

Abstract
Blood-brain barrier (BBB) disruption, mediated through matrix metalloproteinases (MMPs) and other mechanisms, is a critical event during ischemic stroke. Tissue plasminogen activator (tPA) is the only FDA-approved thrombolytic therapy for acute ischemic stroke, but the efficacy and safety of its therapeutic application is limited by narrow treatment time windows and side effects. Thus, there is a pressing need to develop combinational therapy that could offset tPA side effects and improve efficacy in clinical practice. Recent experimental studies indicate that tPA has previously unidentified functions in the brain beyond its well established thrombolytic activity, which might contribute to tPA-related side effects through MMPs (mainly MMP-9) and several signaling pathways involved in LDL receptor-related protein (LRP), activated protein C (APC) and protease-activated receptor 1 (PAR-1), platelet-derived growth factor C (PDGF-C), and N-methyl-D-aspartate (NMDA) receptor. Therapeutic targeting of MMPs and/or tPA-related signaling pathways might offer promising new approaches to combination therapies for ischemic stroke. This review provides an overview of the relationship between structural components and function of the BBB/neurovascular unit with respect to ischemic stroke. We discuss how MMPs and tPA contribute to BBB disruption during ischemic stroke and highlight recent findings of molecular signaling pathways involved in neurotoxicity of tPA therapy.
Keywords: Blood-brain barrier, neurovascular unit, tPA, MMPs, ischemic stroke, signaling pathways
Introduction
The blood-brain barrier (BBB) is primarily formed by specialized brain endothelial cells that are interconnected by well-developed tight junctions and provides a dynamic interface between the blood and the brain (Abbott, et al., 2010). BBB disruption is a critical event in the pathogenesis of acute ischemic stroke, however, the molecular mechanisms involved are not completely understood (Sandoval and Witt, 2008). Emerging studies indicate that matrix metalloproteinases (MMPs) and tissue-type plasminogen activator (tPA) play critical roles in the BBB disruption during acute ischemic stroke (Adibhatla and Hatcher, 2008). Experimental data suggest that MMPs have deleterious roles in the early phase of ischemic stroke, but also have beneficial roles in stroke recovery during the later phase. By degrading neurovascular matrix and disruption of the BBB tight junctions, MMPs (mainly MMP-9) promote BBB damage, brain edema and hemorrhage during acute ischemic stroke (Sandoval and Witt, 2008). tPA is the only thrombolytic drug approved by the U.S. FDA, but the efficacy and safety of its therapeutic application is limited by narrow treatment time windows (within 3h of the onset of stroke symptoms) and side effects on brain edema and hemorrhagic complications (Cronin, 2010; Derex and Nighoghossian, 2008; Gravanis and Tsirka, 2008). Experimental data have shown that tPA have pleiotropic actions in the brain beyond its well established thrombolytic role, including activation of MMPs and other molecular pathways (Adibhatla and Hatcher, 2008; Yepes et al., 2009; Rosell and Lo, 2008). These effects may increase tPA neurotoxicity, further damage the BBB, and worsen edema and cerebral hemorrhage (Adibhatla and Hatcher, 2008; Yepes et al., 2009). Thus, combination therapies targeting MMPs and other tPA-related pathways may limit neurotoxic effects and extend treatment time windows of tPA in ischemic stroke. This review provides an overview of the relationship between structural components and function of the BBB with respect to ischemic stroke. We discuss how MMPs and tPA contribute to BBB disruption during ischemic stroke and highlight recent findings of molecular signaling pathways involved in neurotoxicity of tPA therapy.
1. The blood-brain barrier structural component and functional integrity
The structure and function of the blood-brain barrier (BBB) has been discussed in reviews elsewhere (Sandoval and Witt, 2008; Abbott, et al., 2010). Structural and molecular components of the BBB are summarized in Fig. 1. Briefly, the BBB comprises the tight junctions (TJs) and adherens junctions (AJs). TJs are continuous membrane strands located at the apical site between brain endothelial cells (ECs), which consist of three integral protein types: claudins, occludin, and junctional adhesion molecules (JAMs) (Abbott et al., 2010). TJs also consist of several accessory proteins, including zonula occludens (ZO), cingulin, 7H6, AF-6, and others (Abbott, et al., 2010). These accessory proteins may serve as recognition proteins for tight junctional placement and as a support structure for signal transduction proteins (Hawkins and Davis, 2005; Piontek et al., 2008). While not traditionally considered as a TJ protein, the actin cytoskeleton in brain ECs plays a critical role in modulating BBB permeability (Lai et al., 2005).
Figure 1.

Schematic diagram of the BBB structure that comprises the tight and adherens junctions. The tight junctions consist of occludin, claudins, and junctional adhesion molecules (JAM). The tight junctions also consist of several accessory proteins necessary to form structural support, including the zonula occluden (ZO) proteins, AF6, 7H6, cingulin, and others. Most of the tight junction components (ZO proteins, claudin, and occludin) have the ability to bind to actin cytoskeleton in brain endothelial cells. The adherens junctions consist of vascular endothelial cadherin (VE-cadherin) and catenin proteins and provide structural integrity and attachment between the cells, and are necessary for formation of tight junctions. Updated from Abbott NJ, 2009 and other sources.
Claudins are small tetraspan membrane proteins (20–24 kDa) with two extracellular loops that form dimers and are able to bind homotypically to adjacent endothelial cells to form the primary seal of the tight junctions (Furuse et al., 1999; Piontek et al., 2008). Several claudin family members, such as claudins-3, -5, and -12, have been identified in BBB endothelial cells (Sandoval and Witt, 2008; Hawkins and Davis, 2005). Among the claudin family members, claudin-5 has been shown to be a major cell adhesion molecule of BBB tight junctions (Nitta et al., 2003). Occludin is a 60–65 kDa tetraspan membrane protein that is exclusively localized at TJ strands (Sandoval and Witt, 2008; Hirase et al., 1997). Occludin’s carboxy-terminal binds to zonula occludens (ZO) proteins, which in turn binds to the actin cytoskeleton (Haskins et al., 1998; Fanning et al., 1998; Liu et al., 2008). Although occludin is not required for the formation of TJ strands per se, the presence of occludin in the membrane is associated with increased electrical resistance across the membrane and decreased paracellular permeability (Balda et al., 1996; Saitou et al., 2000; Tsukita et al., 1999). Molecular structure and function of TJ proteins can be regulated by alterations in their expression levels and/or distribution at the BBB under various pathophysiological conditions, including ischemic stroke (Sandoval and Witt, 2008). Experimental studies suggest that phosphorylation represents a critical mechanism regulating distribution/localization and function of BBB TJ proteins (Yamamoto et al., 2008; Feldman et al., 2005). For example, phosphorylation of claudin-5 at Thr207, via the protein kinase-A (PKA) (Soma et al., 2004) and Rho kinase activation (Yamamoto et al., 2008), has been shown to increase BBB permeability. Tyrosine phosphorylation of occludin has been identified with its disassociation from intracellular ZO proteins and increased BBB permeability (Rao et al., 2002; Kale et al., 2003; Kago et al., 2006). More such efforts are needed to better understand how TJ proteins are phosphorylated at the level of specific residues (e.g. serine/threonine/tyrosine) and how their phosphorylation differentially and synergistically contributes to BBB disruption at different stages of stroke.
JAMs are a family of immunoglobulin superfamily proteins (~40 kDa) that localize within the intercellular cleft between brain ECs (Abbott et al., 2010). Heretofore, three JAMs (-A, -B, and –C) have been identified in brain ECs (Abbott et al., 2010). Intriguingly, JAMs have direct inflammatory-related activities because they are also expressed in leukocytes and platelets (Weber et al., 2007). JAM-A is required for neutrophil infiltration in inflammatory or ischemic tissues by controlling beta1-integrin internalization and recycling (Cera et al. 2009). Anti-JAM-A monoclonal antibody (BV11) reduces monocyte transmigration across endothelial cells in vitro and in vivo (Del Maschio et al., 1999; Martin-Padura et al., 1998). JAM-B and -C have an impact on leukocyte adhesion and migration. JAM-B can bind to integrin VLA-4 (α4β1), a leukocyte integrin that contributes to rolling and firm adhesion of lymphocytes to endothelial cells through binding to vascular cell adhesion molecule (VCAM)-1, but this effect requires prior engagement with JAM-C (Cunningham et al., 2002; Ludwig et al., 2009). JAM-C can act as a novel counterreceptor for the integrin Mac-1 (CD11b/CD18) and mediates leukocyte transmigration independently of JAM-B (Santoso et al., 2002; Keiper et al., 2005; Chavakis et al., 2004). Taken together, experimental data suggest that JAMs may play an active role in the regulation of cerebral inflammatory response and BBB permeability during ischemic stroke.
