

Abstract
The protein kinase AKT1 (v-akt murine thymoma viral oncogene homologue 1), also referred to as PKB (protein kinase B), is an essential mediator of the phosphatidylinositol 3-kinase signaling pathway. Elevated activity of AKT1 is common in human cancer. Localization at the plasma membrane, leading to enhanced phosphorylation and activation of AKT1, is an important factor determining the oncogenicity of this kinase. Although the phosphatidylinositol 3-kinase signaling pathway is frequently upregulated in cancer, cancer-specific mutations in AKT1 are not common. Recently, such a mutation has been identified in breast, colon and ovarian cancers. The mutation is located in the pleckstrin homology (PH) domain of AKT1 and results in a glutamic acid to lysine substitution at residue 17. The resultant change in the conformation of the PH domain facilitates membrane binding of the mutant protein. Here we show that exchange of the PH domain leading to preferential binding of phosphatidylinositol 4,5-bisphosphate (PIP2) over phosphatidylinositol 3,4,5-trisphosphate (PIP3) constitutively activates AKT1. AKT1 with this altered PIP affinity induces oncogenic transformation in cultures of chicken embryo fibroblasts and causes neoplastic growth and angiogenesis in the chorioallantoic membrane of the chicken embryo. Gain-of-function mutants of AKT1 may not be affected by PI3K inhibitors that are currently in development. Therefore, AKT1 remains a distinct and important cancer target.
The serine/theronine kinase AKT1 (v-akt murine thymoma viral oncogene homologue 1), also referred to as PKB (protein kinase B), is an important target of PI3K (phosphatidylinositol 3-kinase) and a central mediator of the PI3K signaling pathway. Upon activation of receptor tyrosine kinases or G-protein coupled receptors by growth factors or other extracellular stimuli, PI3K Class I enzymes catalyze the production of phosphatidylinositol 3,4,5-trisphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2). The PH domain of AKT1 binds to PIP3, facilitating the recruitment of the enzyme to the cell membrane. AKT1 then becomes phosphorylated at threonine 308 (T308) and serine 473 (S473) 1–5. The binding of AKT1 to PIP3 induces a conformational change which makes T308 accessible for phosphorylation by PDK1 (3-phosphoinositide-dependent kinase) and is the crucial step in AKT1 activation 6–11. Both phosphorylations at T308 and S473 are essential for a complete activation of the kinase function of AKT1 12.
Enhanced membrane recruitment and elevated AKT1 kinase activity are frequently observed in human cancer. Constitutive activation of AKT1 is commonly induced by recurring mutations in enzymes that directly or indirectly regulate levels of PIP3. Among these are gain-of-function mutations in receptor tyrosine kinases and in the catalytic subunit p110α of PI3K and loss-of-function mutations in PTEN (phosphatase and tensin homologue lipid phosphatase), the antagonist of PI3K 13–20. These genetic changes lead to elevated levels of PIP3 that facilitate increased membrane recruitment and phosphorylation of AKT1. In the absence of enhanced membrane association, overexpressed wild-type AKT1 is not oncogenic in vitro or in vivo. Fusion to a membrane-targeting myristylation signal leads to constitutive activity and oncogenicity21. All isoforms of Akt have been observed to be amplified or overexpressed in human cancer, but this change alone is insufficient to cause transformation21–23.
As an intermediary between mutated upstream regulatory proteins and downstream signaling molecules, AKT1 seemed to have a passive role in human cancer, until Carpten and coworkers discovered a cancer-specific mutation that results in a glutamic acid to lysine substitution at amino acid 17 in the PH domain of AKT124. The E17K mutation alters the affinity of AKT1 for phosphoinositides25. The mutation leads to elevated membrane association of AKT1 and to constitutive activation of the kinase 24–27.
