amplify host-cell responses and defense systems that help to eliminate the bacterial infection. However, uncontrolled and excessive cytokine expression can induce acute or chronic inflammatory processes leading to multi-organ failure and death (Cohen, 2002 and Reddy et al., 2009). Glucose dyshomeostasis has been shown to most injurious metabolic syndrome during endotoxemia and intense hyperglycemia is generally observed prior to death of endotoxemia in animals and humans (Berk et al., 1970; Hinshaw 1976; Filkins, 1978; Fischer et al., 1978 and Fukuzumi et al., 1996). Further, glucose consumption has been shown to be increased in LPS treated animals (Lang et al., 1985; Lang and Dobrescu, 1991a,). Most of the glucose utilization is observed in macrophages rich organs (Lang and Dobrescu; 1991b; Meszaros et al., 1987). Excessive expression of glucose transporters (GLUTs) has been identified as a central mediator to tissue damage and dysfunction associated with inflammation. Further, cytokines, growth factors have been shown to upregulate expression and membrane localization of GLUT-1 and GLUT-3 in different cell types including vascular as well as macrophages (Ahmed et al., 1997; Fu et al., 2004; Phillips et al., 2005; Schuster et al., 2007).
Our recent studies indicate that polyol pathway enzyme aldose reductase (AR) mediates LPS-induced inflammatory signals in macrophages by preventing NF-κB dependent expression of inflammatory cytokines and chemokines (Ramana et al 2006a; Ramana and Srivastava 2006; Ramana et al., 2007; Ramana et al 2009). Further, we have shown that inhibition of AR prevents LPS-induced production of inflammatory cytokines, cardiac dysfunction and cardiomyopathy in mice. AR inhibition also prevents LPS-induced lethality in mice (Ramana et al 2006b). However, the mechanism(s) by which AR inhibition prevents LPS-induced inflammation is not clearly known. We hypothesize that AR mediated regulation of cellular redox state alters the inflammatory signaling pathways responsible for macrophage glucose uptake and the expression of GLUTs in endotoxemia. GLUTs are known to be expressed by the transcriptional control of CREB. Several studies indicate that activation of CREB plays a vital role in a variety of cellular processes including proliferation, differentiation and survival (Barton et al 1996; Fazia et al., 1997). cAMP/PKA pathway activates the CREB which is a member of large super family of DNA-binding proteins, collectively known as the bZIP proteins (Delghandi et al., 2005). Oxidative stress-initiated by LPS increases intracellular cAMP which causes activation of PKA (Osawa et al., 2006). Activated PKA translocated into the nucleus and phosphorylates CREB at ser133 which then binds to cAMP responsive element and causes transcriptional activation of various inflammatory markers including GLUT proteins (Hardy and Shenk, 1998; Hayes et al., 2009; Montminy et al 1990; Thomson et al., 2008). In the present study, we investigated the molecular mechanisms by which AR inhibition regulates glucose uptake and GLUT expression in macrophages. Our results demonstrate that the inhibition of AR prevents glucose uptake by inhibiting the GLUT-3 expression by regulating the activation of cAMP, PKA and CREB in macrophages, indicating that AR inhibition prevents LPS-induced inflammatory response by blocking the glucose uptake in macrophages.
2. Materials and methods
2.1. Materials
Phosphate-buffered saline (PBS), penicillin/streptomycin solution, trypsin, fetal bovine serum (FBS) and Dulbecco's Modified Eagle's Medium (DMEM) were purchased from Life Technologies. Antibodies against p-CREB, CREM-1, PKA, p-PKA, COX-2, iNOS, GLUT-1, GLUT-3, GLUT-4, Actin and GAPDH were purchased from Santa Cruz Biotech inc. USA. cAMP and prostaglandin E2 (PGE2) assay kits were obtained from Cayman chemical company and glutathione assay kit (GSH-400) from OXIS international inc. USA. Consensus oligonucleotides for CREB (5′- AGAGATTGCCTGACGTCAGACAGCTAG-3′) transcription factor was obtained from Promega Corp. Lipopolysaccharide (LPS), 1400W dihydrochloride and H-89 were purchased from Sigma Co. (St. Louis, MO) Deoxy-D-glucose, 2-[1-14C] was purchased from PerkinElmer. AR inhibitor, fidarestat was a gift from Sanwa Kagaku Kenkyusho Co. Ltd. (Tokyo, Japan). All other reagents were of the highest purity available.
2.2. Cell culture
Murine macrophage cell line (RAW264.7) were obtained from American Type Culture Collection - ATCC and maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere of 5% CO2.
2.3. cAMP assay and PGE2
RAW264.7 cells grown to 80% confluent in 6 well plates (1-2×106 cells/well in 6 well plates) were growth arrested in the serum-free medium without or with AR inhibitor (2μM fidarestat) for 24 h followed by incubation with 1 μg/ml LPS for another 0,12 and 24h. We have shown earlier that similar or > 2 μM concentrations of AR inhibitors prevent LPS-induced inflammatory response in RAW264.7 cells as well as isolated mouse peritoneal macrophages. Further, 2 uM of fidarestat prevents >95 % of the AR's enzyme activity in RAW264.7 cells. The cell culture medium was collected from each well after incubation with LPS and analyzed for PGE2 using an Enzyme Immuno Assay kit according to the manufacturer's instructions (Cayman Chemical Co.). For cAMP determinations, cells were treated with 1μg/ml LPS for 0, 1, 3, 6, and 12 h, the reaction was terminated by aspiration of the growth medium and addition of 0.1 N HCl. The cells were scraped into Eppendorff tubes and the suspensions were centrifuged; the supernatants were then neutralized with 10 N NaOH. cAMP levels were determined by an enzyme immunoassay according to manufacturer's instructions (Cayman Chemical Co. USA).
2.4. Gel-shift assay
The cytosolic as well as nuclear extracts were prepared as described before [Montminy et al., 1990; Ramana et al., 2002). Briefly, nuclear extracts prepared from various control and treated cells were incubated with the 32P-labeled consensus oligonucleotides for CREB (5′-AGAGATTGCCTGACGTCAGACAGCTAG-3′) for 15 min at 37 °C, and the DNA-protein complex formed was resolved on 6.5% native polyacrylamide gels. The specificity of binding was examined by competition with excess of unlabeled oligonucleotides. After electrophoresis, the gels were dried by using a vacuum gel dryer and were autoradiographed on x-ray films. The radiolabeled bands were quantified by an Alpha Imager 2000 scanning densitometer equipped with the AlphaEase™ Version 3.3b software.