The adherens junctions (AJs) are mainly composed of VE-cadherin (vascular endothelial cadherin), which are necessary for formation of TJs by forming a continuous belt localized near the apical end of the junctional cleft, just below the TJs (Dejana et al., 2008). While not considered as the primary BBB paracellular barrier, AJs are functionally and structurally linked to TJs and may play an important role in the localization and stabilization of the TJs (Taddei et al., 2008). Experimental data show that endothelial VE-cadherin at AJs upregulates TJ protein claudin-5, suggesting a direct regulation of TJ integrity by AJ proteins (Taddei et al., 2008).
2. Expression, activation, and function of MMPs in BBB disruption in ischemic stroke
Matrix metalloproteinases (MMPs) comprise a family of zinc endopeptidases that can broadly target almost all components of the mammalian central nervous system (CNS). Emerging evidence indicates that MMPs play both detrimental and beneficial roles in ischemic stroke. In the early phase (hours to days) after cerebral ischemia, MMPs disrupt the BBB by degrading the TJ proteins (e.g. occludin and claudin-5) and basal lamina proteins (e.g. fibronectin, laminin, collagen, proteoglycans, and others) and thereby lead to BBB leakage, leukocyte infiltration, brain edema, and hemorrhage (Adibhatla and Hatcher, 2008; Cunningham et al., 2005). In contrast, MMPs may play beneficial roles in stroke recovery by modulating neurovascular remodeling (Cunningham et al., 2005; Zhao et al., 2006).
2.1 Expression and activation of MMPs in the brain during ischemic stroke
Expression of MMPs in the adult brain is very low to undetectable under normal conditions. Clinical and experimental studies have demonstrated that several MMPs such as MMP-2, MMP-3, MMP-7, or MMP-9 are upregulated and activated after ischemic stroke (Clark, et al., 1997; Rosell et al., 2006; Heo et al., 1999; Anthony et al., 1997; Suzuki et al., 2007; Sole et al., 2004). In the brain, MMPs can be expressed by various cell types, including resident cells (endothelial cells, microglia, neurons, and astrocytes) and infiltrating inflammatory cells during cerebral ischemia, but the brain regions and cellular sources of expression differ according to the specific MMPs, as well as the type, severity and duration of injuries (Adibhatla and Hatcher, 2008; Cunningham et al., 2005; Jin et al., 2010). Experimental data indicate that brain microvascular ECs and infiltrating leukocytes (most likely neutrophils) are key cellular sources of brain MMP-9 at least in the early phase (within 24h) after focal cerebral ischemia (Jin et al., 2010; Justicia et al., 2003; Gidday et al., 2005; McColl et al., 2008). By immunostaining and microdissection, clinical data confirm that microvessel endothelium and infiltrating neutrophils are the major source of the increased brain MMP-9 after ischemic and hemorrhagic stroke in humans (Rosell et al., 2006 & 2008b).
MMPs are synthesized and secreted as inactive pro-enzymes that subsequently become proteolytically cleaved and activated. To achieve optimal enzymatic activity, MMPs are tightly controlled at the transcriptional level, as well as at the protein level through activation by their zymogens and inhibition by tissue-specific inhibitors (Cunningham et al., 2005). Potential mechanisms contributing MMP activation are summarized in Fig. 2. During cerebral ischemia, proMMP-2 can be activated by membrane-type MMP (MT1-MMP), and the latter is activated by furin (Candelario-Jalil et al., 2009; Yang et al., 2007), and proMMP-9 can be activated by MMP-3 (stromelysin-1), as well as other mechanisms such as proinflammatory factors (e.g. IL-1β, TNF-α, CD40L, and many others) and reactive oxygen species (ROS) (Gasche et al., 2001; Haorah et al., 2007; Jian et al., 2005). tPA has been shown to activate MMPs through plasmin-dependent and –independent mechanisms (Candelario-Jalil et al., 2009). In the brain, the molecular mechanisms by which MMPs are upregulated and activated during ischemic stroke are not fully understood. It remains largely unclear how the expression and activation of specific MMPs are differentially regulated in different cell types within the neurovascular unit.
Figure 2.
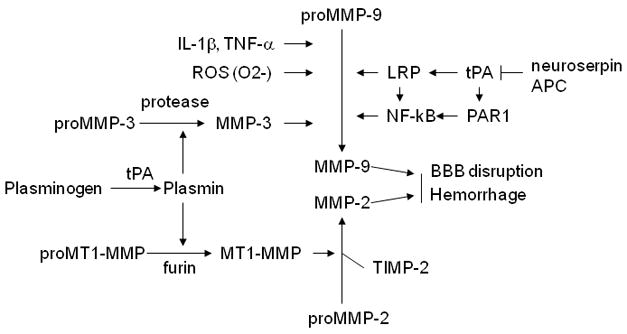
Mechanisms of activation of MMPs. ProMMP-2 can be activated by membrane-type-1 MMP (MT1-MMP/MMP-14), and the latter can be activated by furin. ProMMP-9 can be activated by MMP-3, tissue type plasminogen activator (tPA), proinflammatory factors (e.g. IL-1β and TNF-α) and reactive oxygen species (ROS). Plasmin can activate both MT1-MMP and MMP-3. tPA can activate both MMP-3 and MMP-9 through multiple pathways. tPA activity can be inhibited by neuroserpin and activated protein C (APC).
The neurovascular unit is comprised of the endothelial cells which make up the vessels as well as perivascular neurons, astrocytes, and pericytes (del Zoppo, 2006) (Fig. 3). Over the past decade, an extensive investigation of the BBB in cerebrovascular disease has expanded from consideration of only endothelial cells to include interactions with different types of cells and extracellular matrix in the neurovascular unit. In recent years, the concept of the neurovascular unit has emerged as a new paradigm for stroke investigation and therapy. This concept emphasizes that cell-cell signaling among the various neuronal, glial, and vascular compartments underlies the homeostasis of normal brain function (Guo and Lo, 2009). Conversely, dysfunctional signaling within the neurovascular unit should contribute to disease. There are temporal and spatial changes of MMP-9 within the cells of the neurovascular unit after stroke (Zhao et al., 2006). In a rat model of transient middle cerebral artery occlusion (MCAO), most of the MMP-9 activities colocalized with brain microvessel ECs within 24 hours, but at 7 to 14 days, the MMP-9 signal shifted to the periphery of cortical infarction and was associated mainly with neurons and astrocytes (Zhao et al., 2006). This redistribution within the neurovascular unit likely reflects multiphasic roles of MMP-9 in ischemic stroke, that is, the pathological role in mediating disruption of the BBB, neuronal cell death and hemorrhage early after stroke, and the beneficial role in mediating neurovascular remodeling during the repair phase (Adibhatla and Hatcher, 2008; Zhao et al., 2006; Cunningham, et al., 2005). Inhibition of MMP-9 during the late phase (7 to 14 days) after stroke has been shown to reduce the number of neurons and new vessels that correlated with increased brain injury and impaired functional recovery (Zhao et al. 2006). Thus, it is reasonable to speculate that blocking MMPs at a badly chosen time and in nontarget cell types may result in unwanted side effects (Zlokovic, 2006).
Figure 3.

Schematic diagram of the neurovascular unit that comprises neurons, microvessels (endothelium), astrocytes, and pericytes that reside within the basement membrane. Neurons and microvessels communicate through astrocytes (updated from del Zoppo GJ, 2006 and other sources). There is spatiotemporal change of MMP-9 expression within the neurovascular unit after ischemic stroke. In the acute phase (within 24h), MMP-9 is mainly derived from brain endothelial cells and infiltrating leukocytes (especially neutrophils). In the late phase, MMP-9 is mainly secreted by astrocytes and neurons. Updated from Zlokovic BV, 2006 and other sources.
2.2 Biphasic opening of BBB during ischemic stroke
BBB opening during focal cerebral ischemia/reperfusion (I/R) injury has long been considered to follow a biphasic time course, but considerable discrepancies across studies exist with respect to the timing of the second opening (Belayev et al., 1996; Huang et al., 1999; Kuroiwa et al., 1985; Sharp et al., 2000; Chen et al., 2009a). Morphologically, BBB opening correlates with a redistribution of the TJ and AJ proteins from the plasma membrane to the cytoplasm as well as reorganization of the endothelial actin cytoskeleton (Bolton et al., 1998; McColl et al., 2007; McColl et al., 2008). The extent of BBB disruption is associated with the type and severity and duration of ischemic insults. A mild to moderate opening of the BBB may be partially reversible, and allows plasma constituents to enter brain and possibly damage cells. In contrast, severe BBB disruption after ischemic stroke is unlikely to be reversible and allows even further extravasation of potentially harmful plasma constituents (Chen et al., 2009a). The molecular mechanisms underlying BBB opening and its consequences in ischemic stroke are not fully understood. Several MMPs (especially MMP-9) have been implicated in the regulation of BBB permeability and function during ischemia stroke.