In this study, we determined the properties of the E17K mutation in greater detail and explored the role of the PH domain-PIP interaction in oncogenesis and signaling. The PH domain of wild-type AKT1 was substituted with either the PH domain of general receptor for phosphoinositides-1 (GRP1) that is specific for PIP3 or the PH domain of phospholipase C-delta1 (PLCδ1), specific for PIP2 28, 29. E17K-AKT1, GRP-AKT1 and PLC-AKT1 were studied in tests for oncogenic potential and signaling. Wild-type AKT1 served as negative control, and the constitutively active and oncogenic myristylated AKT1 (myrAKT1) served as positive control 21 (Figure 1a). To facilitate identification, the AKT1 proteins used in this study were fused to a C-terminal HA-tag. We expressed the different proteins from the retroviral vector RCAS in chicken embryonic fibroblasts (CEF). Cells transfected with this vector release infectious avian retrovirus that expresses the insert and infects surrounding cells. Tests for oncogenic potential were carried out on monolayers of primary CEF. In this system, transformed cells, under an overlay of nutrient agar, grow into multilayered microtumors, termed foci 30. E17K-AKT1 induced such foci of transformation in CEF, confirming the oncogenic activity seen in previous experiments with Rat1 cells24. E17K-AKT1 was comparable in oncogenic activity to the positive control, myrAKT1. The PLC-AKT1 chimera was also potently transforming. In contrast, wild-type AKT1 did not induce focus formation within 17 days of incubation, and the oncogenic activity of GRP-AKT1 was marginal, only detectable at high concentrations of transfected DNA (Figure 1b). The efficiencies of transformation for the various constructs were as follows: E17K-AKT1: 400 foci/µg plasmid DNA, PLC-AKT1: 750 foci/µg plasmid DNA, myrAKT1: 650 foci/µg plasmid DNA and GRP-AKT1: 20 foci/µg plasmid DNA.
Figure 1. PIP2-specific AKT1 induces oncogenic transformation in CEF.


(a) Schematic structure of AKT1 constructs.
Human full-length AKT1 (NM_005163.2) was fused to an HA epitope tag on the carboxy terminus. E17K-AKT1 and myrAKT1 were generated using human wild-type AKT1 as template for PCR. The chimeric constructs GRP-AKT1 and PLC-AKT1 were obtained by substituting the PH domain of AKT1 with either the PH domain of GRP1 (NM_004227.3) or PLCδ1 (NM_006225). All constructs were subsequently cloned into the avian retroviral expression vector RCAS(A) 21.
(b) Focus assay with AKT1 constructs.
CEF were transfected with RCAS(A) encoding wild-type AKT1, E17K-AKT1, myrAKT1, GRP-AKT1 or PLC-AKT1 using the dimethyl sulfoxide/polybrene method 45. Cells were overlaid with agar medium every 2–3 days and stained with 2% (w/v) crystal violet in 10% (v/v) methanol after 17 days. A representative experiment out of three independent ones is shown.
Tests for in vivo oncogenicity were carried out on the chorioallantoic membrane (CAM) of the chicken embryo. The CAM is a vascularized membrane, located underneath the shell and engulfing the embryo. It is a readily accessible and sensitive model for the evaluation of angiogenesis and tumor formation in vivo 31–33. The CAMs of 9-day-old chicken embryos were inoculated with the various AKT1 constructs. Inoculation with wild-type AKT1 or empty RCAS vectors served as negative controls, and the oncogenic myrAKT1 served as a positive control. All CAMs inoculated with myrAKT1, E17K-AKT1 or PLC-AKT1 displayed increased vascularization and formation of neoplastic nodules (Figure 2). Areas with abnormal cell growth were marked by elevated angiogenesis. Histological analysis of the tumor confirmed the malignant nature of the growths on E17K-AKT1, PLC-AKT1- and myrAKT1-inoculated CAMs (data not shown). One CAM out of five inoculated with CEF expressing the non-transforming GRP-AKT1 variant showed a small area of hyperplasia that did not reveal malignant features in hematoxylin- and eosin-stained sections, confirming the observations in cell culture.
Figure 2. Tumor induction and angiogenesis induced by PIP2 affine AKT1 constructs in the CAM of the chicken embryo.