2.5. pCRE-hrGFP cis-reporter plasmid transfection and FACS analysis
The effect of AR inhibition on LPS-induced expression of CRE (cAMP Responsive Element) and PKA was analyzed by trasfecting PathDetect® reporter plasmid pCRE-hrGFP (Stratagene, USA) in mouse macrophage cell line RAW264.7 using standard protocols. Briefly, 60% confluent RAW264.7 cells in 6 well plates were replaced with serum-free medium and transfections were performed with the plasmid mixture containing 1ug PathDetect® reporter plasmid pCRE-hrGFP / unrelated control plasmid in 3μl FuGENE6 (Roche Molecular Biochemicals USA) transfection reagent per well containing 2ml medium. After 8-10 h of transfection, cells were replaced with fresh medium with and without ARI/PKA inhibitors (2 uM fidarestat or 50 μg/ml H-89, a known PKA inhibitor) prior to the LPS (1μg/ml) treatments. After 24 h of LPS treatment cells were collected and fixed using ice cold 70% ethanol and analyzed for GFP positive cells using FACSCanto (BD Biosciences USA) and appropriate filters. Percentage of GFP-positive cells was plotted against controls.
2.6. Western blot
To examine the effect of AR inhibition on LPS-induced activation of p-CREB, p-PKA, CREM-1, GLUT-1, GLUT-3 GLUT-4, COX-2 and iNOS, equal amount of protein from macrophages were subjected to SDS-PAGE and Western blotting, and membranes were probed against specific antibodies. The antigen-antibody complexes were detected by enhanced chemiluminescence (Pierce USA). All blots were probed with either GAPDH or Actin as a loading control and densitometric analysis was carried out by using Kodak Image station.
2.7. Reverse transcription –PCR
To determine the effect of AR inhibition on LPS-induced activation of GLUT-3 at the transcription level, RAW264.7 cells were pre-incubated without and with fidarestat (2 μM) for 24 h and treated with 1μg/ml LPS for 0-6 h. Total RNA was isolated by using RNeasy® Mini Kit from Qiagen USA and reaction was followed by Qiagen® one step RT-PCR kit. Gene specific primer sequences are; GLUT-3 forward: 5 CAACTTGCTGGCCATCATTGCG 3′ GLUT-3 reverse: 5′ GCAAAATGGAAGGGCTGCGCTTTG 3′ and GAPDH forward 5′ AGGCCGGTGCTGAGTATGTC 3′ and GAPDH reverse 5′TGCCTGCTTCACCACCTTCT3′, were purchased from sigma (Sigma genosys USA) and reaction was performed for 30 cycles followed by cDNA synthesis according to Qiagen RT-PCR assay kit.
2.8. Glucose uptake
To determine the effect of AR inhibition on LPS-induced glucose uptake, growth arrested RAW264.7 cells were preincubated with 2 μM fidarestat or iNOS inhibitor 1400W (20 μmol) for 24 h and 1 h, respectively followed by 1 μg/ml LPS. Similar studies were performed in siRNA transfected cells. After 6 h of LPS treatment, cells were replaced with fresh medium containing 1.1μci of C14 labeled methyl-D-glucose (Perkline Elmer. USA) and incubated for 0, 30, 60 and 120 min. At the end of the incubations, cells were washed twice with ice cold PBS and cell contents were dissolved in 0.1N NaOH for overnight and neutralized with concentrated hydrochloric acid. Equal amount of cell contents were subjected for radioactivity using scintillation counter (Beckman).
2.9. GSH estimation
To determine the effect of AR inhibition on LPS-induced oxidative stress, growth arrested RAW264.7 cells were preincubated with 2 μM fidarestat or iNOS inhibitor 1400W (20 μmol) for 24 h and 1 h, respectively followed by 1 μg/ml LPS. Similar experiments were performed in AR-siRNA transfected cells. After 24 h of LPS treatments cells were washed twice with ice cold PBS and lysed with metaphosphoric acid solution (50 μM). Cellular GSH content as a marker for oxidative stress was estimated from the cell lysates using BIOXY-400 kit from Oxis international following manufacturer's instructions.
2.10. Statistical analysis
Data represent the mean ± SEM. Statistical analyses were carried out by using Sigma Stat 3.5 (Jandel Scientific, San Rafael, CA). All statistical tests were two-sided, and p<0.05 was considered as statistically significant. The differences between control Vs LPS or LPS Vs ARI groups were analyzed by unpaired two-tailed Student's t-test and one-way ANOVA followed by a Bonferroni t-test for multiple comparisons for studies with increasing dose and time.
3. Results
3.1. Effect of AR inhibition on macrophage glucose uptake
Since during infections, endotoxemia, burn injury, and sepsis macrophages augment their cellular glucose uptake and inflammatory response, we have examined the affect of AR inhibition on LPS-induced glucose uptake in RAW264.7 macrophages. As shown Fig. 1, incubation of macrophages with LPS caused significant (∼6-fold) uptake of glucose in a time dependent manner. However, when the cells were preincubated with AR inhibitor, fidarestat following stimulation with LPS only 2-fold glucose uptake was observed. To rule out nonspecific effects of pharmacological inhibitor, we ablated AR by using specific siRNA and investigated its effect on LPS-induced glucose uptake. Similarly, ablation of AR protein by using AR-siRNA also significantly prevented the LPS-induced glucose uptake. AR inhibitor alone or AR-siRNA alone did not affect the basal levels of glucose uptake in macrophages. In addition, pre-incubation of macrophages with iNOS inhibitor (1440W) also prevented the LPS-induced glucose uptake. These results suggest that AR mediates endotoxin-induced glucose uptake, a major source of energy in inflamed macrophages.
Fig.1. AR inhibition prevents LPS-induced glucose uptake in macrophages.