2.3 Differential roles of MMP-2 and MMP-9 in BBB disruption during ischemic stroke
Among MMPs, MMP-2 and MMP-9 are two of the most widely studied enzymes that have been shown to be critical in regulating BBB permeability during cerebral ischemia. The two enzymes appear to differentially mediate disruption of the BBB and neuronal damage after cerebral ischemia. MMP-2 gene knockout does not provide neuroprotection in mouse models of permanent and transient MCA occlusion (Asahi et al., 2001a). Consistently, in vitro data show that MMP-2 is not toxic to neurons in hippocampal slice preparations (Cunningham, et al., 2005). In contrast, MMP-9 gene knockout provides strong neuroprotection in the same animal models, and in vitro MMP-9 is toxic to neurons in hippocampal slice preparations and in cultured primary cortical neurons (Asahi et al., 2000; Asahi et al., 2001b). In accordance with these findings, a recent clinical study (Lucivero et al., 2007) suggests that MMP-9 and MMP-2 may play different roles in human ischemic stroke. Increase in plasma MMP-2 is observed only in patients with lacunar (mild) stroke early (within 12 h) and related to better outcome. In contrast, increase in plasma MMP-9 seems to be late (at day 7) and related to more severe stroke.
Earlier experimental studies provided indirect evidence that MMP-2 played a key role in the initial opening of the BBB after cerebral ischemia. In a rat model of transient middle cerebral artery occlusion (MCAO), the initial opening at 3 hours correlated with brain MMP-2 levels and was blocked by a synthetic MMP inhibitor (BB-1101) (Rosenberg, et al., 1998). The second, delayed opening (maximal opening at 48 hours) appeared to correlate with brain MMP-9 levels (maximally elevated at 48 hours), but had no response to the MMP inhibitor (Rosenberg, et al., 1998). Increased expression of MMP-2 may contribute to the initial opening of BBB by degrading the basal lamina leading to neuronal injury (Heo et al., 1999; Chang et al., 2003). In the non-human primate brains, MMP-2 increased significantly as early as 1 hour after transient MCAO and was persistently elevated at least 7 days (Heo et al., 1999; Chang et al., 2003). Direct injection of MMP-2 into the rat brain resulted in the disruption of the BBB with subsequent hemorrhage, and this effect was inhibited by co-administration of TIMP-2 (Rosenberg, et al., 1992). A recent study (Yang et al., 2007) suggests a potential mechanism whereby MMPs mediate the BBB disruption during ischemic stroke. In a rat model of transient MCAO, the initial opening of the BBB occurred as early as 3 hours after reperfusion and increased activation of MMP-2 correlated with the early opening of the BBB. Correspondingly, the mRNA expression of claudin-5 and occludin, decreased in both hemispheres, and both proteins degraded or fragmented in ischemic hemispheres after 2–3 h of reperfusion, and treatment with the MMP inhibitor BB-1101 reversed the degradation of the TJ proteins. Thus, the early degradation of the TJ proteins seems to be associated with a marked increase in MMP-2 in the early phase after cerebral ischemia. Experimental data clearly demonstrate greater increase in MMP-2 than in MMP-9 at 3 h, along with increased expression of MMP-2 activators, MT1-MMP and furin. In contrast, there was no increase in MMP-2 mRNA and activity, while MMP-9 mRNA and activity markedly increased at 24 h of reperfusion, suggesting an association between MMP-9 and delayed disruption of the BBB. Immunostaining shows that claudin-5 and occludin, though loosened or fragmented, remained within the endothelial clefts during the initial opening of the BBB. Together, these data suggest that the initial opening is a reversible, which is regulated, at least in part, by the activation of MMP-2.
Recent studies suggest that MMP-9 plays a critical role in mediating the second, delayed opening of BBB after ischemic stroke. Emerging data indicate that MMP-9 is associated with severe BBB disruption by further degrading the tight junctions and basal lamina proteins, substantially contributing to brain infarction, edema, and hemorrhagic transformation (HT) in both animal models (Yang, et al., 2007; Lee et al., 2007; Rosenberg, et al., 2007) and in human stroke patients (Rosell et al., 2008; Montaner et al., 2003; Barr, et al., 2009). MMP-9−/− mice display a significant reduction in BBB disruption and brain edema and this effect is associated with reduced degradation of intracellular ZO-1 as compared to wild-type mice after transient MCAO (Asahi, et al., 2001b). MMP-9 has been shown to degrade TJ proteins (claudin-5, occludin, ZO-1) in cultured brain ECs (Chen et al., 2009b) and in animal models of focal cerebral ischemia (Yang et al., 2007; McColl et al., 2008; Bauer et al., 2009; Liu et al., 2009).
There are a few controversies concerning the time course of the expression and activation of MMP-2 and MMP-9 in the brain after ischemic stroke. Some experimental data suggest that MMP-9 responses appear to dominate in the acute phase, whereas MMP-2 elevations seem to occur in the delayed phase after stroke. In the rat transient MCAO model, Planas et al. (2001) shows that MMP-9 is induced and activated from 4 h to 4 days, but a small increase in MMP-2 is detected at 4 h, while a massive increase in MMP-2 expression and activation by day 4. In the mouse permanent MCAO model, Gasche et al. (1999) shows that expression and activation of MMP-9 in the ischemic brains are induced significantly as early as 2–4 h after ischemia, with increased BBB permeability. In contrast, the pro-MMP-2 is induced significantly only after 24 h of permanent ischemia, and no activated form is observed (Gasche et al., 1999). These data suggest that MMP-9, but not MMP-2, plays an active role in early BBB disruption after ischemic stroke.
Recent studies suggest that MMP-9 may play a more prominent role in the BBB disruption during ischemic stroke under clinical relevant conditions linked to elevated systemic inflammation. Experimental data have shown that systemic inflammation exacerbates neutrophil infiltration into the ischemic brain and thus alters the kinetics of the BBB tight junction disruption after experimental stroke (McColl et al., 2007 & 2008). These studies demonstrate that infiltrating neutrophils are the primary source of increased (5-fold) MMP-9 activity in the ischemic brain of mice challenged with interleukin-1β (IL-1β) at 4, 8 or 24h after focal cerebral ischemia. A transformation from transient to sustained BBB disruption caused by enhanced neutrophil-derived MMP-9 is a critical mechanism underlying the exacerbation of ischemic brain injury by IL-1β-induced systemic inflammation, which is mediated through conversion of a transient to a sustained disruption of the TJ protein (claudin-5) and exacerbated disruption of the cerebrovascular basal lamina protein (collagen-IV) (McColl et al., 2008). This mechanism may contribute to the poor clinical outcome in stroke patients with elevated systemic inflammatory status. In stroke patients with prior infection and atherosclerosis, neutrophil infiltration into the ischemic brain and neutrophil-derived neurovascular MMP-9 are elevated (Buck et al., 2008; Emsley et al., 2003; Zeller et al., 2005).
Taken together, the above findings support the hypothesis that MMP-2 and MMP-9 play important but distinct roles in mediating BBB disruption after ischemic stroke. MMP-2 is preferentially associated with the initial opening of the BBB, while MMP-9 appears to be more important in the delayed BBB damage by mediating a conversion from transient to sustained BBB disruption, in particular associated with elevated systemic inflammation.
In addition to MMP-2/-9, other MMP members may also play active roles in mediating BBB disruption during ischemia stroke. MMP-3 (stromelysin-1) has been shown to mediate BBB opening during neuroinflammation (Gurney et al., 2006). After intracerebral injection of lipopolysaccharide (LPS), MMP-3 knockout mice showed less degradation of the TJ proteins (claudin-5, occludin, laminin-alpha-1) together with reduced neutrophil infiltration, compared with wild-type mice (Gurney et al., 2006). In the rat transient MCAO model, brain MMP-3 is activated as determined by the cleavage of the cerebral matrix agrin, an MMP-3 substrate (Sole et al., 2004). It has been speculated that various MMPs function in a network-like fashion with upstream and downstream proteases being closely coupled (Rosell and Lo, 2008). Future studies need to assess not just MMP-2, -3 and -9, but all family members to truly ‘fingerprint’ the role of various MMPs in ischemic stroke (Rosell and Lo, 2008).
3. Expression and neurotoxicity of tPA in BBB disruption during ischemic stroke
Thrombolytic therapy with tissue-type plasminogen activator (tPA) for ischemic stroke represents a two-edged sword, since tPA promotes desirable (thrombolytic) as well as undesirable (neurotoxic) outcomes during stroke (Adibhatla and Hatcher, 2008). It has been established that exogenous tPA can cross both the intact and the damaged BBB to reach the brain parenchyma (Adibhatla and Hatcher, 2008; Yepes et al., 2009). Thus, both endo- and exogenous tPAs are capable of influencing brain functions and dysfunctions. In addition to its intended role in thrombolysis, tPA also possesses important signaling and protease actions in the neurovascular unit after stroke, some of which may mediate neurotoxicity and hemorrhagic transformation (HT) after tPA therapy. Molecular mechanisms underlying tPA’s neurotoxicity within the neurovascular unit have not been fully understood. Better understanding of the activities and neurotoxicity of tPA in the brain may provide a molecular basis for developing effective and safe tPA combination therapy for acute ischemic stroke.
