

Abstract
Cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-activated chloride channel that is present in a variety of epithelial cell types, and usually expressed in the luminal membrane. In contrast, prestin (SLC26A5) is a voltage-dependent motor protein, which is present in the basolateral membrane of cochlear outer hair cells (OHCs), and plays an important role in the frequency selectivity and sensitivity of mammalian hearing. By using in situ hybridization and immunofluorescence, we found that both mRNA and protein of CFTR are present in OHCs, and that CFTR localizes in both the apical and the lateral membranes. CFTR was not detected in the lateral membrane of inner hair cells (IHCs) or in that of OHCs derived from prestin-knockout mice, i.e., in instances where prestin is not expressed. These results suggest that prestin may interact physically with CFTR in the lateral membrane of OHCs. Immunoprecipitation experiments confirmed a prestin-CFTR interaction. Because chloride is important for prestin function and for the efferent-mediated inhibition of cochlear output, the prestin-directed localization of CFTR to the lateral membrane of OHCs has a potential physiological significance. Aside from its role as a chloride channel, CFTR is known as a regulator of multiple protein functions, including those of the solute carrier family 26 (SLC26). Because prestin is in the SLC26 family, several members of which interact with CFTR, we explored the potential modulatory relationship associated with a direct, physical interaction between prestin and CFTR. Electrophysiological experiments demonstrated that cAMP– activated CFTR is capable of enhancing voltage-dependent charge displacement, a signature of OHC motility, whereas prestin does not affect the chloride conductance of CFTR.
Keywords: CFTR, prestin, SLC26A, chloride, outer hair cells, nonlinear capacitance
INTRODUCTION
Inner (IHCs) and outer hair cells (OHCs) are the two mechanoreceptor cell populations housed in the sensory organ of mammalian hearing: the organ of Corti. IHCs, which convert the mechanical force arising from sound waves into neurotransmitter release, are considered the true sensory receptors for hearing [1]. In contrast, OHCs undergo rapid somatic length changes when the voltage across their basolateral membranes is altered [2–5]. This somatic electromotility is thought to function as a constituent of the cochlear amplifier by providing local mechanical enhancement of the basilar membrane’s vibratory pattern [1, 6]. Prestin is responsible for somatic electromotility of OHCs [7], and is essential for normal hearing sensitivity and frequency selectivity of mammals [8–11]. This OHC motor protein belongs to a distinct anion transporter family called solute carrier protein 26 (SLC26). Although prestin’s anion transport properties are controversial [12–15], anions, principally Cl−, are required for prestin’s activity [16, 17].
Aside from its reported function in efferent-mediated inhibition of cochlear output [18], intracellular Cl− in OHCs is essential for enabling prestin’s function and, thus, cochlear feedback amplification [16, 17, 19]. Consequently, a delineation of Cl− channels and transporters that are native to OHCs is of interest, especially since they have not been fully characterized [20]. At present, it is known that hyperpolarization-activated ClC-2 channels [21, 22] and a non-selective stretch-activated conductance, named GmetL, are present in the lateral plasma membrane of OHCs [16]. However, one ubiquitous Cl− channel, CFTR, has received relatively meager attention in the hearing literature. The cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel that regulates ion balance [23, 24]. The gene coding for CFTR is highly conserved among species, and certain mutations of the gene are responsible for cystic fibrosis (CF) [25]. Because CFTR is present in a wide variety of cell types, it is possible that this chloride channel is also present in hair cells. In a previous study, contribution of the CFTR chloride conductance to the relatively low membrane resistance found in OHCs was tested and questioned [16]. However, this study does not rule out the possibility that CFTR is present and, when activated, influences OHCs, since the regulatory status of CFTR was unknown in this particular experiment. If the channel were inactivated, one would not observe CFTR’s electrophysiological signature. Hence, knowing whether CFTR is present in OHCs is important for understanding the regulatory mechanisms of these cells.
Besides being a chloride channel, CFTR is also a regulator of a wide range of other ion channels and transporters through inhibition or activation [26]. Recently, several laboratories have reported that CFTR can interact with members of the SLC26A family including PAT1 (SLC26A6), DRA (down-regulated in adenoma, SLC26A3), and PDS (Pendred’s syndrome, SLC26A4). This interaction has been shown to affect ion transport by these proteins [27–31]. Inasmuch as prestin belongs to the SLC26 family, it is possible that prestin function is modulated by CFTR or vice versa. Because prestin function is essential for normal hearing in mammals, any of its alterations are of potential physiological significance.
MATERIALS AND METHODS
Chemicals, plasmids, and antibodies
Rabbit polyclonal anti-mPres antibody was raised by immunization to a carboxy terminus motif of mouse prestin, which has been previously characterized [32]. Anti-mPres was used in a 1:2000 dilution in immunofluorescence and Western blot experiments. Anti-CFTR (H-182), anti-CFTR (M-15), anti-CFTR (AB3555), and anti-CFTR (M3A7) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA), Santa Cruz Biotechnology Inc. (Santa Cruz, CA), Millipore Corp. (Bedford, MA), and Upstate Biotechnology (Lake Placid, NY), respectively. Mouse anti-V5 and rabbit anti-V5 were purchased from Invitrogen (Carlsbad, CA) and Sigma (St. Louis, MO), respectively, and used in a 1:5000 dilution in immunofluorescence and Western blot experiments. Texas Red-X phalloidin was purchased from Molecular Probes (Eugene, OR). Secondary antibodies, Alexa 488 and Alexa-546 conjugated to anti-rabbit IgG and anti-donkey IgG were purchased from Molecular Probes (Eugene, OR), Pierce (Rockford, Il) or Jackson Immuno Research (Bar Harbor, ME). Dr. S. Muallem kindly provided the pcDNA3/CFTR and GFP-CFTR plasmid constructs. Prestin-CFP and C-tag-Prestin plasmid constructs have CFP and His-V5 tags attached to the C-terminus of prestin [32, 33]. The plasmid pcDNA3.1-CAT (Choramphenicol acetyl-transferase) was used as a transfection control in the expression studies. ATP, cAMP, forskolin (Fsk), and 3-isobutyl-1-methylxanthine (IBMX) were purchased from Sigma (St. Louis, MO). CFTR chloride conductance blocker, CFTRinh-172, was purchased from Calbiochem (La Jolla, CA).
In situ hybridization
All surgical and experimental procedures were conducted in accordance with the policies of Northwestern University’s Animal Care and Use Committee and the NIH. The detailed procedures for producing whole-mount cochlear samples were described in [34]. Briefly, adult mice were cardiac perfused, first with heparinized saline and then with 4% formaldehyde, followed by at least 48 hrs post-fixation. Cochleae were dissected and decalcified in 10% EDTA for at least 24 hrs. An approximately 300-bp fragment corresponding to the CFTR cDNA (4306–4565 bp) was cloned into pGEM-7Zf (+). Anti-sense mRNA was labeled with Dig-UTP using a T7 promoter from the Dig RNA labeling Kit (Roche Applied Science). Similar length of mRNA probe made from PGEM-7Zf plasmid without CFTR insert was used as a negative control. After purification of RNA probes with ChromaSpin-30 columns (Clontech), the Dig-labeled probe was used to hybridize cochlear tissue. Some samples were mounted on glass slides and viewed with a standard microscope. Images were captured with a CCD camera.
RT-PCR and DNA sequencing
RNA was isolated from intact cochleae of wildtype mice using the Absolutely RNA®RT-PCR Miniprep Kit (Stratagene). 200–300 ng total RNA was reverse transcribed to cDNA using Superscript II, RNase H-Reverse Transcriptase at 42° C for 90 min. After cDNA creation, fifteen primers (forward and backward) were used to detect different coding regions of CFTR. Cycling conditions were as follows: denature at 95° C for 1 min followed by 45 cycles at 95° C for 15 sec, 54–58° C for 30 sec and, finally, 72° C for 1 to 2 min dependent on primer pair. PCR products were visualized on a 1% or 2% agarose/EtBr gel and further purified for DNA sequencing.
Immunofluorescence experiments
Separate procedures were used for anti-CFTR antibodies purchased from different sources. For anti-CFTR (H-182, Santa Cruz Biotechnology Inc.), cochleae were dissected from adult mice in L-15 medium. The bony wall of the cochlear apex was removed to expose the apical turn of the organ of Corti. The cochlea was fixed with 100% methanol at −20°C for 20 minutes. Cochleae were incubated in blocking solution (PBS plus 10% normal goat serum) at room temperature for 1 hr and then with anti-CFTR (1:200 H-182 from Santa Cruz) at 4 °C overnight. The tissue samples were then exposed to anti-rabbit-IgG conjugated with Alexa 488 (1:500) and Texas Red-X phalloidin (1:2000). For immunostaining with anti-CFTR (Millipore) and coimmunostaining with anti-CFTR (M-15, Santa Cruz) and anti-mPres, adult mice were cardiac perfused first with heparinized saline and then with 4% formaldehyde. After ~2 hours post fixation in 4% formaldehyde at room temperature, cochleae were dissected and treated with 0.2% Triton X-100/PBS for 30 min. 1% BSA and 10% normal goat or donkey serum were used to block nonspecific binding. The tissue samples were then exposed to anti-CFTR (Millipore) at 1:100 dilution, or anti-CFTR (M-15, 1:100) plus anti-mPres at 1:10,000 for co-staining. The tissue samples were then exposed to anti-rabbit-IgG conjugated with Alexa 488 at 1:500 dilution, and anti-goat-IgG conjugated with Alexa546 at 1:500 for co-staining. Samples were mounted on glass slides with Fluoromount-G (Southern Biotechnology Associates, Inc., Birmingham, AL) and observed at room temperature by object lenses of either 20× (0.5 numerical aperture) or 63× (1.32 numerical aperture, oil) using a Leica confocal system with a standard configuration DMRXE7 microscope.