Untransfected and AR-siRNA transfected RAW264.7 cells were incubated with and without 2μM fidarestat and iNOS inhibitor (1400W) for 24 h and 30 min, respectively followed by incubation with 1μg/ml LPS. After 6 h of LPS treatments cells were replaced with fresh medium containing C14 labeled methyl-D-glucose for respective time intervals. At the end of the incubations, cells were washed twice with ice cold PBS and cell contents were dissolved in 0.1N NaOH for overnight and neutralized with hydrochloric acid. Equal amount of cell extracts were subjected for radioactivity measurements using scintillation counter. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to control cells (0 min), #P < 0.001 compared to LPS-treated respective groups.
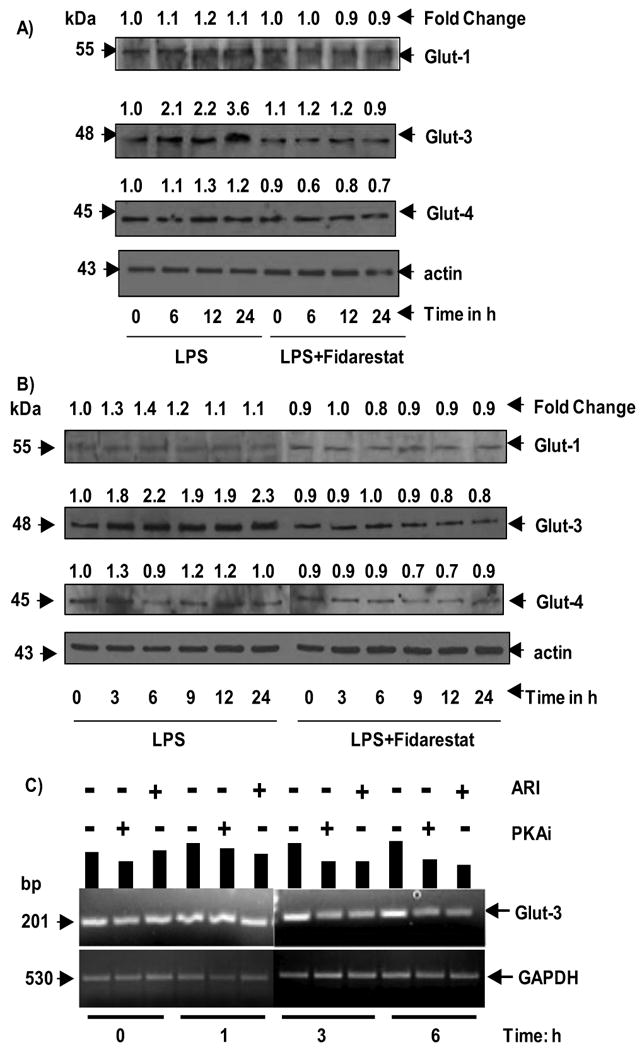
3.2. Effect of AR inhibition on the regulation of glucose transporters
To investigate the mechanism by which AR inhibition prevents macrophage glucose uptake, we have examined the effect of AR inhibition on LPS-induced expression glucose transporters. As shown in Fig.2A and B, incubation of macrophages with LPS increased expression of GLUT-3 levels in a time-dependent manner in the whole cell lysates as well as membrane fractions. A maximum increase of 3.6 and 2.3 -folds were observed at 24 h in total cell extracts and membrane extracts, respectively. The LPS-induced increase in the expression of GLUT-3 was significantly (∼90%) prevented by pre-incubation of macrophages with AR inhibitor, fidarestat. Further, GLUT-1 levels were marginally increased upon LPS stimulation and AR inhibition prevented it. However, GLUT-4 levels were not significantly altered by LPS, suggesting that GLUT-3 was more predominantly expressed in LPS-induced RAW264.7 macrophages as compared to GLUT-1 and GLUT-3. Pre-incubation of AR inhibitor alone did not affect the basal levels of GLUT proteins. Since GLUT-3 protein is predominantly expressed and is significantly inhibited by AR inhibition, we have next examined the effect of AR inhibition on GLUT-3 at the transcriptional level. As shown in Fig. 2C, incubation of macrophages with LPS caused significant increase in the expression of GLUT-3 mRNA and inhibition of AR prevented it, suggesting that AR regulates LPS-induced GLUT-3 expression at the transcriptional level. Therefore, we next examined the molecular mechanism by which AR inhibition prevents GLUT-3 expression.
Fig.2. AR inhibition prevents LPS-induced activation of glucose transporters.

Growth-arrested mouse macrophages (RAW264.7) were incubated with and without 2 μM fidarestat for 24 h and cells were treated with LPS for various time points ranging from 0-24 h. A) whole cell lysates and B) membrane fractions were probed against glucose transporter protein antibodies GLUT-1, GLUT-3 and GLUT-4. C) RT-PCR was performed for GLUT-3 expression using total RNA isolated from treated macrophages as described in methods. Band intensities were measured by densitometry analysis to determine the fold changes. Data shown are representative of three independent experiments.
3.3. Effect of AR inhibition on LPS-induced CREB activation
Since CREB transcription factor has been shown to transcribe GLUT proteins; we next examined the effect of AR inhibition on CREB activation. Nuclear extracts from macrophages treated with LPS without and with AR inhibitor were subjected to EMSA. Results shown in Fig. 3A indicate that a dose-dependent increase in the DNA binding activity of CREB upon LPS stimulus and inhibition of AR with fidarestat prevented it. Further, as shown in Fig. 3B, AR-siRNA transfected cells did not show an increase in the LPS-induced CREB DNA binding activity. AR inhibitor alone or AR-siRNA alone did not affect the CREB activity. Competition experiments with excess of cold oligo suggest validity of our EMSA (data not shown). These results indicate that AR inhibition could prevent GLUT-3 expression by preventing the activation of CREB in LPS-induced macrophages.
Fig.3. AR inhibition prevents LPS-induced nuclear localization of CREB in macrophages.