3.1 Expression and regulation of tPA in systemic circulation and in brain
tPA is a highly specific serine proteinase and is found predominantly in the blood, where its primary function is as a thrombolytic enzyme and its principal substrate is the zymogen plasminogen (Gravanis and Tsirka, 2008). The catalytic activity of tPA is rapidly inactivated in the blood stream through binding of serine protease inhibitor(s), primarily plasminogen activator inhibitor-I (PAI-1). The balance between t-PA and PAI-1 regulates the systemic fibrinolytic potential in the vasculature. The tPA/PAI-1 complex is cleared from the circulation by the liver via a scavenging receptor, the low-density lipoprotein receptor-related protein-1 (LRP-1) (Yepes et al., 2003). By the LRP-mediated clearance, tPA has a 5 to 10 min short half-life in the bloodstream in humans (Gravanis and Tsirka, 2008). In the brain, tPA has been identified mainly in the endothelial cells of the BBB and in the endothelium of the small vessels (Levin and del Zoppo, 1994), where it may regulate BBB permeability and vascular tone. tPA is also expressed and released by neurons and microglia and mediates neuronal death and microglial activation after excitotoxic injury (Siao et al., 2003). tPA is synthesized by neurons and glia in most brain regions, particularly abundant in the hippocampus, hypothalamus, cerebellum, and amygdala (Salles et al., 2002; Sappino et al., 1993). A wide distribution of tPA biosynthesis in the brain is likely to be associated with different actions of tPA, such as promoting synaptic plasticity and regulating the permeability of the neurovascular unit related to cerebral ischemia and seizures (Qian et al., 1993; Seeds et al., 1995; Yepes et al., 2002; Mataga et al., 2002).
In the brain, tPA activity is regulated by neuroserpin, a potent endogenous inhibitor of tPA. Neuroserpin, a member of the serpin (serine proteinase inhibitors) family, is expressed primarily in the brain, where it reacts preferentially with tPA by inhibiting its activity (Miranda et al., 2006). Using transgenic mice overexpressing (~6-fold) neuroserpin in the nervous system, Cinelli et al. (2001) demonstrates that neuroserpin reduces microglial activation and, therefore, the tPA activity and has a neuroprotective role in mice after focal ischemic stroke. In rodent experimental cerebral ischemia, Lebeurrier et al. (2005) demonstrates that administration of exogenous neuroserpin into the brain protects neurons against NMDA-induced excitotoxicity, calcium influx and tissue damage in vitro and in vivo. Together, these data suggest that overexpression or administration of neuroserpin in the brain may limit the deleterious effects of tPA after cerebral ischemia. Indeed, adjuvant treatment with neuroserpin has been shown to significantly reduce BBB leakage, brain edema, and infarct size and extend the therapeutic window for tPA administration in a rat model of embolic stroke (Zhang et al., 2002).
3.2 Neurotoxicity of the tPA-MMP-9 (and MMP-3) pathway during ischemic stroke
The standard hypothesis postulates that MMP-9 play a central role in tPA-mediated neurotoxicity in thrombolytic therapy for acute stroke (Rosell and Lo, 2008). This hypothesis is supported by data from both clinical and experimental studies. Clinical data have shown that plasma and brain levels of MMP-9 are elevated in patients with acute ischemic stroke, and delayed (>3 h of symptoms onset) tPA therapy causes a further increase in plasma MMP-9 in stroke patients (Barr et al., 2010; Castellanos et al., 2003; Castellanos et al., 2007; Horstmann et al., 2003; Kelly et al., 2008; Montaner et al., 2003; Rosell et al., 2006; Rosell et al., 2008b). MMP-9 in blood has emerged as a promising biomarker for human stroke, because plasma levels of MMP-9 correlate with and predict poor neurological outcome and hemorrhagic complications after thrombosis in these clinical trials.
Experimental studies provide direct evidence supporting the tPA-MMP-9 hypothesis. In rodent stroke models, endogenous tPA activity in the brain is induced as early as 1 h (preceding changes in MMP-9 and BBB integrity) after focal cerebral ischemia and is present mainly in the perivascular tissue and vessel wall (Tsuji et al., 2005; Yepes et al., 2003). Genetic deficiency of tPA or inhibition of its activity by neuroserpin has been shown to decrease BBB disruption, edema, neuronal death and brain infarction (Tsuji et al., 2005; Wang et al., 1998). Further, examination of both tPA−/− and wild-type (WT) mice demonstrates that the increased endogenous tPA is required for the initial opening of the BBB after transient MCAO (Yepes et al., 2003). Levels of MMP-9 in ischemic brain tissues are significantly reduced in tPA−/− mice compared with wild-type mice and intravenous administration of exogenous tPA in tPA−/− mice can rebuild the ischemic MMP-9 response back up to WT levels (Tsuji et al., 2005; Wang et al., 1998). Intravenous administration of exogenous tPA also increases MMP-9 levels in ischemic brains in rats after transient MCAO (Yepes et al., 2003). Together, these findings support the hypothesis (Fig. 2) that tPA is a major upstream mediator of MMP-9 induction during ischemic stroke, especially when tPA is exogenously administrated.
Although numerous studies have established the neurotoxicity of tPA in ischemic stroke, some studies demonstrate show neurotorphic effects of tPA on neurons, independent of thrombolysis. An earlier study by Kim et al. (1999) shows that recombinant tPA markedly attenuated zinc-induced neuronal cell death in cortical culture, and, when injected into cerebrospinal fluid, also reduced kainate seizure-induced hippocampal neuronal death in adult rats. A recent study (Lee et al., 2007) by the same group shows that low dose tPA (500 ng/ml) treatment induces neurotrophic effects, promoting neurite elongation and neuronal survival in cortical neuronal cultures, and this neurotrophic effect is mediated by typical trophic signaling kinases such as Raf-K/ERK, PKC, and PI3-K/Akt, but not mediated by its proteolytic action because it is not affected by tPA protease inhibitors, such as PAI-1. In contrast, high-dose tPA (20ug/ml) has been shown to induce neurotoxicity in cortical cultures (Nocole et al., 2001). Nevertheless, it remains unclear if similar effects take place in vivo during ischemic stroke, especially when treated with tPA at therapeutic doses (e.g. 2.5–10mg/kg).
In addition to MMP-9, other MMP members may also play active roles in tPA-mediated neurotoxicity in thrombolytic therapy for acute stroke. A recent study (Suzuki et al., 2007) shows that MMP-3 is critical for intracranial bleeding after t-PA treatment of stroke in mice. MMP-3 expression is significantly enhanced in the ischemic hemisphere, where it is expressed only in neurons, but its expression is up-regulated in endothelial cells with t-PA treatment. MMP-3 knockout mice treated with tPA display significantly reduced hemorrhagic transformation (HT) than wild-type mice treated with tPA. In vitro, t-PA induces MMP-3 in cultured murine brain ECs, and this effect is prevented by inhibition of either LDL receptor-related protein (LRP) or NF-kB activation (Suzuki et al., 2009). Collectively, experimental data indicate that tPA enhances brain MMP-9 (and MMP-3) levels in stroke in vivo, and suggest that combination therapies targeting MMPs may improve tPA therapy.
3.3 Molecular signaling pathways associated with tPA neurotoxicity
The tPA-LRP pathway
The LDL receptor-related protein (LRP) is a member of the LDL receptor gene family that binds several ligands, including tPA (Herz and Strickland, 2001). In the brain, LRP is found in neurons and in perivascular astrocytes (Polavarapu et al., 2007) [134], and the interaction between tPA and LRP has been shown to regulate cerebrovascular tone (Nassar et al., 2002) and BBB permeability (Yepes et al., 2003; Polavarapu et al., 2007). Increased tPA activity in the ischemic brain has been associated with worsening the ischemic lesion (Wang et al., 1998; Yepes et al., 2000), neuronal death (Tsirka et al., 1995; Tsirka et al., 1997), and BBB disruption (Yepes et al., 2003; Polavarapu et al., 2007). Injection of recombinant tPA into the cerebrospinal fluid in the absence of ischemia results in a rapid dose-dependent increase in the BBB permeability (Yepes et al., 2003), and this effect is inhibited by blocking LRP using anti-LRP antibodies or the LRP antagonist called the receptor-associated protein (RAP), suggesting the tPA’s action is mediated via its receptor LRP (Yepes et al., 2003).