Cell culture and transient transfection
Plasmids encoding Prestin and CFTR cDNA were expressed in HEK293T cells in a ratio of 1:1. Cells were transiently transfected using Effectene (Qiagen, Valencia CA) as the transfection reagent as described previously [35]. After 48 hours incubation, the transiently transfected cells were used for NLC measurements or Co-Immunoprecipitation (Co-IP) experiments.
Co-Immunoprecipitation and immunobloting
HEK293T cells were transiently transfected with V5-His tagged prestin and CFTR at a ratio of 1:3. 48 hrs after transfection, some transfected cells were treated with 12.2 μM Fsk and 0.1mM IBMX for 30 min. All cells were lysed in lysis buffer (1× TBS, 0.1% NP40, 1mM EDTA, 10mM phenylmethansulphonylfluoride (PMSF), 1× Protease Inhibitor cocktail pH 7.6) for 1.5 hr at 4°C. Cellular residues were sedimented at 10,000g for 15 min and the supernatant applied to Protein A sepharose beads that had been pre-washed with lysis buffer. 2 μg anti-V5 was added to the solution, and the beads were incubated overnight, rotating, at 4°C. The next day, the beads were washed 4 times with lysis buffer. Prestin was eluted by the addition of 2× LDS buffer with 0.1M DTT and incubated at 37°C for 30 min. Fractions were run on a 7.5% Next gel and blotted with either anti-V5 or anti-CFTR (M3A7). Cochleae derived from adult wildtype and prestin-KO mice [36] were dissected in L-15 or L-15 medium containing 12.2 μM Fsk and 0.1mM IBMX. Procedures for handling cochlear samples were similar to those for HEK293T cells except cell lysates were solubilized with 8% perfluorooctanoate (PFO) for 4 days. The lysates were precipitated by anti-CFTR (H182 or M3A7)/Protein A sepharose. Eluted proteins from Protein A sepharose were run on LDS-PAGE along with reducing reagents β-mercaptoethanol and DTT or EDT [37] and blotted with anti-mPres.
Electrophysiology
Voltage-dependent membrane capacitance was measured from HEK293T cells 48 to 72 hours after transfection or from mouse OHCs within two hours after isolation. For preparing OHCs, adult mice were euthanized with euthasol, and OHCs were isolated in the same way as described before [38]. Whole-cell recordings were performed using the Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) at room temperature. Recording pipettes were pulled from borosilicate glass to achieve initial bath resistances averaging 2–3 MΩ for HEK293T cells and 4-5MΩ for OHCs, and were filled with an intracellular solution (ICS) containing (mM): 140 CsCl, 2 MgCl2, 10 EGTA, and 10 HEPES. Cells were bathed during whole-cell recordings in an extracellular solution (ECS) containing (mM): 120 NaCl, 20 TEA-Cl, 2 CoCl2, 2 MgCl2, 10 HEPES, and 5 glucose. Osmolarity and pH of both ICS and ECS were adjusted to 300 mOsml−1 (with glucose) and 7.3, respectively. Data were collected using jClamp software (SciSoft Company, New Haven, CT). Membrane capacitance was measured by a two-sine voltage stimulus (390.6 and 781.2 Hz with 10 mV amplitude) superimposed onto a sinusoidal stimulus (2.5 Hz with 120 mV amplitude) with holding potential at 0mV. OHCs and HEK293T cells expressing prestin exhibit nonlinear capacitance (NLC), and the voltage-dependent membrane capacitance is described as:
where Qmax is the maximum charge transfer, Vpk is the voltage at which half maximum charge is moved, Clin is the linear capacitance of a cell, and α (= ze/kBT) is the slope factor of the voltage-dependence of charge transfer where kB is Boltzmann’s constant, T is absolute temperature, z is valence, and e is electron charge. GraphPad PRISM software was used for curve fitting analysis of NLC. For activation of CFTR, 0.2mM cAMP, 1mM ATP, and 50μM IBMX were added to the intracellular pipette solution, both in HEK293T cells and OHCs. For recording NLC in HEK293T cells in the presence of cAMP/ATP/IBMX, CFTRinh-172 (less than 10 μM, see below) was included in the ECS to maintain the membrane resistance higher than 50MΩ because low membrane resistance (thus high membrane conductance) interferes with the measurement of NLC. Because DMSO affects prestin function[39], ethanol was used to prepare 1 mM CFTRinh-172 stock solution although ethanol did not completely dissolve CFTRinh-172. After adding 10 μM-equivalent CFTRinh-172 to the ECS, a significant amount of CFTRinh-172 crystal was observed, suggesting that the actual concentration of CFTRinh-172 in ECS was very low (≪10 μM). This explains the slow kinetics of chloride conductance blockage exemplified in Fig. 8.
Fig. 8. Qmax enhancement by activated CFTR with blocked chloride conductance.

Time-dependent NLC was measured on HEK293T cells expressing both prestin-CFP and CFTR-GFP. The intracellular pipette solution contained 0.2mM cAMP, 1mM ATP, and 50μM IBMX for rapid activation of CFTR. The recording started after establishing a whole-cell configuration. A low concentration of CFTRinh-172 (≪ 10μM, see Materials and Methods) was included in the bath solution to block the chloride conductance of CFTR. The initial rapid increment of chloride conductance was not completely blocked by CFTRinh-172, and was followed by a slow attenuation. This is because of the very low concentration of CFTRinh-172 (see Materials and Methods), and because the open state of CFTR is favorable for CFTRinh-172 action [63]. The broken line indicates the initial resistance level. Qualitatively similar results were obtained on different cells (n=3).
Voltage-dependent current was measured from HEK293T cells 48 to 72 hours after transfection in a whole-cell configuration at room temperature. Recording pipettes with initial bath resistance of 2–3 MΩ were filled with ICS containing (mM): 10 CsCl, 87 Na2SO4, 2 MgCl2, 10 EGTA, and 10 HEPES. Cells were bathed during whole cell recordings in ECS containing (mM): 87 Na2SO4, 10 TEA-Cl, 2 CoCl2, 2 MgCl2, and 10 HEPES. Since the chloride conductance upon CFTR activation with the ICS and the ECS used for the NLC measurements was very high, and the contribution of the series resistance (Rs) to the observed conductance was significant in some recordings, reduced chloride solutions were used. Osmolarity and pH of both ICS and ECS were adjusted to 300 mOsml−1 (with glucose) and 7.3, respectively. 0.2mM cAMP, 1mM ATP, and 50μM IBMX were added to the intracellular pipette solution for activation of CFTR. Activation of CFTR was monitored by tracking the reduction of membrane resistance. After the CFTR activation reached a plateau, voltage-dependent current was measured by stepping from −60mV to +60mV (500 msec duration in 20mV increments) at 0mV holding potential. Data were corrected for the Rs. The conductance was obtained from the slope of the current-voltage relationship and divided by the membrane capacitance for comparison of data from different cells.
Statistical analysis
Whether prestin-dependent charge movement is significantly enhanced by the activation of CFTR or not was evaluated by the following t-test using the NLC data obtained at the end of each time course.
Qrel is the relative magnitude of Qmax compared to the Qmax that was measured right after establishing the whole-cell configuration. SEtrans is the standard error of the Qrel value representing the error transmission for calculating Qrel together with the uncertainty of the Qmax determination. The p-values were calculated from the Student’s t-distribution by using the t value defined above. P-values smaller than 0.05 were accepted as significantly different.
RESULTS
CFTR mRNA and protein are expressed in IHCs and OHCs
In situ hybridization is a technique used to detect, visualize and localize specific mRNA at the cellular level. Therefore, we used this method to examine cochlear CFTR mRNA distribution. Fig. 1 shows portions of a whole-mount cochlea stained with a control probe and anti-sense probe for CFTR. As shown in Figs. 1A and 1C, CFTR mRNA (brown color) was detected in both types of hair cells. A negative-control probe produced no staining (Figs. 1B and 1D). These data suggest that CFTR mRNA is expressed in IHCs and OHCs. Consistent with this result, mRNA of CFTR was also detected by PCR in the cochlea (see supplemental material Fig. S1).
Fig. 1. CFTR mRNA expression in the cochlea.

Adult mouse cochleae were treated with either an anti-sense probe for CFTR (A and C) or a control probe (B and D). A–B. Whole-mount cochlea taken with a 20× objective. C–D. Whole-mount cochlear sample taken using a 50× objective. Brown staining indicates the presence of CFTR mRNA.