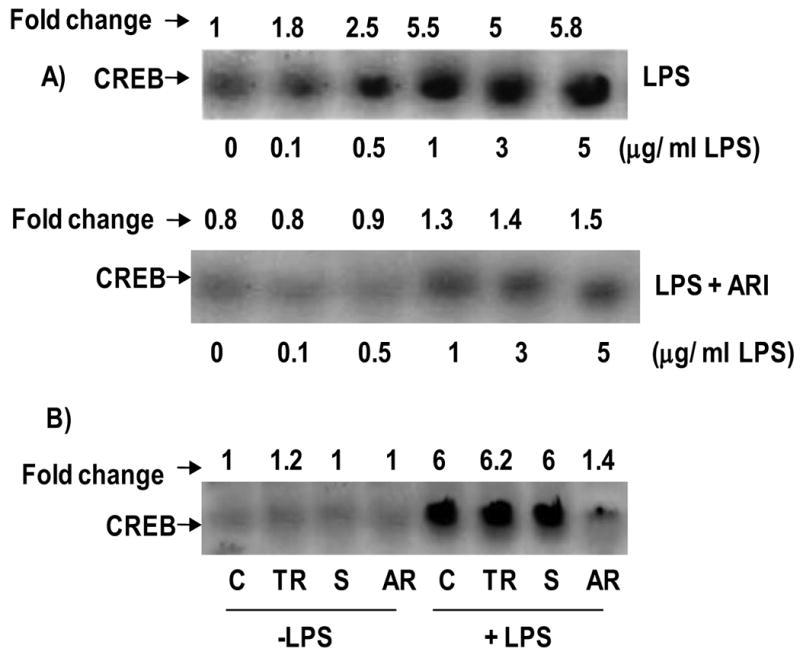
A) Growth-arrested macrophages were pre-incubated with and without AR inhibitor 2 μM fidarestat for 24 h and treated with indicated concentrations of LPS for 2 h. B) The AR siRNA transfected and un-transfected cells were treated with 1 μg/ml of LPS for 2 h. Nuclear extracts were prepared and equal amounts of nuclear extracts were subjected to EMSA for CREB as described in the Methods. Band intensities were measured by densitometry analysis to determine the fold changes. Data shown are representative of three independent experiments. C, Control; TR, Transfection reagent; S, scrambled SiRNA and AR, AR-SiRNA.
3.4. Effect of AR Inhibition on LPS-induced cAMP levels
Since CREB is a cAMP dependent transcription factor; we next examined the effect of AR inhibition on LPS-induced cAMP levels in macrophages. As shown in the Fig. 4, treatment of macrophages with LPS caused a significant increase in cAMP levels in the culture media at 3, 6 and 12 h and maximum 2.5 fold increase was observed at 6 h. AR inhibition significantly prevented the LPS-induced increase in cAMP. The cAMP levels were low but were detectable in controls and AR inhibition alone did not affect the basal levels of cAMP.
Fig.4. AR inhibition prevents LPS-induced cAMP production in macrophages.

Growth-arrested macrophages were pre-incubated with and without AR inhibitor (2 μM fidarestat) for 24 h, and were challenged with LPS (1 μg/ml) for 0,1,3,6 and 12 h. The cell lysates were collected as described in methods and cAMP levels were measured by using specific EIA kit as described in the methods. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to control cells, #P < 0.001 compared to LPS-treated cells
3.5. Effect of AR inhibition on LPS-induced cAMP downstream signals
Since PKA, also known as cAMP-dependent protein kinase, is a well known family of kinases whose activity is dependent on the cAMP levels; we next examined the effect of AR inhibition on PKA activation in LPS-stimulated macrophages. As shown in Fig. 5A, a time - dependent phosphorylation of PKA was observed in LPS-treated macrophages, a maximum increase of 2 fold was observed at 45 min. However, in the cells treated with AR inhibitor, the LPS-induced phosphorylation of PKA was significantly suppressed. Since activation of PKA causes the phosphorylation of CREB at Ser133 that leads to transcription of CREB – dependent genes, we examined the effect of AR inhibition of LPS-induced phosphorylation of CREB. As shown in Fig.4A, a time- dependent phosphorylation of CREB was observed in LPS-treated macrophages, a maximum increase of 3.2 fold was observed at 15 min and inhibition of AR prevented it. To examine the increased phosphorylation is indeed due to activation of PKA, we have examined the effect of PKA inhibitor as well as AR inhibitor on LPS-induced CREB activation. The results obtained by using FACS analysis of the PathDetect® reporter plasmid pCRE-hrGFP assay indicated that with AR inhibitor significantly prevents the LPS-induced activation of CREB in macrophages (Fig. 5B). Similar results were seen with a known PKA inhibitor H-89 used as a positive control.