After acute ischemic stroke, the thrombolytic action of tPA in the intravascular space is beneficial, whereas its effect on neurons in the extravascular space is considered deleterious (Adibhatla and Hatcher, 2008; Yepes et al., 2009; Zhang et al., 2002). It has been postulated that if tPA therapy effectively reverses ischemia promptly and the BBB remains intact, then (exogenous) tPA remains within the vascular space. In contrast, if tPA therapy is ineffective and ischemia is prolonged, then there is the risk that exogenous tPA will across BBB and enter the brain parenchemia and thereby damage both the brain and the neurovascular unit by interacting with LRP (Adibhatla and Hatcher, 2008; Yepes et al., 2009). Indeed, some studies have demonstrated that tPA may cross the intact BBB by LRP-mediated transcytosis (Benchenane et al., 2005) and enter the brain from the intravascular space under ischemic and nonischemic conditions (Benchenane et al., 2005a and 2005b). Desmoteplase, a recombinant form of the plasminogen activator from saliva of the vampire bat Desmodus rotundus, has been shown to antagonize vascular tPA-induced neurotoxicity possibly by competing with tPA for the LRP binding at the BBB and thus effectively blocking tPA access to the brain parenchyma (López-Atalaya et al., 2007). Desmoteplase might offer a safe, effective adjunct therapy for tPA in acute stroke, since it is not neurotoxic and has longer (> 4 hr) half-life in circulation compared to tPA (5–10 min) (López-Atalaya et al., 2007).
Experimental studies have shown that LRP signaling plays an important role in the tPA-induced expression and activation of MMP-9 (and MMP-3) in cell culture and in animal models of stroke (Suzuki et al., 2009; Wang et al., 2003; Yepes et al., 2003). In vitro, recombinant tPA treatment stimulates MMP-9 expression in cultured human brain ECs, and this tPA-induced response is significantly reduced in the ECs treated with siRNA to suppress LRP, and was absent in LRP-deficient MEF cells (Wang et al., 2003). In vivo, direct intraventricular injection of tPA into mouse brain increases BBB permeability and this response is attenuated by LRP antagonists (Yepes et al., 2003). In addition, tPA has been shown to induce MMP-3 (stromelysin-1) in cultured murine brain ECs and this induction by t-PA is prevented by inhibition either of LRP or of nuclear factor (NF)-κB activation (Suzuki et al., 2009). These findings indicate that t-PA promotes BBB damage via MMP-9 (and MMP-3) induction in endothelial cells, which is regulated through the LRP/NF-κB pathway (Fig. 2). Moreover, the tPA-LRP interaction has been shown to induce the expression of iNOS in astrocytes during cerebral ischemia via the activation of the NF-κB pathway (Zhang et al., 2007), which might contribute to increased oxidative stress and BBB damage in ischemic stroke. Accordingly, targeting the tPA-LRP signaling pathway in brain may offer new approaches for decreasing tPA neurotoxicity and improving stroke therapy (Wang et al., 2003).
The tPA-APC/PAR1 pathway
Activated protein C (APC) is a serine protease with anticoagulant, anti-inflammatory and antiapoptotic activities. Experimental studies indicate that APC is neuroprotective during transient cerebral ischemia and promotes activation of antiapoptotic mechanisms in brain cells by acting directly on endothelium and neurons (Bernard et al., 2001; Cheng et al., 2003 & 2006; Feistritzer et al., 2005; Guo et al., 2004; Liu et al., 2004; Thiyagarajan et al., 2008; Zlokovic et al., 2005). In vitro, tPA substantially increases caspase-3 and caspase-8 activity in mouse cortical neurons treated with NMDA, but did not enhance caspase-9 activity. APC inhibits tPA-induced activation of caspase-8 and caspase-3 in human brain ECs and caspase-3-dependent nuclear translocation of apoptosis-inducing factor (AIF) in NMDA-treated neurons (Liu et al., 2004). In hypoxic human brain ECs treated with tPA, blockade of caspase-8, but not MMP-9, effectively inhibited caspase-3 activation, in contrast, in the absence of tPA, blockade of caspase-9, but not MMP-8, effectively inhibited caspase-3 activation. These findings suggest that tPA shifts the apoptotic pathways from the initiator caspase-9 to caspase-8. Both caspase-8 and caspase-3 activation in neurons treated with the combination of NMDA and tPA are substantially reduced (>80%) by APC. In vivo, recombinant APC can markedly reduce tPA-induced cerebral ischemic injury in a mouse model of transient brain ischemia (Liu et al., 2004) [150]. Further, experimental studies indicate that late APC administration (at 6–72 h or 72–144 h post ischemia) is also neuroprotective and mediates brain repair (i.e., neovascularization and neurogenesis) in a mouse model of transient brain ischemia (Thiyagarajan et al., 2008) [146]. These data suggest a significant extension of the therapeutic window for APC intervention in postischemic brain. Moreover, APC is an FDA-approved drug for severe sepsis. APC stabilizes vascular endothelial barriers (Thiyagarajan et al., 2008; Feistritzer et al., 2005) and has a low risk for brain hemorrhage in patients with severe sepsis (Bernard, et al., 2001) and in animal models of stroke (Cheng et al., 2003; Liu et al., 2004; Zlokovic et al., 2005). Experimental studies have shown that APC promotes neovascularization and neurogenesis in postischemic brain and blocks tPA vascular and neuronal toxicities in vitro and in vivo via protease-activated receptor 1 (PAR-1) (Thiyagarajan et al., 2008; Feistritzer et al., 2005; Cheng et al., 2006). For example, Cheng, et al (Cheng et al., 2006) demonstrate that APC inhibits tPA-induced, NF-κB-dependent MMP-9 pathway in ischemic brain endothelium in vivo and in vitro by acting through PAR-1 (Fig. 2). Taken together, these findings suggest that the combination therapy with tPA and APC may hold a great promise for blocking tPA’s neurovascular toxicity and promoting stroke recovery.
The tPA-PDGF-CC pathway
Platelet-derived growth factor C (PDGF-C) is one of four members in the PDGF family, which are known mitogens and survival factors for cells of mesenchymal origin. Experimental data show that tPA is both necessary and sufficient to directly induce opening of the BBB (Yepes et al., 2003), but this effect is independent of plasminogen, implicating other substrate(s) for tPA. Recently, PDGF-CC has been identified as a new substrate for tPA (Fredriksson et al., 2004 & 2005). tPA can activate and cleave latent PDGF-CC by directly interacting with the N-terminal CUB domains in PDGF-CC (Fredriksson et al., 2005; Li et al., 2000). Experimental data show that PDGF-CC is a downstream substrate of tPA within the neurovascular unit (Su et al., 2008). Activation of PDGF-CC by tPA impairs BBB integrity during ischemic stroke. Intracerebral injection of recombinant tPA or active PDGF-CC even in the absence of ischemia results in a significant increase in BBB permeability (Su et al., 2008). In contrast, co-injection of neutralizing antibodies to PDGF-CC with tPA blocks this increased permeability, indicating that PDGF-CC is a downstream substrate of tPA within the neurovascular unit (Su et al., 2008). Further, data show that the interaction between tPA and PDGF-CC is mediated through activation of PDGF-alpha receptors (PDGFR-α) on perivascular astrocytes, and treatment of mice with the PDGFR-alpha antagonist imatinib reduces BBB permeability and hemorrhage associated with late administration of tPA after ischemic stroke (Su et al., 2008). Taken together, these findings demonstrate that the tPA-PDGFR-α/PDGF-CC pathway regulates BBB permeability and suggest potential new therapeutic strategies for decreasing tPA neurotoxicity and improving stroke therapy.
The tPA-NMDA receptor pathway
The N-methyl-D-aspartate (NMDA) receptor, a glutamate receptor, is the predominant molecular device for controlling synaptic plasticity and memory function. Glutamate is the main excitatory neurotransmitter in the brain. Glutamate levels increase dramatically in cerebral ischemia and stroke. This may lead to opening of the BBB and induce further brain damage. The mechanisms of glutamate-induced disruption of the BBB integrity are not fully understood. Experimental data show that the NMDA receptors are involved in glutamate-induced alterations of the TJ molecule occludin expression and phosphorylation in brain ECs (András et al., 2007). Treatment with glutamate increases tyrosine phosphorylation and decreases threonine phosphorylation of occludin. Inhibition of the NMDA receptors by MK-801 partially protects against glutamate-induced elevation of occludin tyrosine phosphorylation. Inhibition of the NMDA receptors also attenuates glutamate-induced changes in occludin redistribution but not in the total protein levels (András et al., 2007). These findings suggest that glutamate-induced occludin phosphorylation and redistribution, leading to disruption of the BBB functions are regulated by the NMDA receptors.