We further examined CFTR protein expression in the organ of Corti using immunofluorescence. As shown in Fig. 2, green fluorescence (indicating CFTR protein) is found mostly in OHCs and IHCs (Fig. 2A). These data suggest that CFTR protein is expressed in IHCs and OHCs, consistent with the CFTR mRNA expression pattern (Fig. 1). High magnification immunofluorescence images reveal CFTR protein expression in the cytoplasm and the lateral membrane of OHCs as indicated by arrowheads in Fig. 2C and Fig. 3. In contrast, there is no staining in the lateral membrane of IHCs, i.e., the labeling predominates at the apex of the cell as shown in Fig. 2E–2K, which are images taken from the same cells. Fig. 2J and 2K show three IHCs, as examples, where no green stain (CFTR label) was found in the lateral membrane (indicated by small arrowheads). Thus, the subcellular localization of CFTR is different for OHCs and IHCs.
Fig. 2. CFTR protein expression in the cochlea.

CFTR is stained with anti-CFTR (green color, H182, Santa Cruz). Actin is stained with Texas Red-X phalloidin (red color, Molecular Probes). A–B. Immunofluorescence (A) and the corresponding phase-contrast images (B) of a whole-mount cochlea taken at a low magnification (20× objective). C–D. Immunofluorescence (C) and the corresponding phase-contrast images of a whole-mount cochlea show a cross section of OHCs. The lateral membrane of OHCs is indicated by arrowheads. E–I. Immunofluorescence (E and F) and the corresponding phase-contrast images (H) of a whole-mount cochlea. Mechanical manipulation was used to adjust cochlear samples, allowing some hair cells to lie flat on the slide. As a result, a quasi side-sectional image of IHCs was obtained. G. Superimposed images from E (green) and F (red). I. Superimposed images from E, F and H (phase contrast). J–K. These images correspond to the locations marked with “*” and are given at higher magnification for better examination of the lateral membrane of IHCs (arrowheads). Bar length: 20 μm (A, C, and E), 7 μm (J).
Fig. 3. CFTR localizes to the basolateral membrane of wildtype OHCs.

Immunofluorescent images of mouse OHCs derived from wildtype and prestin-KO mice are stained with anti-CFTR (for CFTR, green) and Texas Red-X phalloidin (for actin, red). A–H. Immunofluorescent and corresponding phase-contrast images taken at the cell-body position from whole-mount cochlear preparations. A–D: wildtype cochlea. E–H: prestin-KO cochlea. A and E were stained with anti-CFTR (against 1–182 a.a of CFTR, from Santa Cruz). C and G were stained with anti-CFTR (against 1468–1480 a.a of CFTR, from Millipore). I–P Immunofluorescent and corresponding phase-contrast images taken from radial views of OHCs stained with anti-CFTR (Santa Cruz) and Texas Red-X phalloidin. I–L: wildtype OHCs. M–P: prestin-KO OHCs. Green (CFTR) and red (actin) images were superimposed in K and O. Bar length: 12 μm.
CFTR and prestin protein are co-localized at the lateral membrane of wildtype OHCs
CFTR is an apical-membrane protein expressed in various epithelial cells [40]. It is uncommon for CFTR to be seen in the lateral membrane of polarized epithelial cells, such as OHCs [41]. It is, therefore, possible that a CFTR isoform exists in OHCs and that this isoform prefers the lateral membrane location. This possibility was investigated by using RT-PCR to explore the presence of a splicing isoform of CFTR, using organ of Corti cDNA as template material. Fifteen primer pairs, covering the entire CFTR coding region, were used for RT-PCR experiments. The PCR products were further sequenced to confirm the DNA content. No CFTR isoform was discovered.
Because it appears likely that interaction between prestin and CFTR changes CFTR’s location, we examined CFTR expression in wildtype OHCs and OHCs derived from prestin-KO mice, in which the prestin protein is absent [9, 10]. As shown in Fig. 3, CFTR protein is clearly expressed in the apical surfaces of both wildtype OHCs (Fig. 3I–3L) and prestin-KO OHCs (Fig. 3M–3P). However, CFTR is not found in the lateral membrane of OHCs derived from prestin-KO mice (Fig. 3E and Fig. 3G), even though the typical “ring” staining pattern (shown by arrowheads), is consistently found in OHCs derived from wildtype mice (Fig. 3A and Fig. 3C). Since the ring-like pattern could conceivably result from exclusion of the stain by the nucleus (unstained center = nucleus), we also examined OHC CFTR staining patterns from a radial view. OHCs were manipulated to lie flat on a slide. Radial views of OHCs also showed that CFTR staining was found in wildtype OHC lateral membranes (Fig. 3I–3L), but not in OHCs from prestin-KO (Fig. 3M–3P). Because false positive staining is often associated with an antibody’s lack of specificity, we used two antibodies against different antigens located at different locations of CFTR: 1–182 a.a. (H-182) (see Fig. 3A and 3E) and 1468–1480 a.a. (AB3555) (see Fig. 3C and 3G) in order to control for this potential error. We demonstrated that these two anti-CFTR antibodies do not cross react with prestin protein (see supplemental material Fig. S2). Thus, experimental results using either antibody suggest that CFTR is found in the apical regions of both OHC genotypes (WT: Fig. 3I–3L, KO: Fig. 3M–3P), but only in basolateral membranes of wildtype OHCs where prestin is abundantly expressed. To further test for the coexistence of CFTR and prestin in the lateral membrane of OHCs, we co-immunostained CFTR and prestin in OHCs (Fig. 4). As expected, the ring-like staining pattern of CFTR was evident (Fig. 4A), which overlaps with the prestin staining in the lateral membrane (Fig. 4B and C). These results confirm that CFTR is indeed present in the lateral membrane of OHCs and that this expression is associated with the presence of prestin.
Fig. 4. Co-immunostaining of CFTR and prestin in OHCs.

CFTR and prestin were co-stained with anti-CFTR (red, M-15, Santa Cruz) and anti-mPres (green), respectively. A. CFTR staining. B. prestin staining. C. Superimposed image from A and B. D. Phase-contrast image. The ring-like staining pattern for CFTR was not observed in OHCs derived from prestin-KO mice (not shown). The results are consistent with the observations in the preceding figures (Fig. 2 and 3).
Prestin binds CFTR
Given that the presence of prestin in OHCs changes the distribution of CFTR membrane targeting in polarized cells, it is of interest to determine whether prestin and CFTR bind together. Therefore, cochlear samples collected from both wildtype mice and prestin-KO mice were used for co-immunoprecipitation experiments with anti-CFTR/Protein A sepharose. Because the interaction between CFTR and DRA (SLC26A3) is known to be controlled by the regulatory state of CFTR [30], cochlear samples were dissected both in the presence and absence of forskolin (Fsk) and 3-isobutyl-1-methylxanthine (IBMX). The immunoprecipitated proteins were separated on LDS-PAGE gel and blotted with anti-mPres. As shown in Fig. 5A, the prestin monomer band (slightly less than 100 kD) was found in wildtype cochleae, suggesting that prestin can bind activated-CFTR. Prestin coimmunoprecipitation with anti-CFTR/Protein A sepharose was also observed without Fsk/IBMX treatment (data not shown). The results suggest that prestin can bind CFTR irrespective of the regulatory status of CFTR. We confirmed the prestin-CFTR binding in an in vitro system, whereby HEK293T cells were co-transfected with the V5-His tagged prestin and CFTR (Fig. 5B). The CFTR-prestin interaction was analyzed by a co-immunoprecipitation assay using anti-V5/Protein A sepharose (Fig. 5B). Multiple prestin bands were due to oligomerization/glycosylation of prestin that were repeatedly observed in mammalian cell lines [37, 42]. CFTR protein (~170kD [43]) coprecipitated with prestin irrespective of the Fsk/IBMX treatment, reaffirming the observation on isolated OHCs that CFTR binds to prestin regardless of its regulatory state. The prestin-CFTR binding was further confirmed using anti-CFTR/Protein A sepharose (data not shown). The results are consistent with the colocalization of CFTR and prestin observed in the lateral membrane of OHCs (Fig. 2 and Fig. 3). To exclude possible artifacts caused by the in vitro overexpression system, prestin was also co-expressed with a non-interacting protein, α-tectorin, in HEK293T cells. We confirmed that α-tectorin is not co-immunoprecipitated with prestin (data not shown).
Fig. 5. Prestin binds CFTR.