Fig.5. AR inhibition prevents LPS-induced phosphorylation of CREB and PKA in macrophages.
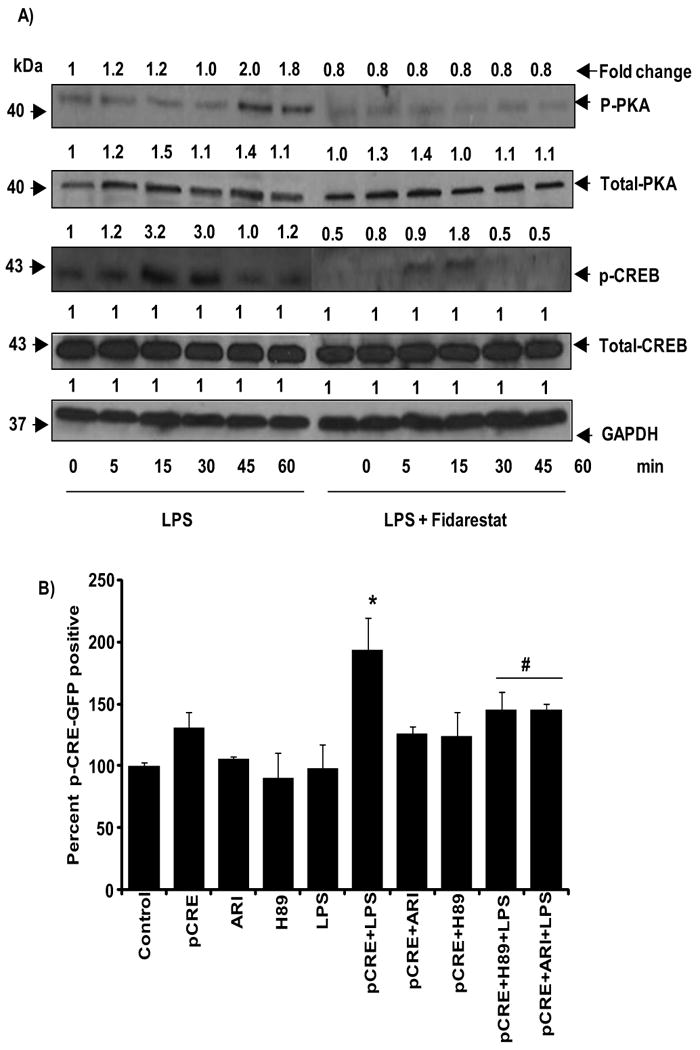
A) Growth-arrested macrophages were pre-incubated with and without AR inhibitors (2 μM fidarestat) for 24 h followed by the incubation with LPS (1 μg/ml) for 0, 5, 15, 30, 45 and 60 min. The whole cell lysates were subjected to Western blot analysis and probed with p-CREB, CREB, p-PKA, PKA and GAPDH antibodies. Fold change was calculated based on the densitometric analysis using Kodak Image station. Data shown are representative of three independent experiments. B) RAW264.7 cells were transfected with PathDetect® reporter plasmid pCRE-hrGFP as described in methods and treated with and without 2 μM fidarestat or 50 μg/ml H-89 (PKA inhibitor) for an hour prior to treatment with LPS 1μg/ml for 24 h. Ethanol-fixed cells were washed twice in cold PBS and analyzed for GFP-positive cells using FACSCanto. Percentage of positive cells was plotted. Experiment was carried with triplicates. All the data are expressed as percentage of Mean ± SEM (N = 4). *P < 0.001 as compared to pCRE control cells, #P < 0.001 compared to LPS+pCRE-treated cells
3.6. Effect of AR inhibition on LPS-induced CREB downstream signals
In addition to GLUT proteins, CREB also regulates a wide variety of genes by binding to cyclic AMP response element (CRE) in the promoter region, including Cox-2 and iNOS, we next examined the effect of fidarestat on LPS-induced Cox-2 and iNOS expression in macrophages. As shown in Fig. 6A and B, LPS-caused respectively 2.5 fold and 2.1 fold inductions of Cox-2 and iNOS proteins and fidarestat significantly prevented it.
Fig.6. AR inhibition prevents LPS-induced inflammatory markers.
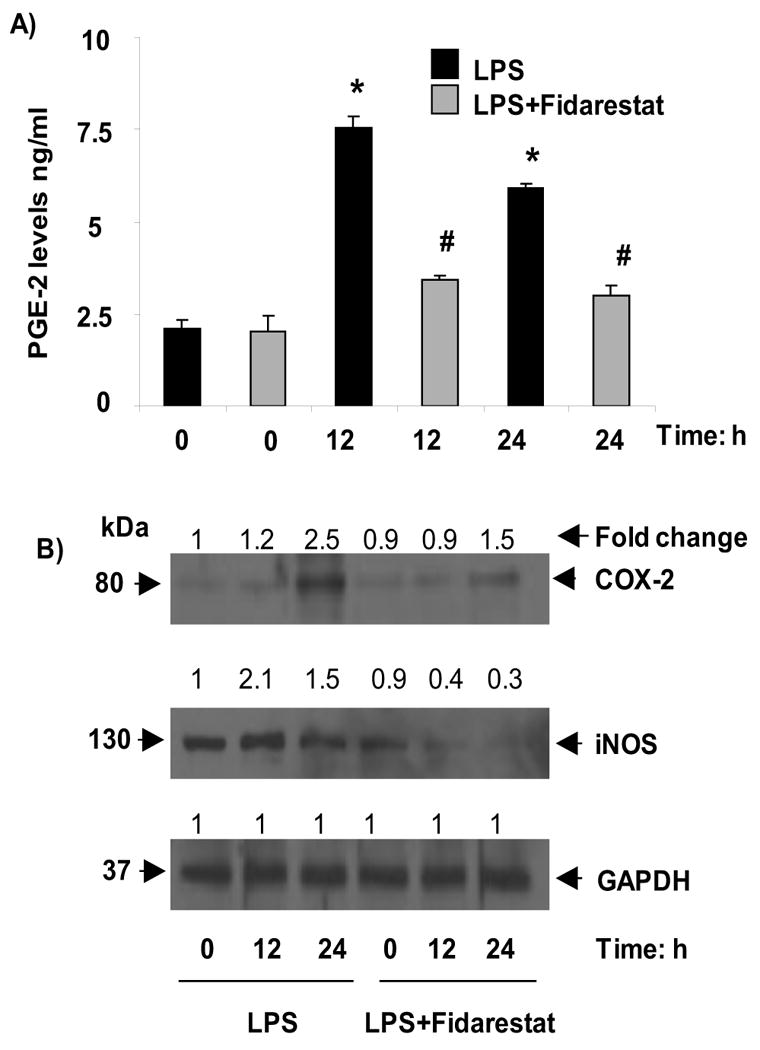
Growth-arrested RAW264.7 cells were incubated with and without 2μM fidarestat for 24 h and then treated with 1μg/ml LPS for 0, 12 and 24 h. A) PGE-2 was measured in cell culture medium and B) whole cell lysates were used for Westren blot analysis and probed with COX-2, iNOS and GAPDH antibodies. Band intensities were measured by densitometric analysis to determine the fold change. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to control cells, #P < 0.001 compared to LPS-treated cells
3.7. Effect of AR inhibition on cAMP-dependent repression of CREM-1
Since cAMP responsive element modulator-1 (CREM-1) is also a bZIP transcription factor that binds to cAMP responsive elements and acts as a transcriptional repressor rather than activator (Molina et al., 1993), we next examined the effect of AR inhibition on LPS-induced CREM expression in macrophages. As shown in Fig. 7, treatment of macrophages with LPS down-regulated the expression of CREM-1 and inhibition of AR significantly reversed the LPS-induced down regulation of CREM-1.
Fig.7. AR inhibition restores CREM expression in macrophages.