Several studies have highlighted a role for tPA in modulating glutamatergic neurotransmission through a direct interaction with NMDA receptors, findings suggesting a potential mechanism directly involved in the ability of tPA to promote excitotoxic neuronal death both in vitro and in vivo (Nicole et al., 2001). tPA is recognized as a modulator of glutamatergic neurotransmission (Samson et al., 2006). In vitro, tPA increases NMDA-receptor-mediated calcium influx by interacting with, and then cleaving, the NR1 subunit within its N-terminal domain, and in vivo, blocking the tPA-NR1 interaction prevents permanent cerebral ischemia and reduces the severity of excitotoxic neuronal death in mouse brains (Lopez-Atalaya et al., 2008; Centonze et al., 2003; Benchenane et al., 2007; Kvajo et al., 2004). MMP-9 has emerged as a physiological regulator of NMDA receptor-dependent synaptic plasticity and memory and increases surface trafficking of the NR1-NMDA receptor through integrin β1 signaling (Michaluk et al., 2009). Taken together, these findings suggest a molecular link between tPA →MMP-9→NR1 and targeting this pathway may hold a great promise for blocking tPA’s neurotoxicity.
Concluding Remarks
Cerebral I/R injury induces dynamic changes in the BBB permeability, but the underlying mechanisms have remained largely unknown. More efforts are needed to better understand the molecular mechanism underlying the degradation and redistribution of various protein components of the BBB according to the type, severity and duration of cerebral ischemic insults. Since clinical trials indicates that a singular focus on saving neurons alone does not work for stroke, in recent years the concept of the neurovascular unit has emerged as a new paradigm for stroke investigation and therapy. More effective stroke treatment should rescue the integrity of interactions among all cell types within the entire neurovascular unit.
Abundant evidence indicates that MMPs contribute to the BBB disruption during the early phase of stroke. The major challenge with therapeutic interventions of MMPs remains how to accomplish temporal and spatial control of their activity in the brain. In experimental stroke, the therapeutic window for MMP-9 inhibition seems to be narrower than previously estimated and may not extend beyond 24 hours, mainly because a delayed inhibition of MMP-9 exacerbates stroke pathology. Blocking MMPs at a badly chosen time and in nontarget cell types may result in unwanted side effects.
Emerging evidence strongly suggests that MMPs (mainly MMP-9) play a central role in tPA-mediated neurotoxicity in thrombolytic therapy for acute stroke. Thus, pharmacological inhibition of MMPs (at an appropriate time) may hold promise as a safe and effective adjunct therapy for tPA in acute ischemic stroke. In addition to MMPs, several other molecular pathways also have been implicated in tPA-induced neurotoxicity. As discussed above, tPA neurotoxicity is blocked either by co-treatment with neuroserpin or APC or by inhibiting LRP, PDGF-CC, or NMDA receptors during ischemic stroke. Targeting these pathways might offer new combinational therapies for decreasing tPA neurotoxicity and improving stroke therapy. More efforts are needed to characterize both beneficial or detrimental effects of these pathways at different stages of stroke and to establish the time window for tPA co-treatment of acute ischemic stroke.
Acknowledgments
The work was supported by the National Institutes of Health Grant HL087990 (Dr Li) and by a Scientist Development Grant (0530166N (Dr Li) from American Heart Association. We gratefully thank our colleagues for many helpful discussions.
Footnotes
AUTHORSHIP
The concept and design and writing of the manuscript (Dr. Li).
The literature search and discussion of the manuscript (Drs. Jin and Yang, equally contributed to this work)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott NJ, et al. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Adibhatla RM, Hatcher JF. Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke: therapeutic strategies. CNS Neurol Disord Drug Targets. 2008;7:243–53. doi: 10.2174/187152708784936608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- András IE, et al. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J Cereb Blood Flow Metab. 2007;27:1431–43. doi: 10.1038/sj.jcbfm.9600445. [DOI] [PubMed] [Google Scholar]
- Anthony DC, et al. Differential matrix metalloproteinase expression in cases of multiple sclerosis and stroke. Neuropathol Appl Neurobiol. 1997;23:406–415. [PubMed] [Google Scholar]
- Asahi M, et al. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Asahi M, et al. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. NeuroReport. 2001a;12:3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]
- Asahi M, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001b;21:7724–7732. doi: 10.1523/JNEUROSCI.21-19-07724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balda MS, et al. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134:1031–1049. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr TL, et al. Blood-brain barrier disruption in humans is independently associated with Increased matrix metalloproteinase-9. Stroke. 2010;41:E123–128. doi: 10.1161/STROKEAHA.109.570515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AT, et al. Matrix metalloproteinase-9 mediates hypoxia-induced vascular leakage in the brain via tight junction rearrangement. Cereb Blood Flow Metab. 2009 Dec 9; doi: 10.1038/jcbfm.2009.248. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belayev L, et al. Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 1996;739:88–96. doi: 10.1016/s0006-8993(96)00815-3. [DOI] [PubMed] [Google Scholar]
- Benchenane K, et al. Tissue-type plasminogen activator crosses the intact blood-brain barrier by low-density lipoprotein receptor-related protein-mediated transcytosis. Circulation. 2005a;111:2241–2249. doi: 10.1161/01.CIR.0000163542.48611.A2. [DOI] [PubMed] [Google Scholar]
- Benchenane K, et al. Oxygen glucose deprivation switches the transport of tPA across the blood-brain barrier from an LRP-dependent to an increased +LRP-independent process. Stroke. 2005b;36:1065–1070. doi: 10.1161/01.STR.0000163050.39122.4f. [DOI] [PubMed] [Google Scholar]
- Benchenane K, et al. Anti-NR1 N-terminal-domain vaccination unmasks the crucial action of tPA on NMDA-receptor-mediated toxicity and spatial memory. J Cell Sci. 2007;120:578–585. doi: 10.1242/jcs.03354. [DOI] [PubMed] [Google Scholar]
- Bernard GR, et al. Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- Bolton SJ, et al. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood–brain barrier breakdown in vivo. Neuroscience. 1998;86:1245–1257. doi: 10.1016/s0306-4522(98)00058-x. [DOI] [PubMed] [Google Scholar]
- Buck BH, et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke. 2008;39:355–360. doi: 10.1161/STROKEAHA.107.490128. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, et al. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983–994. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos M, et al. Plasma metalloproteinase-9 concentration predicts hemorrhagic transformation in acute ischemic stroke. Stroke. 2003;34:40–46. [PubMed] [Google Scholar]
- Castellanos M, et al. Serum cellular fibronectin and matrix metalloproteinase-9 as screening biomarkers for the prediction of parenchymal hematoma after thrombolytic therapy in acute ischemic stroke: a multicenter confirmatory study. Stroke. 2007;38:1855–1859. doi: 10.1161/STROKEAHA.106.481556. [DOI] [PubMed] [Google Scholar]
- Centonze D, et al. Tissue plasminogen activator is required for corticostriatal long-term potentiation. Eur J Neurosci. 2002;16:713–721. doi: 10.1046/j.1460-9568.2002.02106.x. [DOI] [PubMed] [Google Scholar]
- Cera MR, et al. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci. 2009;122:268–277. doi: 10.1242/jcs.037127. [DOI] [PubMed] [Google Scholar]
- Chang DI, et al. Activation systems for latent matrix metalloproteinase-2 are upregulated immediately after focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23:1408–1419. doi: 10.1097/01.WCB.0000091765.61714.30. [DOI] [PubMed] [Google Scholar]
- Chavakis T, et al. The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J Biol Chem. 2004;279:55602–55608. doi: 10.1074/jbc.M404676200. [DOI] [PubMed] [Google Scholar]
- Chen B, et al. Severe blood-brain barrier disruption and surrounding tissue injury. Stroke. 2009a;40:e666–674. doi: 10.1161/STROKEAHA.109.551341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, et al. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology. 2009b;50:1914–1923. doi: 10.1002/hep.23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, et al. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- Cheng T. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- Cinelli P, et al. Neuroserpin, a neuroprotective factor in focal ischemic stroke. Mol Cell Neurosci. 2001;18:443–457. doi: 10.1006/mcne.2001.1028. [DOI] [PubMed] [Google Scholar]
- Clark AW, et al. Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett. 1997;238:53–56. doi: 10.1016/s0304-3940(97)00859-8. [DOI] [PubMed] [Google Scholar]
- Cronin CA. Intravenous Tissue Plasminogen Activator for Stroke: A Review of the ECASS III Results in Relation to Prior Clinical Trials. J Emerg Med. 2010;38:99–105. doi: 10.1016/j.jemermed.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Cunningham SA, et al. JAM2 interacts with alpha4beta1: facilitation by JAM3. J Biol Chem. 2002;277:27589–27592. doi: 10.1074/jbc.C200331200. [DOI] [PubMed] [Google Scholar]