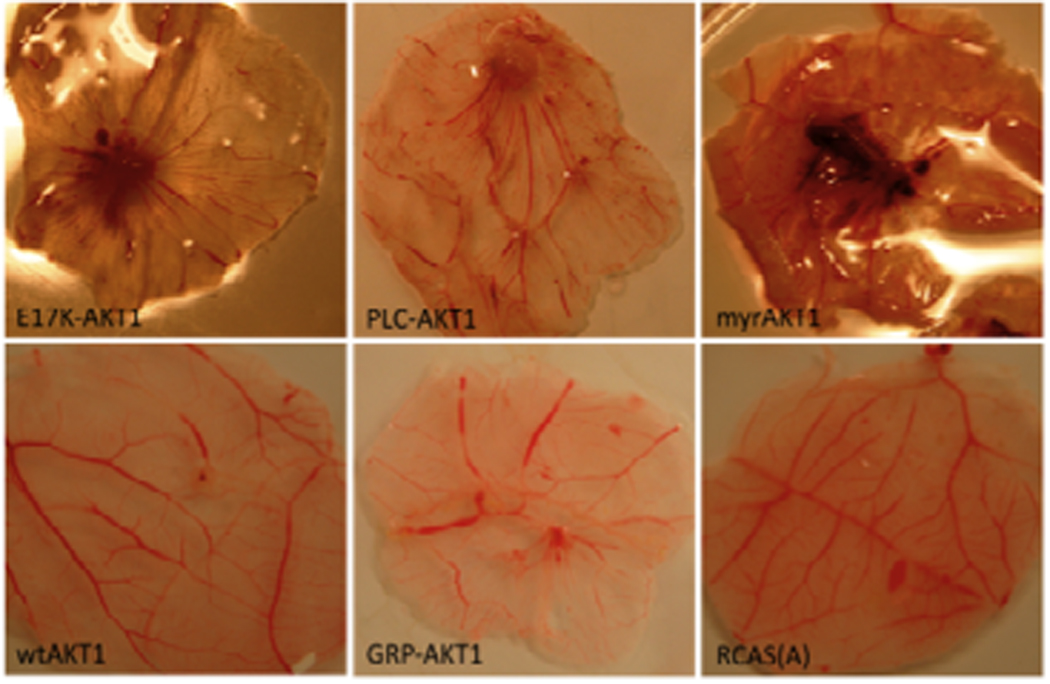
CAM assays were performed with fertilized chicken eggs (White Leghorn) obtained from Charles Rivers Breeding Laboratories (Wilmington, MA). Eggs were incubated at 37.5 °C and 70% humidity for 9 days. Eggs were candled to confirm embryo growth and to locate vascularized areas of the CAM. Two holes were drilled, one into the air sac at the bottom of the eggs, another one at the lateral end above the blood vessels, penetrating through the outer shell membrane without injuring the CAM. Applying suction to the air sac exposed the CAM. CAMs were inoculated with AKT1, E17K-AKT1, myrAKT1, GRP-AKT1, PLC-AKT1 or empty vector. Eggs were sealed and incubated for another 9 days. A representative experiment out of three independent ones is shown.
Activated AKT1 is phosphorylated at T308 and S473 and catalyzes the phosphorylation of numerous downstream targets. Among them glycogen synthase kinase-3 β (GSK3β), a substrate of AKT1, and ribosomal protein S6, a substrate of p70S6K which is activated downstream of AKT1. In CEF expressing E17K-AKT1, PLC-AKT1 or myrAKT1, AKT1 was highly phosphorylated at T308 and S473 in the presence of serum and under serum-starved conditions. Wild-type AKT1- or GRP-AKT1-expressing cells showed only low levels of AKT1 phosphorylation in the presence of serum, and under serum-starved conditions, phosphorylation of AKT1 was barely detectable despite efficient expression of the proteins (Fig. 3). The downstream targets GSK3β and S6 were preferentially phosphorylated in E17K-AKT1-, PLC-AKT1- or myrAKT1-expressing cells and were not phosphorylated in CEF expressing wild-type AKT1 or GRP-AKT1. Wild-type AKT1 and GRP-AKT1 were, however, functionally intact, because they were capable of being phosphorylated in the presence of an upstream activating signal such as serum stimulation as shown in figure 3 and can be transformed by overexpression of p110δ (data not shown). GRP-AKT1 also does not function as a dominant negative in cells transformed by gain-of-function mutants of AKT1 (data not shown).