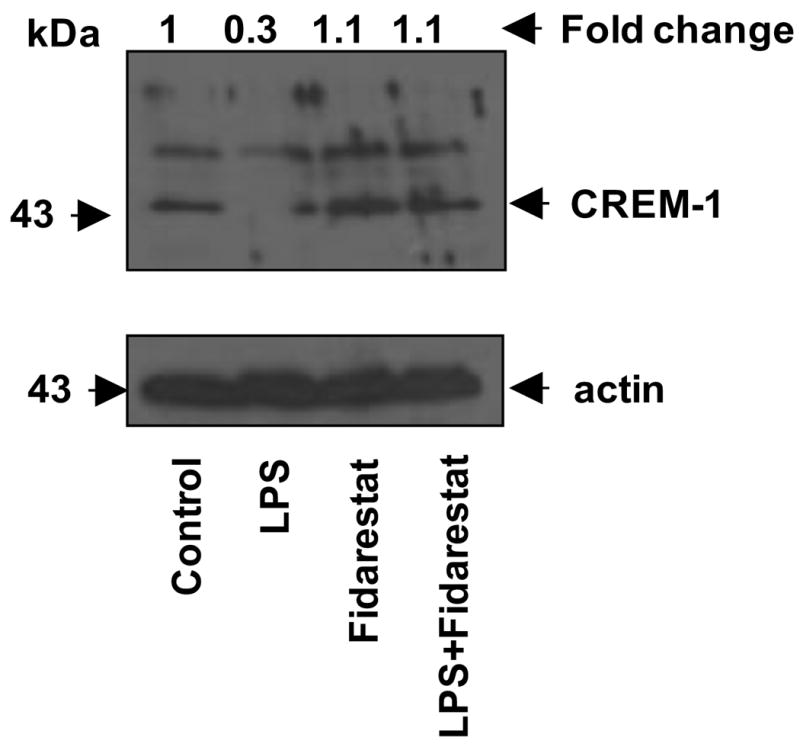
Growth arrested RAW264.7 cells were incubated with and without 2 μM fidarestat for 24 h and the cells were treated with 1 μg/ml LPS for 24 h. The whole cell lysates were probed against CREM-1 antibodies. Band intensities were measured by densitometry analysis to determine the fold change.
3.8. Effect of AR inhibition on LPS-induced oxidative stress
Since increased oxidative stress –induced reactive oxygen species (ROS) has been shown to increase activation of CREB and glucose-uptake, we next examined the effect of AR inhibition on LPS-induced oxidative stress, by measuring cellular GSH levels as a marker for oxidative stress. As shown in Fig.8, incubation of macrophages with LPS decreased the cellular GSH, indicating increased oxidative stress. Pre-incubation of AR inhibitor restored the LPS depleted GSH levels to normal, suggesting that inhibition of AR could prevent LPS-induced oxidative stress which is responsible for inflammatory changes.
Fig. 8. AR inhibition restores LPS –depleted GSH levels in macrophages.

Untransfected and AR-siRNA transfected RAW264.7 cells were incubated with and without 2 μM fidarestat and iNOS inhibitor (1400W) for 24 h and 30 min, respectively followed by incubation with 1μg/ml LPS. After 24 h of LPS treatment, GSH content was estimated from the cell lysates using BIOXY-400 kit following manufacturer's instructions. All the data are expressed as Mean ± SEM (N = 4). *P < 0.001 as compared to control cells, #P < 0.001 compared to LPS-treated cells
4. Discussion
During infections- and burn injury- induced severe inflammation and sepsis, there is a profound change in carbohydrate metabolism characterized by enhanced glucose utilization in macrophage-rich tissues, which could be responsible for increased whole body glucose disposal. Glucose transport proteins, belongs to a family of glucose transporter genes (GLUT 1–5) with variable tissue-specific expression, subcellular localization, and kinetics for glucose uptake, are the key functional units of the cellular glucose transport system (Crane, 1960; Fu et al., 2004; Meszaros et al., 1987). However, regulation of glucose uptake and glucose transporter expression in the cells insensitive to insulin is poorly understood. Oxidative stress induced by a variety of reagents such as growth factors and cytokines stimulate glucose transport by translocating transporters from intracellular sites to the plasma membrane or by increasing transporter expression (Crane, 1960; Ebeling et al., 1998; Hotamisligil et al., 1993; McGowan et al., 1997).
Binding of LPS to its receptors trigger multiple signaling events that cause tissue dysfunction, damage leading to multi-organ failure and sepsis (Cohen, 2002; Miller et al., 2005; Palsson-McDermott et al., 2004). Recent studies indicate that glucose dyshomeostasis leading to hypoglycemia is commonly observed in endotoxemia (Filkin, 1978). LPS administration has been shown to increase glucose consumption significantly (Lang et al., 1985). Further, glucose consumption in the body is generally observed in the tissues rich in macrophages and thereby macrophages contribute to observed hypoglycemia (Fukuzumi et al., 1996; Lang and Dobrescu, 1991b). However, how LPS induced expression of GLUT proteins are regulated in macrophages is not clearly known. Our recent studies indicate that inhibition of AR prevents LPS-induced inflammatory response in RAW264.7 macrophages as well as in isolated mouse peritoneal macrophages. Further, we have also shown that inhibition of AR prevents LPS-induced inflammatory cytokines and chemokines in mice serum, liver, heart and spleen. Most importantly, inhibition of AR also prevented the LPS-induced cardiomyopathy in mouse model of endotoxemia. Although, we have shown that inhibition of AR prevents LPS-induced inflammatory response via inhibiting NF-kB–dependent inflammatory signals, still it is not clear how AR inhibition prevents LPS-induced inflammation. In order, to identify if AR regulates LPS-induced macrophage glucose uptake which is responsible for inflammation and cytotoxicity, we have examined the mechanisms by which AR regulates glucose uptake in macrophages. Our results demonstrate that inhibition of AR prevents LPS-induced glucose uptake. Further, we have also demonstrated that AR mediates the expression of GLUT proteins specifically GLUT3 in LPS- stimulated macrophages. These results identify a novel mechanism by which AR inhibition could prevent inflammatory response in inflammatory cells. Since, CREB transcribes the GLUT proteins, we next examined if AR inhibition prevents LPS-induced cAMP signals leading to activation of transcription factor CREB in murine macrophages. Our results indicate that AR inhibition prevents LPS-induced CREB due to attenuation of phosphorylation of PKA and CREB. Inhibition of AR also prevented the LPS -induced decrease in the abundance of CREM-1 and the up-regulation of GLUT proteins indicating that AR inhibitor, fidarestat profoundly affects the cAMP signaling in response to LPS. We have shown earlier that inhibition of AR blocks NF-κB activation via PKC/PLC prevents apoptosis of macrophages (Ramana et al., 2007, Ramana et al 2006a, Ramana and Srivastava 2006). Our current observations indicate that inhibition of AR prevents LPS -induced PKA phosphorylation and CREB activation via cAMP. Collectively, these results provide comprehensive support to the notion that AR-sensitive events are upstream to the dichotomy between CREB and NF-κB activation and that AR-catalysis is essential for the manifestation of the pleiotrophic effects of LPS. The