- Cunningham LA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329–39. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- Del Zoppo GJ. Stroke and neurovascular protection. N Engl J Med. 2006;354:553–555. doi: 10.1056/NEJMp058312. [DOI] [PubMed] [Google Scholar]
- Dejana E, et al. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–2122. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- Del Maschio A, et al. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM) J Exp Med. 1999;190:1351–1356. doi: 10.1084/jem.190.9.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derex L, Nighoghossian N. Intracerebral haemorrhage after thrombolysis for acute ischaemic stroke: an update. J Neurol Neurosurg Psychiatry. 2008;79:1093–1099. doi: 10.1136/jnnp.2007.133371. [DOI] [PubMed] [Google Scholar]
- Emsley HC, et al. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J Neuroimmunol. 2003;139:93–101. doi: 10.1016/s0165-5728(03)00134-6. [DOI] [PubMed] [Google Scholar]
- Fanning AS, et al. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998;273:29745–29753. doi: 10.1074/jbc.273.45.29745. [DOI] [PubMed] [Google Scholar]
- Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- Feldman GJ, et al. Occludin: structure, function and regulation. Adv Drug Deliv Rev. 2005;57:883–917. doi: 10.1016/j.addr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Fredriksson L, et al. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004;23:3793–3802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson L, et al. Structural requirements for activation of latent platelet-derived growth factor CC by tissue plasminogen activator. J Biol Chem. 2005;280:26856–26862. doi: 10.1074/jbc.M503388200. [DOI] [PubMed] [Google Scholar]
- Furuse M, et al. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999;147:891–903. doi: 10.1083/jcb.147.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasche Y, et al. Early appearance of activated matrix metalloproteinase-9 after focal cerebral ischemia in mice: a possible role in blood-brain barrier dysfunction. J Cereb Blood Flow Metab. 1999;119:1020–1028. doi: 10.1097/00004647-199909000-00010. [DOI] [PubMed] [Google Scholar]
- Gasche Y, et al. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- Gidday JM, et al. Leukocyte-derived matrix metalloproteinase-9 mediates blood–brain barrier breakdown and is proinflammatory after transient focal cerebral ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H558–H568. doi: 10.1152/ajpheart.01275.2004. [DOI] [PubMed] [Google Scholar]
- Gravanis I, Tsirka SE. Tissue-type plasminogen activator as a therapeutic target in stroke. Expert Opin Ther Targets. 2008;12:159–170. doi: 10.1517/14728222.12.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, et al. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- Guo S, Lo EH. Dysfunctional Cell-Cell Signaling in the Neurovascular Unit as a Paradigm for Central Nervous System Disease. Stroke. 2009;40:S4–7. doi: 10.1161/STROKEAHA.108.534388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney KJ, et al. Blood-brain barrier disruption by stromelysin-1 facilitates neutrophil infiltration in neuroinflammation. Neurobiol Dis. 2006;23:87–96. doi: 10.1016/j.nbd.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Haorah J, et al. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem. 2007;101:566–576. doi: 10.1111/j.1471-4159.2006.04393.x. [DOI] [PubMed] [Google Scholar]
- Haskins J, et al. ZO-3, a novel member of the MAGUK protein family found at the tight junction, interacts with ZO-1 and occludin. J Cell Biol. 1998;1412:199–208. doi: 10.1083/jcb.141.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Heo JH, et al. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999;19:624–633. doi: 10.1097/00004647-199906000-00005. [DOI] [PubMed] [Google Scholar]
- Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108:779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirase T, et al. Occludin as a possible determinant of tight junction permeability in endothelial cells. J Cell Sci. 1997;110:1603–1613. doi: 10.1242/jcs.110.14.1603. [DOI] [PubMed] [Google Scholar]
- Horstmann S, et al. Profiles of matrix metalloproteinases, their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke. 2003;234:2165–2170. doi: 10.1161/01.STR.0000088062.86084.F2. [DOI] [PubMed] [Google Scholar]
- Huang ZG, et al. Biphasic opening of the blood-brain barrier following transient focal ischemia: effects of hypothermia. Can J Neurol Sci. 1999;26:298–304. doi: 10.1017/s0317167100000421. [DOI] [PubMed] [Google Scholar]
- Jian Liu K, Rosenberg GA. Matrix metalloproteinases and free radicals in cerebral ischemia. Free Radic Biol Med. 2005;39:71–80. doi: 10.1016/j.freeradbiomed.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Jin R, et al. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010 Feb 3; doi: 10.1189/jlb.1109766. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justicia C, et al. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2003;23:1430–1440. doi: 10.1097/01.WCB.0000090680.07515.C8. [DOI] [PubMed] [Google Scholar]
- Kago T, et al. Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. 2006;339:1197–1203. doi: 10.1016/j.bbrc.2005.11.133. [DOI] [PubMed] [Google Scholar]
- Kale G, et al. Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem Biophys Res Commun. 2003;302:324–329. doi: 10.1016/s0006-291x(03)00167-0. [DOI] [PubMed] [Google Scholar]
- Keiper T, et al. The role of junctional adhesion molecule-C (JAM-C) in oxidized LDL-mediated leukocyte recruitment. FASEB J. 2005;19:2078–2080. doi: 10.1096/fj.05-4196fje. [DOI] [PubMed] [Google Scholar]
- Kelly PJ, et al. Oxidative stress and matrix metalloproteinase-9 in acute ischemic stroke: the Biomarker Evaluation for Antioxidant Therapies in Stroke (BEAT-Stroke) study. Stroke. 2008;39:100–104. doi: 10.1161/STROKEAHA.107.488189. [DOI] [PubMed] [Google Scholar]
- Kim YH, et al. Nonproteolytic neuroprotection by human recombinant tissue plasminogen activator. Science. 1999;284:647–665. doi: 10.1126/science.284.5414.647. [DOI] [PubMed] [Google Scholar]
- Kuroiwa T, et al. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol 68. 1985;68:122–129. doi: 10.1007/BF00688633. [DOI] [PubMed] [Google Scholar]
- Kvajo M, et al. Regulation of brain proteolytic activity is necessary for the in vivo function of NMDA receptors. J Neurosci. 2004;24:9734–9743. doi: 10.1523/JNEUROSCI.3306-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CH, et al. Critical role of actin in modulating BBB permeability. Brain Res Brain Res Rev. 2005;50:7–13. doi: 10.1016/j.brainresrev.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Lebeurrier N, et al. The brain-specific tissue-type plasminogen activator inhibitor, neuroserpin, protects neurons against excitotoxicity both in vitro and in vivo. Mol Cell Neurosci. 2005;30:552–558. doi: 10.1016/j.mcn.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Lee CZ, et al. Matrix metalloproteinase-9 inhibition attenuates vascular endothelial growth factor-induced intracerebral hemorrhage. Stroke. 2007;38:2563–2568. doi: 10.1161/STROKEAHA.106.481515. [DOI] [PubMed] [Google Scholar]
- Lee HY, et al. Non-proteolytic neurotrophic effects of tissue plasminogen activator on cultured mouse cerebrocortical neurons. J Neurochem. 2007;101:1236–1247. doi: 10.1111/j.1471-4159.2007.04417.x. [DOI] [PubMed] [Google Scholar]
- Levin EG, del Zoppo GJ. Localization of tissue plasminogen activator in the endothelium of a limited number of vessels. Am J Pathol. 1994;144:855–861. [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. PDGF-C is a new protease-activated ligand for the PDGF-α receptor. Nat Cell Biol. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- Liu D, et al. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med. 2004;10:1379–1383. doi: 10.1038/nm1122. [DOI] [PubMed] [Google Scholar]
- Liu LB, et al. Bradykinin increases blood-tumor barrier permeability by down-regulating the expression levels of ZO-1, occludin, and claudin-5 and rearranging actin cytoskeleton. J Neurosci Res. 2008;86:1153–1168. doi: 10.1002/jnr.21558. [DOI] [PubMed] [Google Scholar]
- Liu W, et al. Normobaric hyperoxia attenuates early blood-brain barrier disruption by inhibiting MMP-9-mediated occludin degradation in focal cerebral ischemia. J Neurochem. 2009;108:811–820. doi: 10.1111/j.1471-4159.2008.05821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Atalaya JP, et al. Recombinant Desmodus rotundus salivary plasminogen activator crosses the blood-brain barrier through a low-density lipoprotein receptor-related protein-dependent mechanism without exerting neurotoxic effects. Stroke. 2007;38:1036–1043. doi: 10.1161/01.STR.0000258100.04923.84. [DOI] [PubMed] [Google Scholar]
- Lopez-Atalaya JP, et al. Toward safer thrombolytic agents in stroke: molecular requirements for NMDA receptor-mediated neurotoxicity. J Cereb Blood Flow Metab. 2008;28:1212–1221. doi: 10.1038/jcbfm.2008.14. [DOI] [PubMed] [Google Scholar]
- Lucivero V, et al. Different roles of matrix metalloproteinases-2 and -9 after human ischaemic stroke. Neurol Sci. 2007;28:165–170. doi: 10.1007/s10072-007-0814-0. [DOI] [PubMed] [Google Scholar]