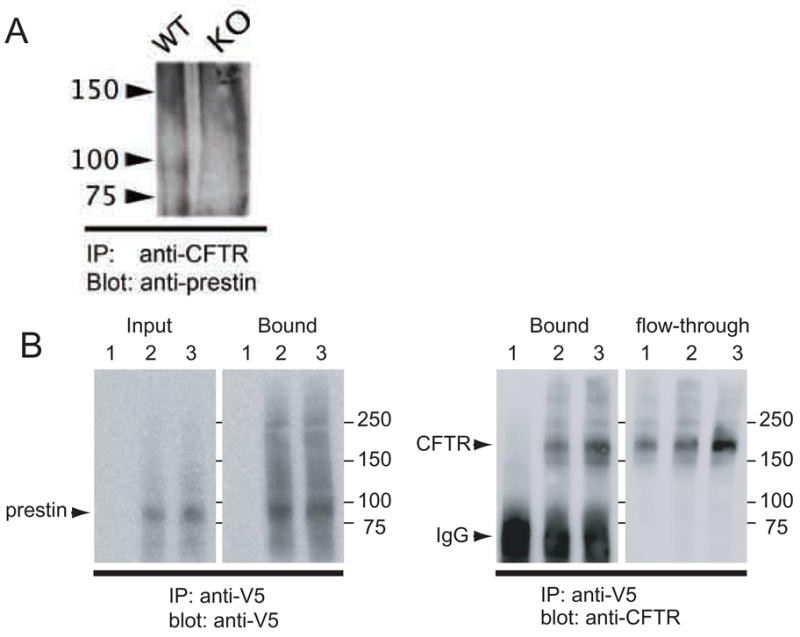
(A). Co-IP experiment: Cochlear samples derived from wildtype (WT) and prestin KO (KO) were treated with Fsk/IBMX, and were incubated with anti-CFTR/Protein A sepharose. The immunoprecipitated proteins were then subjected to LDS-PAGE, and the membrane was blotted with anti-mPres. A prestin band (slightly less than100 kDa) was found in WT, but not in KO samples. A similar result was obtained in the absence of Fsk/IBMX treatment (data not shown). (B). Pull down experiment: Detergent-solubilized fraction of HEK293T cells transfected with V5-His tagged prestin and CFTR were incubated with anti-V5/Protein A sepharose. Precipitated proteins were separated on SDS-PAGE, and blotted with anti-V5 (for prestin) and anti-CFTR, respectively. 1: HEK293T cells transfected with CFTR. 2: HEK293T cells transfected with V5-His tagged prestin and CFTR. 3: HEK293T cells transfected with V5-His tagged prestin and CFTR that were treated with 12.2 μM FSK and 0.1mM of IBMX.
Modulation of prestin function by CFTR
Cl− and OH−/HCO3− transport by members of the SLC26 family, including DRA (SLC26A3), PDS (SLC26A4), and PAT1 (SLC26A6), is known to be activated by CFTR [31]. In this study, we found that another SLC26 family member, prestin (SLC26A5), also interacts with CFTR. Thus, it is of interest to test if prestin’s function is modulated by CFTR. To this end, we measured NLC of HEK293T cells expressing either prestin or prestin/CFTR. For identifying cells expressing these proteins, we used CFP-tagged prestin and GFP-tagged CFTR. The heterologous expression of prestin conferred typical nonlinear capacitance (NLC) on the HEK293T cells as previously reported [7], suggesting that the CFP tag does not interfere with the function of prestin (Fig. 6A). Cells expressing both prestin and CFTR also showed NLC, which was similar to that of cells expressing only prestin (Fig. 6B). Because individual HEK293T cells differ in size, and because larger cells tend to express a larger number of prestin molecules, we calculated the charge density (the maximum charge displacement (Qmax) divided by the linear capacitance of a cell (Clin, which is proportional to the surface area of the cell), and used this metric for comparison (Fig. 6C). There was no statistically significant difference (p=0.71) in charge density between cells expressing only prestin (n=11) and cells expressing both prestin and CFTR (n=11), suggesting that the mere presence of CFTR has no modulatory effect on the charge displacement function of prestin. As a control, we also measured voltage-dependent capacitance of cells expressing only CFTR, and found that mere expression of CFTR did not confer NLC on the cells. This latter result was found for both activated and inactive CFTR (data not shown).
Fig. 6. Quiescent CFTR does not affect prestin function.

(A, B) Examples of NLC measured on HEK293T cells expressing either prestin-CFP (A) or both prestin-CFP and CFTR-GFP (B). (C) Summary of charge density. Bars indicate mean and standard deviation.
The effect of activated CFTR on prestin function was also studied. In these experiments, the cell was dialyzed in the whole-cell configuration, with cAMP/ATP/IBMX-containing intracellular solution in order to activate CFTR. As expected, cells overexpressing CFTR showed drastic decrements in membrane resistance, due to increased chloride conductance, as exemplified in the inset of Fig. 7A. We measured NLC of individual cells expressing both prestin and CFTR at different time points during the CFTR activation (Fig. 7A). As summarized in Fig. 7C, Qmax (the maximum charge translocated) showed an increase. In control cells overexpressing only prestin (Fig. 7B and 7D), neither a rapid decrement in the membrane resistance, nor an increment in Qmax was observed. The Qmax enhancements seen in cells overexpressing both prestin and activated CFTR were statistically significant (p<0.05) for all recordings (Fig. 7C, n=5, see Materials and Methods), whereas those without CFTR coexpression were not (p= 0.33–0.94) (Fg. 5D, n=6). Statistically significant difference was also manifested on the mean relative Qmax value taken at the end of each time course between cells overexpressing both prestin and CFTR vs. cells overexpressing only prestin (p=0.016) (Fig. 7E). These results strongly suggest that activated CFTR enhances the function of prestin, as found for other members in the SLC26 family, including DRA and PAT1 [31]. This enhancement is manifested as increased ion transport for other SLC26A members, but as increased charge transfer (motility) for prestin (SLC26A5). An alternative interpretation of the data might be that the increase in Qmax resulted from an increase in the number of prestin molecules in the cell membrane due to exocytic membrane trafficking of vesicles containing extra prestin/CFTR molecules during the measurement. Although exocytic vesicle transport induced by CFTR activation is a contentious issue [41, 44], we did not observe any increment in linear membrane capacitance upon cAMP/ATP/IBMX stimulation in HEK293T cells expressing CFTR (data not shown). It should be noted that the kinetics of CFTR exocytic membrane transport [45–49] is much slower than that of the Qmax enhancement (Fig. 7C), indicating that the Qmax change was not induced by CFTR-dependent membrane transport if, in fact, any occurred. Furthermore, the possibility of cAMP-dependent exocytic vesicle transport is not supported by our control measurements (Fig. 7B and Fig. 7D), in which we observed neither a rapid decrement in the membrane resistance, nor an increment in Qmax. These results indicate that the increasing Qmax is indeed activated-CFTR dependent, i.e., it is not caused by extra prestin molecules being transported to the cell membrane during the measurement. The control experiment is also contrary to the possibility that the Qmax change was induced by equilibration of cells with the intracellular solution during the measurements. The degree of Qmax enhancement was highly variable among cells. This likely reflects differences in the molar ratio of prestin and CFTR expressed in individual transfected cells. If the activation of prestin were achieved by a stoichiometric prestin-CFTR physical interaction, then the degree of change in Qmax should depend on the availability of CFTR to each prestin molecule: too much prestin over CFTR would attenuate the overall apparent relative Qmax change because of the scarcity of CFTR/prestin associated molecular pairs. Estimation of the prestin/CFTR expression in the plasma membrane of individual cells was difficult because fluorescence was often found in the cytosolic compartment due to overexpression of the proteins. Separating fluorescence signals in the plasma membrane from those in the cytosol was not feasible with the epi-fluorescence microscope used for the electrophysiological recordings.
Fig. 7. Effect of CFTR activation on prestin function.
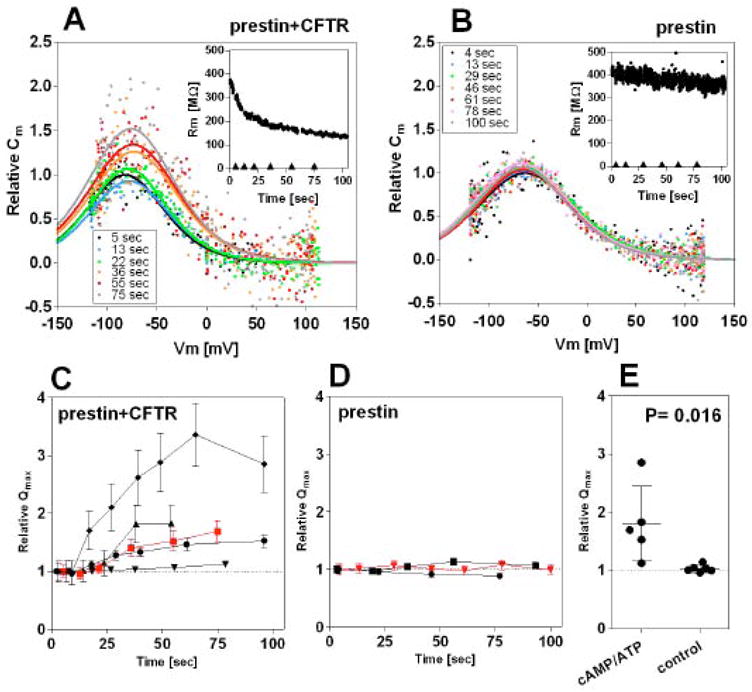
The intracellular pipette solution contained 0.2mM cAMP, 1mM ATP, and 50μM IBMX for activation of CFTR. Time-dependent NLC was measured on HEK293T cells expressing either prestin-CFP (B and D) or both prestin-CFP and CFTR-GFP (A and C). (A, B) Examples of the time-dependent NLC measurements. The insets show time course of the membrane resistance (Rm) changes. The NLC recordings were performed at the time points indicated (◆) after establishing whole-cell configuration. (C, D) Time-dependent relative Qmax values with respect to the Qmax measured at the beginning of the time course. Each symbol represents a different cell. The error bars indicate the accuracies of the relative Qmax values calculated from the standard errors of the curve fittings of the original NLC data, some of which are too small to be seen. Only three representative results (out of six recordings) are shown in D for clarity. The red symbols show the data points obtained from the time-dependent NLC recordings shown in A and B. (E) Summary of the Qmax change. The data point at the end of each time course was summarized for comparison. The average relative Qmax value was 1.81 ± 0.64 (mean ± s.d., n=5) for cells overexpressing both prestin and CFTR, and 1.03 ± 0.06 (mean ± s.d., n=6) for cells overexpressing only prestin. Student’s t-test found a significant difference (p= 0.016) in the average relative Qmax, but not for α and Vpk values (p>0.05). In fact, all Qmax increments observed in individual cells overexpressing both prestin and CFTR were statistically significant (p<0.05), while all of those observed in cells overexpressing only prestin were not.