mechanisms by which inhibition of AR prevents CREB remain unclear, but may relate to AR-induced changes in the cellular redox state (Oates, 2008; Kaneko et al 2005; Muthenna et al., 2009). Indeed, we have shown that inhibition of AR prevents LPS-induced oxidative stress by increasing the cellular GSH levels depleted by LPS. Previous studies have shown that the activation of CREB by LPS, in part, due to increased generation of ROS in the mitochondria. The critical role of redox changes in LPS signaling is suggested by data showing that treatment with antioxidants simultaneously blocks LPS-induced NF-κB and CREB activation (Ryu et al., 2005). Our previous studies suggest that inhibition of AR prevents LPS-induced ROS generation and formation of toxic lipid peroxidation derived aldehydes in macrophages (Ramana et al., 2007, Ramana et al 2006a, Ramana and Srivastava 2006). Further, our previous studies also indicate that AR catalyzed reduction product GS-DHN mediates oxidative stress-induced inflammatory signals via activation of PKC (Ramana et al., 2007). Recent studies demonstrate that PKC and ROS regulate the cAMP levels in human eosinophils (Ezeamuzie and Taslim, 2006). However, further studies are required to investigate how AR inhibition prevents LPS-induced cAMP levels in macrophages. In conclusion, the present study indicates that the inhibition of AR diminishes the LPS-induced glucose uptake and expression of glucose transporter proteins in macrophages by preventing the signals upstream to CREB activation such as cAMP/PKA and CREM. These results indicate a novel mechanism for the anti-inflammatory role of AR. Further evaluation of role of AR pathway and assessment of its significance in LPS-mediated inflammatory events will help in developing better anti-inflammatory treatments for endotoxemia and sepsis.
Acknowledgments
This work was supported by NIH grants GM71036 (KVR) and DK36118 (SKS)
Abbreviations
- cAMP
cyclic Adenosine mono phosphate
- PKA
protein kinase A
- CREB
cAMP responsive element binding protein
- LPS
lipopolysaccharide
- GSH
glutathione
- GLUT
glucose transporter protein
- COX2
cyclooxygenase-2
- iNOS
inducible nitric oxide synthase
- PGE2
prostaglandin E2
- TNF-α
tumor necrosis factor-alpha
- IL
Interleukine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed N, Kansara M, Berridge MV. Acute regulation of glucose transport in a monocyte-macrophage cell line: Glut-3 affinity for glucose is enhanced during the respiratory burst. Biochem J. 1997;327:369–75. doi: 10.1042/bj3270369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton K, Muthusamy N, Chanyangam M, Fischer C, Clendenin C, Leiden JM. Defective thymocyte proliferation and IL-2 production in transgenic mice expressing a dominant-negative form of CREB. Nature. 1996;379:81–5. doi: 10.1038/379081a0. [DOI] [PubMed] [Google Scholar]
- Berk JL, Hagen JF, Beyer WH, Gerber MJ. Hypoglycemia of shock. Ann Surg. 1970;171:400–8. doi: 10.1097/00000658-197003000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–91. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- Crane RK. Intestinal absorption of sugars. Physiological Reviews. 1960;40:789–825. doi: 10.1152/physrev.1960.40.4.789. [DOI] [PubMed] [Google Scholar]
- Delghandi MP, Johannessen M, Moens U. The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells Cellular Signalling. 2005;17:1343–51. doi: 10.1016/j.cellsig.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Ebeling P, Koistinen HA, Koivisto VA. Insulin-independent glucose transport regulates insulin sensitivity. FEBS Letters. 1998;436:301–3. doi: 10.1016/s0014-5793(98)01149-1. [DOI] [PubMed] [Google Scholar]
- Ezeamuzie CI, Taslim N. Reactive oxygen species mediate phorbol ester-stimulated cAMP response in human eosinophils. European Journal of Pharmacology. 2006;543:174–80. doi: 10.1016/j.ejphar.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Fazia MD, Servillo G, Sassone-Corsi P. Cyclic AMP signalling and cellular proliferation: regulation of CREB and CREM. FEBS Let. 1997;410:22–4. doi: 10.1016/s0014-5793(97)00445-6. [DOI] [PubMed] [Google Scholar]
- Filkins JP. Phase of glucose dyshomeostasis in endotoxicosis. Circ Shock. 1978;5:347–55. [PubMed] [Google Scholar]
- Fischer K, Lees J, Newman L. Hypoglycemia in hospitalized patients: causes and outcomes. N Engl J Med. 1986;315:1245–50. doi: 10.1056/NEJM198611133152002. [DOI] [PubMed] [Google Scholar]
- Fu Y, Maianu L, Melbert BR, Garvey WT. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: a role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol Dis. 2004;32:182–90. doi: 10.1016/j.bcmd.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-Induced Enhancement of Glucose Influx into Murine Peritoneal Macrophages via GLUT1. Infection and Immunity. 1996;64:108–12. doi: 10.1128/iai.64.1.108-112.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw LB. The role of glucose in endotoxin shock. Circ Shock. 1976;3:1–10. [Google Scholar]
- Hardy S, Shenk T. Adenoviral control regions activated by E1A and the cAMP response element bind to the same factor. Proc Natl Acad Sci USA. 1998;85:4171–5. doi: 10.1073/pnas.85.12.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Bence KK, Grill HJ. Dorsal Hindbrain 5′-Adenosine Monophosphate-Activated Protein Kinase as an Intracellular Mediator of Energy Balance. Endocrinology. 2009;150:2175–82. doi: 10.1210/en.2008-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Bucciarelli L, Hwang YC, Lee L, Yan SF, Schmidt AM, et al. Aldose reductase and AGE-RAGE pathways: key players in myocardial ischemic injury. Ann N Y Acad Sci. 2005;1043:702–9. doi: 10.1196/annals.1333.081. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Lang CH, Dobrescu C. Gram-negative infective infection increases noninsulin-mediated glucose disposal. Endocrinology. 1991a;128:645–53. doi: 10.1210/endo-128-2-645. [DOI] [PubMed] [Google Scholar]