- Ludwig RJ, et al. Junctional adhesion molecule (JAM)-B supports lymphocyte rolling and adhesion through interaction with alpha4beta1 integrin. Immunology. 2009;128:196–205. doi: 10.1111/j.1365-2567.2009.03100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Padura I, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataga N, et al. Permissive proteolytic activity for visual cortical plasticity. Proc Natl Acad Sci U S A. 2002;99:7717–7721. doi: 10.1073/pnas.102088899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, et al. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, et al. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaluk P, et al. Matrix metalloproteinase-9 controls NMDA receptor surface diffusion through integrin beta1 signaling. J Neurosci. 2009;29:6007–6012. doi: 10.1523/JNEUROSCI.5346-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda E, Lomas DA. Neuroserpin: a serpin to think about. Cell Mol Life Sci. 2006;63:709–722. doi: 10.1007/s00018-005-5077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaner J, et al. Matrix metalloproteinase-9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation. 2003;107:598–603. doi: 10.1161/01.cir.0000046451.38849.90. [DOI] [PubMed] [Google Scholar]
- Murakami T, et al. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284:21036–2146. doi: 10.1074/jbc.M109.016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar T, et al. Binding of urokinase to low density lipoprotein-related receptor (LRP) regulates vascular smooth muscle cell contraction. J Biol Chem. 2002;277:40499–40504. doi: 10.1074/jbc.M207172200. [DOI] [PubMed] [Google Scholar]
- Nicole O, et al. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- Nitta T, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piontek J, et al. Formation of tight junction: determinants of homophilic interaction between classic claudins. Faseb J. 2008;22:146–158. doi: 10.1096/fj.07-8319com. [DOI] [PubMed] [Google Scholar]
- Planas AM. Expression and activation of matrix metalloproteinase-2 and -9 in rat brain after transient focal cerebral ischemia. Neurobiol Dis. 2001;8:834–846. doi: 10.1006/nbdi.2001.0435. [DOI] [PubMed] [Google Scholar]
- Polavarapu R, et al. Tissue-type plasminogen activator-mediated shedding of astrocytic low-density lipoprotein receptor-related protein increases the permeability of the neurovascular unit. Blood. 2007;109:3270–3278. doi: 10.1182/blood-2006-08-043125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, et al. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- Rao RK, et al. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. 2002;368:471–481. doi: 10.1042/BJ20011804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell A, et al. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke. 2006;37:1399–1406. doi: 10.1161/01.STR.0000223001.06264.af. [DOI] [PubMed] [Google Scholar]
- Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr Opin Pharmacol. 2008;1:82–89. doi: 10.1016/j.coph.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Rosell A, et al. Mechanisms and markers for hemorrhagic transformation after stroke. Acta Neurochir Suppl. 2008a;105:173–178. doi: 10.1007/978-3-211-09469-3_34. [DOI] [PubMed] [Google Scholar]
- Rosell A, et al. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008b;39:1121–1126. doi: 10.1161/STROKEAHA.107.500868. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, et al. TIMP-2 reduces proteolytic opening of blood-brain barrier by type IV collagenase. Brain Res. 1992;576:203–207. doi: 10.1016/0006-8993(92)90681-x. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, et al. Matrix metalloproteinases and TIMPs are associated with blood–brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Yang Y. Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus. 2007;22:E4. doi: 10.3171/foc.2007.22.5.5. [DOI] [PubMed] [Google Scholar]
- Saitou M, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell. 2000;11:4131–4142. doi: 10.1091/mbc.11.12.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salles FJ, Strickland S. Localization and regulation of the tissue plasminogen activator-plasmin system in the hippocampus. J Neurosci. 2002;22:2125–2134. doi: 10.1523/JNEUROSCI.22-06-02125.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson AL, Medcalf RL. Tissue-type plasminogen activator: a multifaceted modulator of neurotransmission and synaptic plasticity. Neuron. 2006;50:673–678. doi: 10.1016/j.neuron.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Santoso S, et al. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med. 2002;196:679–691. doi: 10.1084/jem.20020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappino AP, et al. Extracellular proteolysis in the adult murine brain. J Clin Invest. 1993;92:679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeds NW, et al. Tissue plasminogen activator induction in Purkinje neurons after cerebellar motor learning. Science. 1995;270:1992–1994. doi: 10.1126/science.270.5244.1992. [DOI] [PubMed] [Google Scholar]
- Sharp FR, et al. Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:1011–1032. doi: 10.1097/00004647-200007000-00001. [DOI] [PubMed] [Google Scholar]
- Siao CJ, et al. Cell type-specific roles for tissue plasminogen activator released by neurons or microglia after excitotoxic injury. J Neurosci. 2003;23:3234–3242. doi: 10.1523/JNEUROSCI.23-08-03234.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sole S, et al. Activation of matrix metalloproteinase-3 and agrin cleavage in cerebral ischemia/reperfusion. J Neuropathol Exp Neurol. 2004;63:338–349. doi: 10.1093/jnen/63.4.338. [DOI] [PubMed] [Google Scholar]
- Soma T, et al. Thr(207) of claudin-5 is involved in size-selective loosening of the endothelial barrier by cyclic AMP. Exp Cell Res. 2004;300:202–212. doi: 10.1016/j.yexcr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Su EJ, et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat Med. 2008;14:731–737. doi: 10.1038/nm1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, et al. Stromelysin-1 (MMP-3) is critical for intracranial bleeding after t-PA treatment of stroke in mice. J Thromb Haemost. 2007;5:1732–1739. doi: 10.1111/j.1538-7836.2007.02628.x. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, et al. Tissue-type plasminogen activator (t-PA) induces stromelysin-1 (MMP-3) in endothelial cells through activation of lipoprotein receptor-related protein. Blood. 2009;114:3352–3358. doi: 10.1182/blood-2009-02-203919. [DOI] [PubMed] [Google Scholar]
- Taddei A, et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934. doi: 10.1038/ncb1752. [DOI] [PubMed] [Google Scholar]
- Thiyagarajan M, et al. Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci. 2008;28:12788–12797. doi: 10.1523/JNEUROSCI.3485-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirka SE, et al. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, et al. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji K, et al. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke. 2005;36:1954–1959. doi: 10.1161/01.STR.0000177517.01203.eb. [DOI] [PubMed] [Google Scholar]
- Tsukita S, Furuse M. Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol. 1999;9:268–273. doi: 10.1016/s0962-8924(99)01578-0. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. Lipoprotein receptor–mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat Med. 2003;9:1313–1317. doi: 10.1038/nm926. [DOI] [PubMed] [Google Scholar]
- Wang YF, et al. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- Weber C, et al. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7:467–477. doi: 10.1038/nri2096. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, et al. Phosphorylation of claudin-5 and occludin by rho kinase in brain endothelial cells. Am J Pathol. 2008;172:521–533. doi: 10.2353/ajpath.2008.070076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]
- Yepes M, et al. Neuroserpin reduces cerebral infarct volume and protects neurons from ischemia-induced apoptosis. Blood. 2000;96:569–576. [PubMed] [Google Scholar]
- Yepes M, et al. Regulation of seizure spreading by neuroserpin and tissue-type plasminogen activator is plasminogen-independent. J Clin Invest. 2002;109:1571–1578. doi: 10.1172/JCI14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes M, et al. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest. 2003;112:1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes M, et al. Tissue-type plasminogen activator in the ischemic brain: more than a thrombolytic. Trends Neurosci. 2009;32:48–55. doi: 10.1016/j.tins.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Zeller JA, et al. Platelet-leukocyte interaction and platelet activation in acute stroke with and without preceding infection. Arterioscler Thromb Vasc Biol. 2005;25:1519–1523. doi: 10.1161/01.ATV.0000167524.69092.16. [DOI] [PubMed] [Google Scholar]
- Zhang X, et al. Tissue-type plasminogen activator and the low-density lipoprotein receptor-related protein mediate cerebral ischemia-induced nuclear factor-kappaB pathway activation. Am J Pathol. 2007;171:1281–1290. doi: 10.2353/ajpath.2007.070472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, et al. Adjuvant treatment with neuroserpin increases the therapeutic window for tissue-type plasminogen activator administration in a rat model of embolic stroke. Circulation. 2002;106:740–745. doi: 10.1161/01.cir.0000023942.10849.41. [DOI] [PubMed] [Google Scholar]
- Zhao BQ, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV, et al. Functional recovery after embolic stroke in rodents by activated protein C. Ann Neurol. 2005;58:474–477. doi: 10.1002/ana.20602. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Remodeling after stroke. Nat Med. 2006;12:390–391. doi: 10.1038/nm0406-390. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]