Another characteristic aspect of the Qmax enhancement is its delay. The Qmax change did not occur contemporaneously with the membrane resistance change. The membrane resistance plummeted immediately after establishing the whole-cell configuration, whereas the relative Qmax did not noticeably increase for approximately 10 seconds after establishing the whole-cell configuration (Fig. 7C). This suggests that the increment in Qmax is a result of sequential cellular events, probably consisting of rapid increment of cAMP, PKA activation by cAMP, phosphorylation of the R domain of CFTR, and interaction of the activated CFTR with prestin. Hence, the delay is inconsistent with the idea that the increased Qmax was somehow related to the rapidly decreasing membrane resistance during the NLC measurements. To further check the influence of rapidly decreasing membrane resistance on the time-dependent NLC measurements, we monitored NLC over a longer time course during which chloride conductance was blocked by the CFTR-specific chloride conductance blocker, CFTRinh-172 (Fig. 8). As blocking of the CFTR chloride conductance increased over time, the membrane resistance eventually returned to its initial level. The Qmax, however, stayed higher than its initial value, supporting the validity of our NLC measurements (Fig. 7) and the sequence of biochemical events associated with the direct, physical interaction between prestin and CFTR.
Effect of prestin on CFTR activity
It has been reported that DRA and PAT1 potentiate the conductivity of CFTR by increasing the channel open probability. Together with the evidence of prestin-CFTR interaction, it is of interest to see if prestin is also capable of reciprocally modulating the function of CFTR. To this end, we measured the conductance of cells expressing only CFTR and cells expressing both CFTR and prestin. Conductance was determined from the voltage-dependent current measured after CFTR activation by the intracellularly applied cAMP/ATP/IBMX (Fig. 9). For proper comparison (i.e., to account for differing cell sizes), the conductance was normalized by the membrane capacitance of the cells. As shown in Fig. 9C, a statistically significant difference was not evident between the two groups (n=30 for CFTR, n=34 for CFTR/prestin p=0.21), suggesting that prestin has little modulatory effect on the chloride conductance of CFTR.
Fig. 9. Effect of prestin on CFTR.
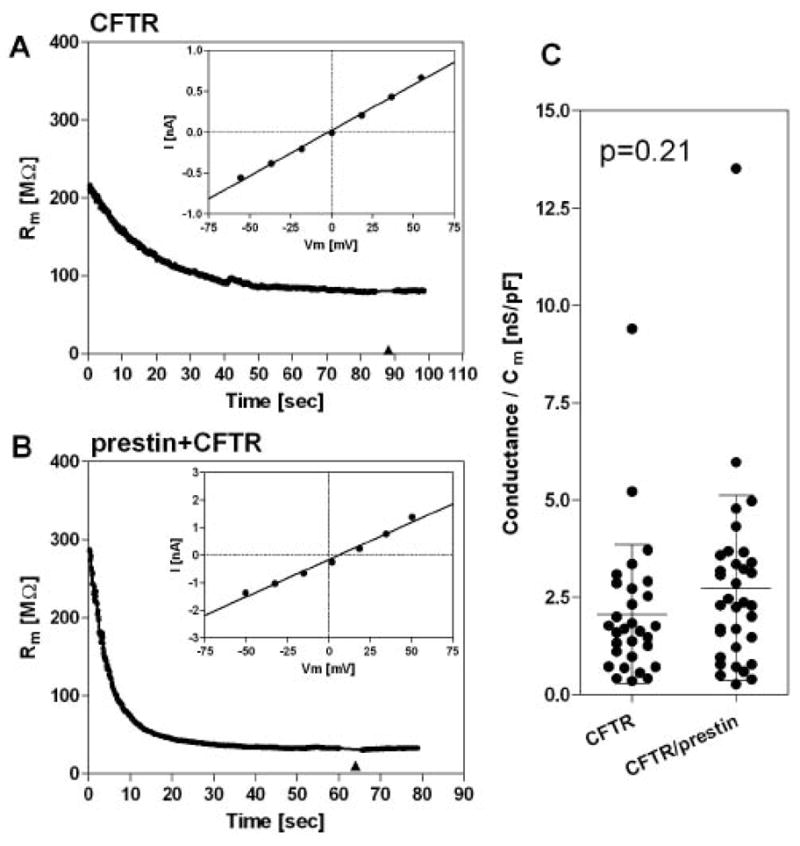
The intracellular pipette solution contained 0.2mM cAMP, 1mM ATP, and 50μM IBMX for activation of CFTR. Voltage-dependent current was measured on HEK293T cells expressing either CFTR-GFP (A) or both CFTR-GFP and prestin-CFP (B) at time points indicated (◆). Although the initial membrane resistance levels varied among recordings, a statistically significant difference was not observed between (A) and (B). (C) Statistics of the measurements. Conductance was calculated from the I–V slope corrected by the series resistance (Rs), and divided by the membrane capacitance (Cm). Each symbol represents an individual recording. Bars indicate mean and standard deviation.
Effect of CFTR activation on prestin in OHCs
Effect of CFTR activation on charge movement was also studied in isolated OHCs using the same experimental procedure as for the HEK293T cell recording described above. As expected, reduction of membrane resistance was observed upon PKA activation (Fig. 10A, inset) though the degree of the decrement was smaller than that observed for HEK cells in which CFTR was overexpressed (Fig. 7A and Fig. 8). In some OHCs, however, reduction of the membrane resistance was not evident (7 out of 20 recordings). Because it is known that phosphorylation of CFTR by PKC modulates the activation of CFTR by PKA [50], this may reflect a lack of basal PKC phosphorylation of CFTR in those cells. Reduction of the membrane resistance was not observed for control recordings without cAMP in the pipette solution (Fig. 10B, inset). Time-dependent NLC was measured to monitor prestin activity with and without cAMP/ATP/IBMX stimulation as exemplified in Fig. 10A and Fig. 10B. The lower noise of the NLC measurements is mainly due to 5–10 times higher prestin expression density in OHCs compared to HEK cells. Vpk shift during the first 10–20 sec was observed in most recordings, which is likely due to dialysis of the cytosol with the pipette solution and/or due to negative pressure [51] transiently applied for establishing the whole-cell configuration. The Vpk shift was not evident in most HEK cells probably due to use of recording pipettes with wider tips (see Materials and Methods). Because the Vpk shift was also observed without CFTR activation (Fig. 10B), this initial transient Vpk shift is not related to prestin-CFTR interaction. Contrary to the results on HEK293T cells, drastic Qmax enhancements were not observed in OHCs with cAMP/ATP stimulation. We could not observe a statistically significant Qmax enhancement for OHC recordings regardless of the magnitude of the membrane resistance reduction. For comparing the relative Qmax changes induced by cAMP/ATP stimulation to those without stimulation, we excluded recordings that did not show decrease in membrane resistance with application of cAMP/ATP (7 out of 20 recordings). Statistically significant difference was not observed (Fig. 10D). This negative result may imply that additional phosphorylation (or dephosphorylation) of CFTR is required for prestin activation (see DISCUSSION).
Fig. 10. Effect of CFTR activation on prestin function in isolated OHCs.