- Lang CH, Dobrescu C. Sepsis-induced increases in glucose uptake by macrophage-rich tissues persist during hypoglycemia. Metabolism. 1991b;40:585–93. doi: 10.1016/0026-0495(91)90048-2. [DOI] [PubMed] [Google Scholar]
- Lang CH, Bagby GJ, Spitzer JJ. Glucose kinetics and body temperature after lethal and nonlethal doses of endotoxin. Am J Physiol. 1985;248:R471–8. doi: 10.1152/ajpregu.1985.248.4.R471. [DOI] [PubMed] [Google Scholar]
- Liu L, Wang Y, Fan Y, Li C, Chang Z. IFN-γ Activates cAMP/PKA/CREB Signaling Pathway in Murine Peritoneal Macrophages Journal of Interferon & Cytokine Research. Journal of Interferon & Cytokine Research. 2004;24:334–2. doi: 10.1089/107999004323142196. [DOI] [PubMed] [Google Scholar]
- McGowan KM, Police S, Winslow JB, Pekala PH. Tumor Necrosis Factor-alpha Regulation of Glucose Transporter (GLUT1) mRNA Turnover. Contribution of the 3prime-untranslated region of the Glut1 message. J Biol Chem. 1997;272:1331–7. doi: 10.1074/jbc.272.2.1331. [DOI] [PubMed] [Google Scholar]
- Meszaros K, Lang CH, Bagby GJ, Spitzer JJ. Contribution of different organs to increased glucose consumption after endotoxin. J Biol Chem. 1987;262:10965–70. [PubMed] [Google Scholar]
- Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P. Inducibility and negative autoregulation of CREM: An alternative promoter directs the expression of ICER, an early response repressor. Cell. 1993;75:875–86. doi: 10.1016/0092-8674(93)90532-u. [DOI] [PubMed] [Google Scholar]
- Montminy MR, Gonzalez GA, Yamamoto KK. Regulation of camp-inducible genes by CREB. Trends in Neurosciences. 1990;13:84–188. doi: 10.1016/0166-2236(90)90045-c. [DOI] [PubMed] [Google Scholar]
- Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- Muthenna P, Suryanarayana P, Gunda SK, Petrash JM, Reddy GB. Inhibition of aldose reductase by dietary antioxidant curcumin: mechanism of inhibition, specificity and significance. FEBS Lett. 2009;583:3637–42. doi: 10.1016/j.febslet.2009.10.042. [DOI] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;420:846–52. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- Oates PJ. Aldose Reductase, Still a compelling target for diabetic neuropathy. Curr Drug Targets. 2008;9:14–36. doi: 10.2174/138945008783431781. [DOI] [PubMed] [Google Scholar]
- Osawa Y, Lee HT, Hirshman CA, Xu D, Emala CW. Lipopolysaccharide-induced sensitization of adenylyl cyclase activity in murine macrophages. Am J Physiol Cell Physiol. 2006;290:C143–51. doi: 10.1152/ajpcell.00171.2005. [DOI] [PubMed] [Google Scholar]
- Palsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–62. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips T, Ferraz I, Bell S, Clegg PD, Carter SD, Mobasheri A. Differential regulation of the GLUT1 and GLUT3 glucose transporters by growth factors and pro-inflammatory cytokines in equine articular chondrocytes. Vet J. 2005;169:216–22. doi: 10.1016/j.tvjl.2004.01.026. [DOI] [PubMed] [Google Scholar]
- Ramana KV, Chandra D, Srivastava S, Bhatnagar A, Aggarwal BB, Srivastava SK. Aldose reductase mediates mitogenic signaling in vascular smooth muscle cells. J Biol Chem. 2002;30:32063–70. doi: 10.1074/jbc.M202126200. [DOI] [PubMed] [Google Scholar]
- Ramana KV, Fadl AA, Tammali R, Reddy AB, Chopra AK, Srivastava SK. Aldose reductase mediates the lipopolysaccharide-induced release of inflammatory mediators in RAW264.7 murine macrophages. J Biol Chem. 2006a;281:33019–29. doi: 10.1074/jbc.M603819200. [DOI] [PubMed] [Google Scholar]
- Ramana KV, Srivastava SK. Mediation of aldose reductase in lipopolysaccharide-induced inflammatory signals in mouse peritoneal macrophages. Cytokine. 2006;36:115–122. doi: 10.1016/j.cyto.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana KV, Willis MS, White MD, Horton JW, DiMaio JM, Srivastava D, et al. Endotoxin-induced cardiomyopathy and systemic inflammation in mice is prevented by aldose reductase inhibition. Circulation. 2006b;24:1838–46. doi: 10.1161/CIRCULATIONAHA.106.630830. [DOI] [PubMed] [Google Scholar]
- Ramana KV, Reddy AB, Tammali R, Srivastava SK. Aldose reductase mediates endotoxin-induced production of nitric oxide and cytotoxicity in murine macrophages. Free Radic Biol Med. 2007;15:1290–302. doi: 10.1016/j.freeradbiomed.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramana KV, Srivastava SK. Aldose reductase: A novel therapeutic target for inflammatory pathologies. Int J Biochem Cell Biol. 2009;42:17–20. doi: 10.1016/j.biocel.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AB, Srivastava SK, Ramana KV. Anti-Inflammatory effect of aldose reductase inhibition in murine polymicrobial sepsis. Cytokine. 2009;48:170–6. doi: 10.1016/j.cyto.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee J, Impey S, Ratan RR, Ferrante RJ. Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mit. Proc Natl Acad Sci U S A. 2005;102:13915–20. doi: 10.1073/pnas.0502878102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster DP, Brody SL, Zhou Z, Bernstein M, Arch R, Link D, et al. Regulation of lipopolysaccharide-induced increases in neutrophil glucose uptake. Am J Physiol Lung Cell Mol Physiol. 2007;292:L845–51. doi: 10.1152/ajplung.00350.2006. [DOI] [PubMed] [Google Scholar]
- Thomson DM, Herway ST, Fillmore N, Kim H, Brown JD, Barrow JR, et al. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104:429–38. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]