Figure 3. Phosphorylation events associated with expression of Akt constructs.

CEF transfected with RCAS constructs encoding wtAKT, E17K-AKT, myrAKT, GRP-AKT, PLC-AKT or empty vector were incubated under normal and serum starved condition. Whole cell lysates were electrophoresed and analysed by immunoblotting with anti-phospho-AKT (S473), anti-phospho-AKT (T308), anti-phospho-S6 (S235/S236), anti-phospho-GSK-3b (S9), anti-HA and anti-β-Actin, all obtained from Cell Signaling Technology® (Danvers, MA).
PLC-AKT1 expression leads to constitutive signaling and cellular transformation, possibly by constitutive membrane localization. The PH domain of PLCδ1 is a high affinity and high specificity binder of PIP2 in the plasma membrane. Furthermore, the PH domain of PLCδ1 is especially attracted to particular arrangements of PIP2 found in caveolae34. In order to document the preferential localization of PLC-AKT1 to specific domains of the plasma membranes, PLC-AKT1- and GRP-AKT1-infected cells were seeded onto glass coverslips, serum starved, fixed with formaldehyde, probed using the Anti-HA antibody and visualized with FITC labeled anti-mouse antibody (Sigma, St Louis, MO). PLC-AKT1 localized primarily to apical sites of the fibroblasts which may be enriched in the PIP2-concentrating structures described recently34. In contrast, GRP-AKT1 did not localize strongly to these areas or the plasma membrane in general (Fig. 4). There is increased association of GRP1-Akt with the nucleus or nuclear membrane. This localization pattern has been previously noted, but the origin of this distribution pattern is unknown35.
Figure 4. Intracellular localization of Akt constructs.

CEF transfected with PLC-AKT1 (A–C) or GRP-AKT1 (D–F) constructs were incubated under serum starved conditions. Cells were fixed with formaldehyde and probed using an Anti-HA antibody. Images were taken at 100× maginification on an Axiovert 100TV fluorescence microscope.
E17K-AKT1, PLC-AKT1 and myrAKT1 induce oncogenic transformation of CEF as well as tumor formation and angiogenesis in CAMs of chicken embryos. These constructs also initiate constitutive downstream signaling under serum-starved conditions. In contrast, the mere overexpression of wild-type AKT1 is insufficient to induce oncogenesis and fails to generate a significant downstream signal in the absence of growth factors. Constitutive localization at the plasma membrane and the resultant elevated phosphorylation and kinase activity are essential requirements for the oncogenicity of AKT1. This study shows that changes in the PH domain that enhance membrane recruitment can trigger the latent oncogenic potential of AKT1. It is not known to what extent the gain of function observed with the AKT1 constructs studied here is caused by an enhanced interaction with all phosphoinositides or with the more abundant PIP2 specifically. In wild-type AKT1, the E17 occupies the phosphoinositide-binding pocket. Upon binding to PIP3 or PIP2, E17 swings away from the pocket. The E17K mutation changes the conformation and surface charge of the PH domain; K17 does not occupy the binding pocket, but forms a hydrogen bond with Y18 24. The mutation leads to constitutive membrane localization of AKT1, possibly because of an increased affinity in the AKT-phosphoinositide interaction or a reduced off rate 24, 25. The oncogenic activity of PLC-AKT1 is mediated by its ability to bind to PIP2. The cellular concentrations of PIP2 are higher than those of PIP3, and binding to PIP2 may effectively cause constitutive membrane localization but may also involve changes in affinity 29, 36. GRP-AKT1 shows marginal oncogenic activity and is inactive in signaling, probably because it binds exclusively to the much less abundant PIP3 37. In this construct, the PH domain may not occlude T308 as effectively as the native PH domain of wild-type AKT1. A low level of PDK1-mediated phosphorylation of T308 may thus occur constitutively and may account for the marginal oncogenicity of GRP-AKT1. AKT1 from which the PH domain is deleted is also constitutively active38, 39. PDK1 is largely but not entirely membrane-associated. Its PH domain interacts with high affinity with PIP3 and with lower affinity with PIP2, and it can activate AKT1 under serum-starved conditions 29.