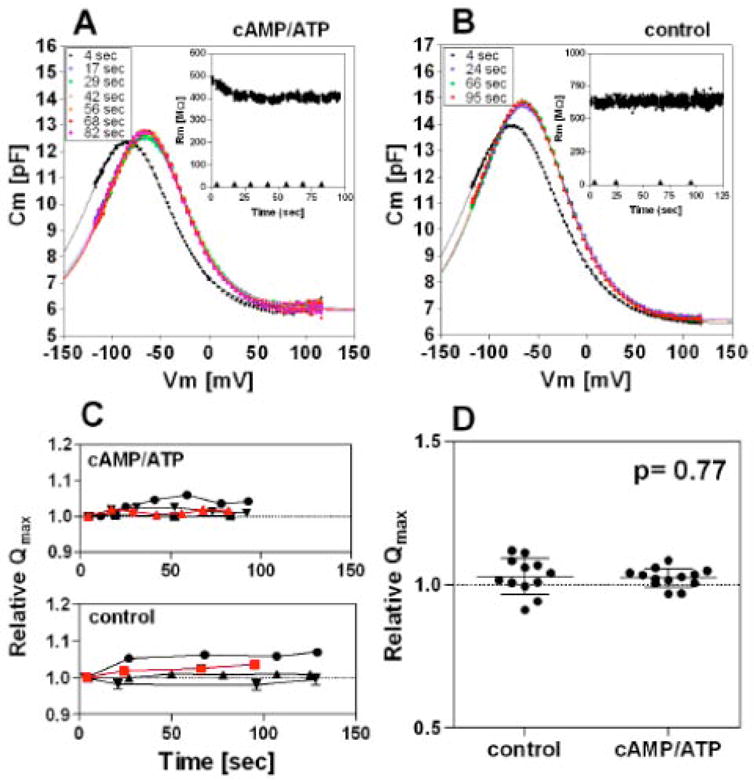
Time-dependent NLC was measured on isolated OHCs. (A) An example of the time-dependent NLC measurements when the intracellular pipette solution contained 0.2mM cAMP, 1mM ATP, and 50μM IBMX for activation of CFTR. The inset shows the time course of the membrane resistance (Rm). After establishing a whole-cell configuration, recordings were taken at the time points indicated (◆). (B) An example of the time-dependent NLC measurements without CFTR activation. (C) Time-dependent relative Qmax values with respect to the Qmax measured at the beginning of the time course. Four results are shown for each condition. Error bars indicate variability of the relative Qmax values calculated from the standard errors of the curve fittings of the original NLC data, many of which are too small to be seen. The red symbols show the data points obtained from the time-dependent NLC recordings shown in A and B. (D) Summary of the Qmax change. The data point at the end of each time course was chosen for comparison. For results obtained with CFTR activation, only results showing decrease in membrane resistance were used for the statistical analysis (13 out of 20). The average relative Qmax values were 1.03 ± 0.01 (mean ± s.d., n=13) for recordings with CFTR activation, and 1.03 ± 0.06 (mean ± s.d., n=12) for recordings without CFTR activation. The Student’s t-test failed to find a significant difference (p= 0.77). A statistically significant difference was also not found for α or Vpk values (p>0.05).
DISCUSSION
In the present study, we found that CFTR is present in both the apical and the basolateral membranes of OHCs, and that CFTR physically associates with prestin. Although CFTR typically resides in the apical membrane in epithelial cells, CFTR has been reported to localize to the basolateral membrane. For example, CFTR is targeted to either apical or basal gill membranes, as well as the intestinal epithelia of teleost fish depending on the salinity of the cell’s environment [52]. These data suggest that the final location of CFTR can be modified by other factors. It is likely that the basolateral localization of CFTR in OHCs is directed by binding of CFTR to prestin, which is exclusively present in the lateral wall. The difference in the immunofluorescence labeling observed between IHCs (apical only), OHCs (both apical and basolateral) and prestin-KO OHCs (apical only) supports this contention (Fig. 2 and Fig. 3).
Since intracellular chloride is essential for prestin to generate significant nonlinear charge movement [17], the presence of CFTR in the close vicinity of prestin has an immediate physiological implication, i.e., regulating prestin’s function by controlling local chloride concentration around the prestin molecules. Although the intracellular chloride concentration in OHCs is estimated to be 10mM or less [19], this value may be much lower in the subplasmalemmal space, given the anatomical architecture of the OHC’s cortical lattice [53]. Because prestin’s chloride binding affinity is 3~6 mM [17, 19, 54], there is a potential for indirectly modulating prestin’s function by local alteration of intracellular chloride concentration [16]. For example, activation of CFTR in the OHC’s lateral membrane could increase chloride concentration around prestin molecules so that prestin exerts a larger mechanical response to the same voltage stimulus. However, experimental proof of this scenario is not readily feasible because replacement of chloride ions with other anions, while maintaining ionic strength and minimizing junction potential during measurement, affects the NLC profile of prestin [16, 55]. In other words, anions that simply replace chloride without affecting prestin’s state-probability function have not been found. Because of the lack of knowledge as to how different anions interact with prestin, one would not be able to correctly attribute an apparent change in the magnitude of nonlinear charge movement to change in local chloride concentration. Therefore, in this report, NLC measurements were performed under saturated and equal chloride concentrations across the cell membrane. This allows us to investigate the effects of a direct, physical interaction between CFTR and prestin molecules. In other words, our study does not exclude the possibility that regulation of [Cl−]i by CFTR indirectly modifies prestin’s functions under physiological conditions.
Another implication of the prestin-directed CFTR localization in the lateral membrane is its potential physiological role in the OHC’s response to efferent signals. It is known that high chloride found in perilymph is important for the efferent-mediated inhibition of cochlear output [18]. In OHCs, the lateral membrane, but not the apical membrane, faces perilymph. Thus, the prestin-directed lateral membrane localization of CFTR found in OHCs is interesting. Besides acetylcholine and γ-aminobutyric acid (GABA), calcitonin gene-related peptide (CGRP) is known as an efferent neurotransmitter for OHCs. It has been demonstrated that activation of CGRP receptors results in activation of CFTR by increasing intracellular cAMP level [56]. Therefore, it is possible that CFTR in the lateral membrane of OHCs is activated by the efferent signal. Although a recent study has questioned the participation of CGRP in cochlear suppression[57], this does not rule out the possibility that CFTR is involved in some, as yet to be elucidated CGRP-related OHC response to efferent signals.
Aside from being a chloride channel, CFTR is also known as a regulator of dozens of proteins including members in the SLC26 family. For SLC26 members, the interaction-binding site for CFTR’s R domain is the STAS domain located at the C-terminus [30]. Upon activation by PKA-dependent phosphorylation, the R domain of CFTR binds to the STAS domain of SLC26 transporters. Like other SLC26 members, prestin also has a STAS domain [33] that could bind to the activated R domain of CFTR. The present study demonstrated that CFTR binds to prestin regardless of Fsk treatment, suggesting that phosphorylation of the R domain is not required for the CFTR-prestin binding (Fig. 5). This may not be unique to CFTR-prestin binding because phosphorylation independent binding of the R domain and the STAS domain of SLC26A3 has been reported recently[58]. The phosphorylation independent CFTR-prestin binding is compatible with the idea that CFTR is situated in the basolateral membrane for some yet to be appreciated physiological purpose. Interaction of R and STAS domains results in a marked and mutual activation of CFTR and the SLC26 transporters [30]. For example, regulatory interaction between the R domain of CFTR and the STAS domain of DRA increases CFTR’s open probability and DRA activity 5–7 fold [30].
For our study of prestin-CFTR interaction, NLC was used as an index of prestin’s function. The results indicate that mere co-expression of CFTR does not affect prestin’s NLC function. However, with activation of CFTR, charge displacement by prestin is significantly enhanced in a heterologous expression system. Because the NLC measurements were performed under saturated and equal chloride concentrations across the cell membrane, it is unlikely that the increased charge movement is indirectly caused by a change in local chloride concentration due to the activation of CFTR. In spite of the fact that prestin did not activate CFTR’s chloride conductance, the results are at least reminiscent of the modulatory interaction with CFTR reported for other SLC26 family members.
Although CFTR mRNA and protein have been expressed in various tissues, and CFTR is reported to regulate the functions of several different proteins [26, 30], mutations of the human CFTR gene mainly cause only two disease states: cystic fibrosis (CF) and congenital bilateral aplasia of the vas deferens. Even so, it is pertinent for this report to document the incidence of sensorineural hearing impairment in CF patients. It is known that bilateral loss of sensitivity may develop during CF treatment, especially at high frequencies. However, this hearing loss is believed to result from damage to hair cells by aminoglycoside antibiotics such as gentamycin and tobramycin [59–61] and not from the disease itself. Patients with a severe case of CF, where a modulatory influence of CFTR might be observed, receive aggressive aminoglycoside therapy and, until recently, they usually had short life spans. This makes it difficult to learn if the CFTR/prestin interaction influences peripheral auditory function, especially if the effects were to be subtle. However, it should also be emphasized that no significant enhancement in charge movement was observed in isolated OHCs, even though activated CFTR was shown to enhance prestin’s function in transfected mammalian cells. It is possible that the insignificant Qmax enhancement observed in isolated OHCs may be attributable to a difference in the basal phosphorylation status of CFTR that may occur in HEK293T vs. OHCs. There are more than ten phosphorylation sites in CFTR that are regulated by PKA, PKC, and PKG [23, 26]. Thus, it is possible that PKC/PKG phosphorylation (or dephosphorylation) of CFTR is also required for the modulatory effect on prestin function. We did not pursue this scenario because direct phosphorylation of prestin by PKG[35] and PKC (unpublished observations) also affects prestin function, and consequently, it is not feasible to separate these concurrent phosphorylation effects. Alternatively, the insignificant increment of Qmax observed in isolated OHCs might be explained by a mechanism of prestin activation by the stoichiometric prestin-CFTR interaction suggested above. If the expression of prestin is much higher than that of CFTR, which is likely in OHCs, most prestin molecules will not be activated by CFTR because they lack their binding partner. It is also possible that prestin is more likely to interact with itself via its C-terminal STAS domain [62], than with CFTR’s R domain. If true, prestin activation by CFTR might be a vestige of prestin’s evolution from transporter to motor protein. Because it is difficult to generalize from in vitro experiments demonstrating protein-protein interactions with potential modulatory roles in vivo, the impact of the a direct CFTR/prestin association on peripheral auditory function requires further study.
Although the physiological relevance of prestin activation by CFTR is not clear, the fact that prestin’s nonlinear charge displacement is enhanced by interaction with CFTR is important for understanding how the molecule functions as a motor. Knowledge of the molecular mechanism underlying prestin’s nonlinear charge movement and force generation is a key to comprehending how prestin functions as a voltage-dependent cochlear amplifier.
Supplementary Material
Acknowledgments
Our colleague Ms. R. Edge significantly contributed to the anatomical imaging. We thank Dr. J. Bartles for his comments on the manuscript. Dr. W. Russin at the Biological Imaging Facility of Northwestern University is thanked for his help in image processing and Dr. S. Muallem for providing the GFP-CFTR plasmid construct. This work is supported in part by NIH grants DC00089 (to P. Dallos), DC006412 (to J. Zheng) and by the Hugh Knowles Center.
Abbreviations
- CFTR
cystic fibrosis transmembrane conductance regulator
- Fsk
forskolin
- IBMX
3-isobutyl-1-methylxanthine
- IHC
inner hair cell
- NLC
nonlinear capacitance
- OHC
outer hair cell
- SLC26
solute carrier protein 26
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dallos P. The active cochlea. J Neurosci. 1992;12:4575–4585. doi: 10.1523/JNEUROSCI.12-12-04575.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santos-Sacchi J, Dilger JP. Whole cell currents and mechanical responses of isolated outer hair cells. Hear Res. 1988;35:143–150. doi: 10.1016/0378-5955(88)90113-x. [DOI] [PubMed] [Google Scholar]
- 3.Ashmore JF. J Physiol. Vol. 388. Lond: 1987. A fast motile response in guinea-pig outer hair cells: the cellular basis of the cochlear amplifier; pp. 323–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kachar B, Brownell WE, Altschuler R, Fex J. Electrokinetic shape changes of cochlear outer hair cells. Nature. 1986;322:365–368. doi: 10.1038/322365a0. [DOI] [PubMed] [Google Scholar]
- 5.Brownell WE, Bader CR, Bertrand D, de Ribaupierre Y. Evoked mechanical responses of isolated cochlear outer hair cells. Science. 1985;227:194–196. doi: 10.1126/science.3966153. [DOI] [PubMed] [Google Scholar]
- 6.Ashmore JF. Cochlear outer hair cell motility. Physiol Rev. 2008;88:173–210. doi: 10.1152/physrev.00044.2006. [DOI] [PubMed] [Google Scholar]
- 7.Zheng J, Shen W, He DZ, Long KB, Madison LD, Dallos P. Prestin is the motor protein of cochlear outer hair cells. Nature. 2000;405:149–155. doi: 10.1038/35012009. [DOI] [PubMed] [Google Scholar]
- 8.Dallos P, Wu X, Cheatham MA, Gao J, Zheng J, Anderson CT, Jia S, Wang X, Cheng WH, Sengupta S, He DZ, Zuo J. Prestin-based outer hair cell motility is necessary for mammalian cochlear amplification. Neuron. 2008;58:333–339. doi: 10.1016/j.neuron.2008.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheatham MA, Huynh KH, Gao J, Zuo J, Dallos P. Cochlear function in Prestin knockout mice. J Physiol. 2004;560:821–830. doi: 10.1113/jphysiol.2004.069559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liberman MC, Gao J, He DZ, Wu X, Jia S, Zuo J. Prestin is required for electromotility of the outer hair cell and for the cochlear amplifier. Nature. 2002;419:300–304. doi: 10.1038/nature01059. [DOI] [PubMed] [Google Scholar]
- 11.Liu X, Ouyang XM, Xia XJ, Zheng J, Pandya A, Li F, Du LL, Welch KO, Petit C, Smith RJ, Webb BT, Yan D, Arnos KS, Corey DP, Dallos P, Nance WE, Chen Z. Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss. Hum Mol Genet. 2003;12:1155–1162. doi: 10.1093/hmg/ddg127. [DOI] [PubMed] [Google Scholar]
- 12.Bai JP, Surguchev A, Montoya S, Aronson PS, Santos-Sacchi J, Navaratnam D. Prestin’s anion transport and voltage-sensing capabilities are independent. Biophys J. 2009;96:3179–3186. doi: 10.1016/j.bpj.2008.12.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaechinger TJ, Oliver D. Nonmammalian orthologs of prestin (SLC26A5) are electrogenic divalent/chloride anion exchangers. Proc Natl Acad Sci U S A. 2007;104:7693–7698. doi: 10.1073/pnas.0608583104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muallem DR, Ashmore JF. An anion antiporter model of prestin, the outer hair cell motor protein. Biophys J. 2006;90:4035–4045. doi: 10.1529/biophysj.105.073254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chambard JM, Ashmore JF. Sugar transport by mammalian members of the SLC26 superfamily of anion-bicarbonate exchangers. J Physiol. 2003;550:667–677. doi: 10.1113/jphysiol.2003.039321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rybalchenko V, Santos-Sacchi J. Cl- flux through a non-selective, stretch-sensitive conductance influences the outer hair cell motor of the guinea-pig. J Physiol. 2003;547:873–891. doi: 10.1113/jphysiol.2002.036434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliver D, He DZ, Klöcker N, Ludwig J, Schulte U, Waldegger S, Ruppersberg JP, Dallos P, Fakler B. Intracellular anions as the voltage sensor of prestin, the outer hair cell motor protein. Science. 2001;292:2340–2343. doi: 10.1126/science.1060939. [DOI] [PubMed] [Google Scholar]
- 18.Desmedt JE, Robertson D. J Physiol. Vol. 247. Lond: 1975. Ionic mechanism of the efferent olivo-cochlear inhibition studied by cochlear perfusion in the cat; pp. 407–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santos-Sacchi J, Song L, Zheng J, Nuttall AL. Control of mammalian cochlear amplification by chloride anions. J Neurosci. 2006;26:3992–3998. doi: 10.1523/JNEUROSCI.4548-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frolenkov GI. J Physiol. Vol. 576. Lond: 2006. Regulation of electromotility in the cochlear outer hair cell; pp. 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawasaki E, Hattori N, Miyamoto E, Yamashita T, Inagaki C. mRNA expression of kidney-specific ClC-K1 chloride channel in single-cell reverse transcription-polymerase chain reaction analysis of outer hair cells of rat cochlea. Neurosci Lett. 2000;290:76–78. doi: 10.1016/s0304-3940(00)01310-0. [DOI] [PubMed] [Google Scholar]
- 22.Kawasaki E, Hattori N, Miyamoto E, Yamashita T, Inagaki C. Single-cell RT-PCR demonstrates expression of voltage-dependent chloride channels (ClC-1, ClC-2 and ClC-3) in outer hair cells of rat cochlea. Brain Res. 1999;838:166–170. doi: 10.1016/s0006-8993(99)01730-8. [DOI] [PubMed] [Google Scholar]
- 23.Vaandrager AB, Smolenski A, Tilly BC, Houtsmuller AB, Ehlert EM, Bot AG, Edixhoven M, Boomaars WE, Lohmann SM, de Jonge HR. Membrane targeting of cGMP-dependent protein kinase is required for cystic fibrosis transmembrane conductance regulator Cl- channel activation. Proc Natl Acad Sci U S A. 1998;95:1466–1471. doi: 10.1073/pnas.95.4.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- 25.Welsh MJ, Smith AE. Cystic fibrosis. Sci Am. 1995;273:52–59. doi: 10.1038/scientificamerican1295-52. [DOI] [PubMed] [Google Scholar]
- 26.Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- 27.Simpson JE, Gawenis LR, Walker NM, Boyle KT, Clarke LL. Chloride conductance of CFTR facilitates basal Cl-/HCO3- exchange in the villous epithelium of intact murine duodenum. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1241–1251. doi: 10.1152/ajpgi.00493.2004. [DOI] [PubMed] [Google Scholar]
- 28.Chernova MN, Jiang L, Friedman DJ, Darman RB, Lohi H, Kere J, Vandorpe DH, Alper SL. Functional comparison of mouse slc26a6 anion exchanger with human SLC26A6 polypeptide variants: Differences in anion selectivity, regulation, and electrogenicity. J Biol Chem. 2004 doi: 10.1074/jbc.M411703200. [DOI] [PubMed] [Google Scholar]
- 29.Chernova MN, Jiang L, Shmukler BE, Schweinfest CW, Blanco P, Freedman SD, Stewart AK, Alper SL. Acute regulation of the SLC26A3 congenital chloride diarrhea anion exchanger (DRA) expressed in Xenopus oocytes. J Physiol. 2003;549:3–19. doi: 10.1113/jphysiol.2003.039818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ, Muallem S. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol. 2004;6:343–350. doi: 10.1038/ncb1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ko SB, Shcheynikov N, Choi JY, Luo X, Ishibashi K, Thomas PJ, Kim JY, Kim KH, Lee MG, Naruse S, Muallem S. A molecular mechanism for aberrant CFTR-dependent HCO(3)(−) transport in cystic fibrosis. Embo J. 2002;21:5662–5672. doi: 10.1093/emboj/cdf580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng J, Long KB, Shen W, Madison LD, Dallos P. Prestin topology: localization of protein epitopes in relation to the plasma membrane. Neuroreport. 2001;12:1929–1935. doi: 10.1097/00001756-200107030-00032. [DOI] [PubMed] [Google Scholar]