Our study confirms the potent oncogenic activity of the E17K mutation described by Carpten and coworkers 24 and suggests that other changes in the PH domain of AKT1 could lead to a gain of function and to oncogenicity. In human cancer, E17K would likely function as a driver mutation, determining the neoplastic phenotype of the cancer cell24, 27. Current research efforts to control PI3K-AKT signaling focus heavily on inhibitors of the catalytic subunit of PI3K 40–44. Such inhibitors would not affect gain-of-function mutations in AKT1. AKT1 therefore remains an important therapeutic cancer target.
Acknowledgments
This work is supported by grants from the National Cancer Institute and by the Stein Endowment Foundation. This is manuscript number 19904 of The Scripps Research Institute.
References
- 1.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 2.Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol. 1997;17:338–344. doi: 10.1128/mcb.17.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 4.Bozulic L, Hemmings BA. PIKKing on PKB: regulation of PKB activity by phosphorylation. Curr Opin Cell Biol. 2009;21:256–261. doi: 10.1016/j.ceb.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Calleja V, Laguerre M, Larijani B. 3-D structure and dynamics of protein kinase B-new mechanism for the allosteric regulation of an AGC kinase. J Chem Biol. 2009;2:11–25. doi: 10.1007/s12154-009-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 7.Currie RA, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, Cohen P, Alessi DR, Lucocq J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J. 1999;337(Pt 3):575–583. [PMC free article] [PubMed] [Google Scholar]
- 8.Filippa N, Sable CL, Hemmings BA, Van Obberghen E. Effect of phosphoinositide-dependent kinase 1 on protein kinase B translocation and its subsequent activation. Mol Cell Biol. 2000;20:5712–5721. doi: 10.1128/mcb.20.15.5712-5721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McManus EJ, Collins BJ, Ashby PR, Prescott AR, Murray-Tait V, Armit LJ, Arthur JS, Alessi DR. The in vivo role of PtdIns(3,4,5)P3 binding to PDK1 PH domain defined by knockin mutation. EMBO J. 2004;23:2071–2082. doi: 10.1038/sj.emboj.7600218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, Van Aalten DM. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J. 2003;375:531–538. doi: 10.1042/BJ20031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road to PKB. Biochem J. 2000;346(Pt 3):561–576. [PMC free article] [PubMed] [Google Scholar]
- 12.Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science. 1997;277:567–570. doi: 10.1126/science.277.5325.567. [DOI] [PubMed] [Google Scholar]
- 13.Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103:1475–1479. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carson JD, Van Aller G, Lehr R, Sinnamon RH, Kirkpatrick RB, Auger KR, Dhanak D, Copeland RA, Gontarek RR, Tummino PJ, Luo L. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J. 2008;409:519–524. doi: 10.1042/BJ20070681. [DOI] [PubMed] [Google Scholar]
- 15.Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–4567. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- 16.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 18.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 19.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 20.Sugita H, Dan S, Kong D, Tomida A, Yamori T. A new evaluation method for quantifying PI3K activity by HTRF assay. Biochem Biophys Res Commun. 2008;377:941–945. doi: 10.1016/j.bbrc.2008.10.083. [DOI] [PubMed] [Google Scholar]
- 21.Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK. The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci U S A. 1998;95:14950–14955. doi: 10.1073/pnas.95.25.14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci U S A. 1992;89:9267–9271. doi: 10.1073/pnas.89.19.9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci U S A. 1987;84:5034–5037. doi: 10.1073/pnas.84.14.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 25.Landgraf KE, Pilling C, Falke JJ. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry. 2008;47:12260–12269. doi: 10.1021/bi801683k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malanga D, Scrima M, De Marco C, Fabiani F, De Rosa N, de Gisi S, Malara N, Savino R, Rocco G, Chiappetta G, Franco R, Tirino V, et al. Activating E17K mutation in the gene encoding the protein kinase AKT in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2007;7 doi: 10.4161/cc.7.5.5485. [DOI] [PubMed] [Google Scholar]
- 27.Shoji K, Oda K, Nakagawa S, Hosokawa S, Nagae G, Uehara Y, Sone K, Miyamoto Y, Hiraike H, Hiraike-Wada O, Nei T, Kawana K, et al. The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br J Cancer. 2009;101:145–148. doi: 10.1038/sj.bjc.6605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DiNitto JP, Cronin TC, Lambright DG. Membrane recognition and targeting by lipid-binding domains. Sci STKE. 2003;2003:re16. doi: 10.1126/stke.2132003re16. [DOI] [PubMed] [Google Scholar]
- 29.DiNitto JP, Lambright DG. Membrane and juxtamembrane targeting by PH and PTB domains. Biochim Biophys Acta. 2006;1761:850–867. doi: 10.1016/j.bbalip.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Temin HM, Rubin H. Characteristics of an assay for Rous sarcoma virus and Rous sarcoma cells in tissue culture. Virology. 1958;6:669–688. doi: 10.1016/0042-6822(58)90114-4. [DOI] [PubMed] [Google Scholar]
- 31.Eliceiri BP, Klemke R, Stromblad S, Cheresh DA. Integrin alphavbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J Cell Biol. 1998;140:1255–1263. doi: 10.1083/jcb.140.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang BH, Zheng JZ, Aoki M, Vogt PK. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A. 2000;97:1749–1753. doi: 10.1073/pnas.040560897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scher C, Haudenschild C, Klagsbrun M. The chick chorioallantoic membrane as a model system for the study of tissue invasion by viral transformed cells. Cell. 1976;8:373–382. doi: 10.1016/0092-8674(76)90149-5. [DOI] [PubMed] [Google Scholar]
- 34.Fujita A, Cheng J, Tauchi-Sato K, Takenawa T, Fujimoto T. A distinct pool of phosphatidylinositol 4,5-bisphosphate in caveolae revealed by a nanoscale labeling technique. Proc Natl Acad Sci U S A. 2009;106:9256–9261. doi: 10.1073/pnas.0900216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray A, Van Der Kaay J, Downes CP. The pleckstrin homology domains of protein kinase B and GRP1 (general receptor for phosphoinositides-1) are sensitive and selective probes for the cellular detection of phosphatidylinositol 3,4-bisphosphate and/or phosphatidylinositol 3,4,5-trisphosphate in vivo. Biochem J. 1999;344(Pt 3):929–936. [PMC free article] [PubMed] [Google Scholar]
- 36.Vadnal RE, Parthasarathy R. The identification of a novel inositol lipid, phosphatidylinositol trisphosphate (PIP3), in rat cerebrum using in vivo techniques. Biochem Biophys Res Commun. 1989;163:995–1001. doi: 10.1016/0006-291x(89)92320-6. [DOI] [PubMed] [Google Scholar]
- 37.Corbin JA, Dirkx RA, Falke JJ. GRP1 pleckstrin homology domain: activation parameters and novel search mechanism for rare target lipid. Biochemistry. 2004;43:16161–16173. doi: 10.1021/bi049017a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- 39.Okuzumi T, Fiedler D, Zhang C, Gray DC, Aizenstein B, Hoffman R, Shokat KM. Inhibitor hijacking of Akt activation. Nat Chem Biol. 2009;5:484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong D, Yamori T. Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci. 2008;99:1734–1740. doi: 10.1111/j.1349-7006.2008.00891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- 42.Maira SM, Voliva C, Garcia-Echeverria C. Class IA phosphatidylinositol 3-kinase: from their biologic implication in human cancers to drug discovery. Expert Opin Ther Targets. 2008;12:223–238. doi: 10.1517/14728222.12.2.223. [DOI] [PubMed] [Google Scholar]
- 43.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–5526. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 44.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Kawai S, Nishizawa M. New procedure for DNA transfection with polycation and dimethyl sulfoxide. Mol Cell Biol. 1984;4:1172–1174. doi: 10.1128/mcb.4.6.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]