- 33.Zheng J, Du GG, Matsuda K, Orem A, Aguiñaga S, Deák L, Navarrete E, Madison LD, Dallos P. The C-terminus of prestin influences nonlinear capacitance and plasma membrane targeting. J Cell Sci. 2005;118:2987–2996. doi: 10.1242/jcs.02431. [DOI] [PubMed] [Google Scholar]
- 34.Judice TN, Nelson NC, Beisel CL, Delimont DC, Fritzsch B, Beisel KW. Cochlear whole mount in situ hybridization: identification of longitudinal and radial gradients. Brain Res Brain Res Protoc. 2002;9:65–76. doi: 10.1016/s1385-299x(01)00138-6. [DOI] [PubMed] [Google Scholar]
- 35.Deák L, Zheng J, Orem A, Du GG, Aguiñaga S, Matsuda K, Dallos P. Effects of cyclic nucleotides on the function of prestin. J Physiol. 2005;563:483–496. doi: 10.1113/jphysiol.2004.078857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheatham MA, Zheng J, Huynh KH, Du GG, Edge R, Anderson CT, Zuo J, Ryan AD, Dallos P. Evaluation of an independent prestin mouse model derived from the 129S1 strain. Audiol Neurootol. 2007;12:378–390. doi: 10.1159/000106481. [DOI] [PubMed] [Google Scholar]
- 37.Zheng J, Du GG, Anderson CT, Keller JP, Orem A, Dallos P, Cheatham MA. Analysis of the oligomeric structure of the motor protein prestin. J Biol Chem. 2006;281:19916–19924. doi: 10.1074/jbc.M513854200. [DOI] [PubMed] [Google Scholar]
- 38.Cheatham MA, Zheng J, Huynh KH, Du GG, Gao J, Zuo J, Navarrete E, Dallos P. Cochlear function in mice with only one copy of the prestin gene. J Physiol. 2005;569:229–241. doi: 10.1113/jphysiol.2005.093518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szönyi M, He DZ, Ribari O, Sziklai I, Dallos P. Cyclic GMP and outer hair cell electromotility. Hear Res. 1999;137:29–42. doi: 10.1016/s0378-5955(99)00127-6. [DOI] [PubMed] [Google Scholar]
- 40.Crawford I, Maloney PC, Zeitlin PL, Guggino WB, Hyde SC, Turley H, Gatter KC, Harris A, Higgins CF. Immunocytochemical localization of the cystic fibrosis gene product CFTR. Proc Natl Acad Sci USA. 1991;88:9262–9266. doi: 10.1073/pnas.88.20.9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertrand CA, Frizzell RA. The role of regulated CFTR trafficking in epithelial secretion. Am J Physiol Cell Physiol. 2003;285:C1–18. doi: 10.1152/ajpcell.00554.2002. [DOI] [PubMed] [Google Scholar]
- 42.Matsuda K, Zheng J, Du GG, Klocker N, Madison LD, Dallos P. N-linked glycosylation sites of the motor protein prestin: effects on membrane targeting and electrophysiological function. J Neurochem. 2004;89:928–938. doi: 10.1111/j.1471-4159.2004.02377.x. [DOI] [PubMed] [Google Scholar]
- 43.Farinha CM, Mendes F, Roxo-Rosa M, Penque D, Amaral MD. A comparison of 14 antibodies for the biochemical detection of the cystic fibrosis transmembrane conductance regulator protein. Mol Cell Probes. 2004;18:235–242. doi: 10.1016/j.mcp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 44.Bradbury NA. Intracellular CFTR: localization and function. Physiol Rev. 1999;79:S175–191. doi: 10.1152/physrev.1999.79.1.S175. [DOI] [PubMed] [Google Scholar]
- 45.Weber WM, Cuppens H, Cassiman JJ, Clauss W, Van Driessche W. Capacitance measurements reveal different pathways for the activation of CFTR. Pflugers Arch. 1999;438:561–569. doi: 10.1007/s004249900086. [DOI] [PubMed] [Google Scholar]
- 46.Lehrich RW, Aller SG, Webster P, Marino CR, Forrest JN. Vasoactive intestinal peptide, forskolin, and genistein increase apical CFTR trafficking in the rectal gland of the spiny dogfish, Squalus acanthias. Acute regulation of CFTR trafficking in an intact epithelium. J Clin Invest. 1998;101:737–745. doi: 10.1172/JCI803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi A, Watkins SC, Howard M, Frizzell RA. CFTR-dependent membrane insertion is linked to stimulation of the CFTR chloride conductance. Am J Physiol. 1996;271:C1887–1894. doi: 10.1152/ajpcell.1996.271.6.C1887. [DOI] [PubMed] [Google Scholar]
- 48.Schwiebert EM, Gesek F, Ercolani L, Wjasow C, Gruenert DC, Karlson K, Stanton BA. Heterotrimeric G proteins, vesicle trafficking, and CFTR Cl- channels. Am J Physiol. 1994;267:C272–281. doi: 10.1152/ajpcell.1994.267.1.C272. [DOI] [PubMed] [Google Scholar]
- 49.Lewarchik CM, Peters KW, Qi J, Frizzell RA. Regulation of CFTR trafficking by its R domain. J Biol Chem. 2008;283:28401–28412. doi: 10.1074/jbc.M800516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia Y, Mathews CJ, Hanrahan JW. Phosphorylation by Protein Kinase C Is Required For Acute Activation of Cystic Fibrosis …. Journal of Biological Chemistry. 1997 doi: 10.1074/jbc.272.8.4978. [DOI] [PubMed] [Google Scholar]
- 51.Kakehata S, Santos-Sacchi J. Effects of salicylate and lanthanides on outer hair cell motility and associated gating charge. J Neurosci. 1996;16:4881–4889. doi: 10.1523/JNEUROSCI.16-16-04881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marshall WS, Singer TD. Cystic fibrosis transmembrane conductance regulator in teleost fish. Biochim Biophys Acta. 2002;1566:16–27. doi: 10.1016/s0005-2736(02)00584-9. [DOI] [PubMed] [Google Scholar]
- 53.Holley MC, Kalinec F, Kachar B. Structure of the cortical cytoskeleton in mammalian outer hair cells. J Cell Sci. 1992;102(Pt 3):569–580. doi: 10.1242/jcs.102.3.569. [DOI] [PubMed] [Google Scholar]
- 54.Song L, Seeger A, Santos-Sacchi J. On membrane motor activity and chloride flux in the outer hair cell: lessons learned from the environmental toxin tributyltin. Biophys J. 2005;88:2350–2362. doi: 10.1529/biophysj.104.053579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rybalchenko V, Santos-Sacchi J. Anion control of voltage sensing by the motor protein prestin in outer hair cells. Biophys J. 2008;95:4439–4447. doi: 10.1529/biophysj.108.134197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luebke AE, Dahl GP, Roos BA, Dickerson IM. Identification of a protein that confers calcitonin gene-related peptide responsiveness to oocytes by using a cystic fibrosis transmembrane conductance regulator assay. Proc Natl Acad Sci USA. 1996;93:3455–3460. doi: 10.1073/pnas.93.8.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maison SF, Emeson RB, Adams JC, Luebke AE, Liberman MC. Loss of alpha CGRP reduces sound-evoked activity in the cochlear nerve. J Neurophysiol. 2003;90:2941–2949. doi: 10.1152/jn.00596.2003. [DOI] [PubMed] [Google Scholar]
- 58.Dorwart MR, Shcheynikov N, Baker JM, Forman-Kay JD, Muallem S, Thomas PJ. Congenital chloride-losing diarrhea causing mutations in the STAS domain result in misfolding and mistrafficking of SLC26A3. J Biol Chem. 2008;283:8711–8722. doi: 10.1074/jbc.M704328200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stavroulaki P, Vossinakis IC, Dinopoulou D, Doudounakis S, Adamopoulos G, Apostolopoulos N. Otoacoustic emissions for monitoring aminoglycoside-induced ototoxicity in children with cystic fibrosis. Arch Otolaryngol Head Neck Surg. 2002;128:150–155. doi: 10.1001/archotol.128.2.150. [DOI] [PubMed] [Google Scholar]
- 60.Ozcelik T, Ozgirgin N, Ozcelik U, Gocmen A, Gurcan B, Kiper N. Auditory nerve-brainstem responses in cystic fibrosis patients. Int J Pediatr Otorhinolaryngol. 1996;35:165–169. doi: 10.1016/0165-5876(95)01321-0. [DOI] [PubMed] [Google Scholar]
- 61.McRorie TI, Bosso J, Randolph L. Aminoglycoside ototoxicity in cystic fibrosis. Evaluation by high-frequency audiometry. Am J Dis Child. 1989;143:1328–1332. doi: 10.1001/archpedi.1989.02150230086028. [DOI] [PubMed] [Google Scholar]
- 62.Pasqualetto E, Seydel A, Pellini A, Battistutta R. Expression, purification and characterization of the C-terminal STAS domain of the SLC26 anion transporter prestin. Protein Expr Purif. 2008;58:249–256. doi: 10.1016/j.pep.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 63.Kopeikin Z, Li M, Hwang TC. On the Mechanism of CFTR Inhibition by CFTRinh-172. Biophysical Journal. 2009;96:469a. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.