

Abstract
Epidemiological and clinical studies suggest that an increased intake of dietary selenium significantly reduces overall cancer risk, but the anti-cancer mechanism of selenium is not clear. In the present study, we fed intestinal cancer mouse model Muc2/p21 double mutant mice with a selenium-enriched (sodium selenite) diet for 12 weeks or 24 weeks, and found that sodium selenite significantly inhibited intestinal tumor formation in these animals (p<0.01), which was associated with phosphorylation of JNK1 and suppression of β-catenin and COX2. In vitro studies showed that sodium selenite promoted cell apoptosis and inhibited cell proliferation in human colon cancer cell lines HCT116 and SW620. These effects were dose- and time course- dependent, and were also linked to an increase of JNK1 phosphorylation and suppression of β-catenin signaling. Reduced JNK1 expression by small RNA interference abrogated sufficient activation of JNK1 by sodium selenite, leading to reduced inhibition of the β-catenin signaling, resulting in reduced efficacy of inhibiting cell proliferation. Taken together, our data demonstrate that sodium selenite inhibits intestinal carcinogenesis in vivo and in vitro through activating JNK1 and suppressing β-catenin signaling, a novel anti-cancer mechanism of selenium.
Keywords: selenium, JNK1, β-catenin, intestinal cancer
Introduction
Selenium is an important trace element that is involved in different physiological functions of human body. Epidemiological and clinical studies reported that inadequate status of selenium increased risk of cancer. Basic research and clinical trials strongly support the protective role of selenium against various types of cancer, the effect was most pronounced in prostate and colorectal cancers 1-7. Unfortunately, a recent report of the Selenium and Vitamin E Cancer Prevention Trial (SELECT) demonstrated that selenium or vitamin E, alone or in combination did not prevent prostate cancer and secondary cancers (including colorectal cancer) 8. However, the failure of one form of selenium does not disprove of other forms of selenium. In addition, the efficacy of selenium against cancer will also depend on the form of selenium and the dosage, organ site specificity of carcinogenesis, and on the baseline of selenium status and genotypes (e.g. polymorphims) of glutathione peroxidase 1, a selenium enzyme, of the subjects. The anticancer effects of selenium has been postulated to link to inhibition of cell proliferation and induction of apoptosis through different signaling pathways, particularly the anti-oxidative and anti-inflammatory effects mediated through the activity of selenoenzymes 7. However, the targets and underlying mechanism of anticancer action by selenium are largely unknown.
Wnt/β-catenin signal pathway plays a critical role in embryonic development, tissue homeostasis and carcinogenesis 9-11. Oncogenic activation of Wnt/β-catenin signaling pathway by mutation of adenomatous polyposis coli (APC) or/and CTNNB1 interacts with TCF/LEF transcription factors that activate transcription of genes, such as c-myc and cyclin D1, and increases prevalence of many kinds of human carcinogenesis, especially colorectal cancer 12, 13. Therefore, targeting β-catenin signaling will be an effective approach for colorectal cancer prevention. Indeed, non-steroidal anti-inflammatory drugs (e.g. aspirin, sulindac, celecoxib, rofecoxib), vitamins (e.g. vitamin A, D and their derivatives), have shown their anticancer effects through inhibiting Wnt/β-catenin signal pathway 14-16. In addition, sulindac inhibits intestinal tumor formation by activating c-Jun NH2-ternimal kinase 1 (JNK1) 17, a member of mitogen-activated protein kinase (MAPK) family, which plays a critical role in the regulation of cell proliferation, differentiation and apoptosis 18-20. Recently we demonstrated that activated JNK1 interacts with and downregulates β-catenin signaling 21. Therefore, JNK1 and Wnt/β-catenin signaling could be promising targets for chemoprevention, chemotherapy and further anticancer drug discovery.
Using the Muc2/p21 mouse model of intestinal cancer 22, 23 and colon cancer cell lines, we found that a selenium-enriched (sodium selenite) diet significantly inhibited intestinal tumorigenesis, and that sodium selenite promoted colon cancer cell apoptosis and inhibited cell proliferation, by activating JNK1 and suppressing β-catenin signaling.
Materials and methods
Animal study and mouse intestinal epithelial cell isolation
As described previously 23, Muc2−/− mice (C57/BL6 background) 22 were mated with p21−/− mice (C57/BL6 background) 24 to generate Muc2−/−,p21+/− double mutant mice 23, in which more intestinal tumors (including colorectal tumors) were expected. After weaning, the Muc2/p21 mice were randomly grouped and fed an AIN-76 control diet (fat 5%, from corn oil) , a Western-style high-risk diet (fat 20%), as reported recently 25, 26, or the Western-style diet supplemented with 0.00004% (4.0 ppm) sodium selenite (a 20 fold increase in dietary selenium level compared to that in the Western-style diet) for 12 weeks or 24 weeks, respectively. All diets were made by the Research Diets, Inc. (New Brunswick, NJ). At the endpoint, the animals were sacrificed, and tumorigenesis was evaluated as described 23. Intestinal epithelial cells from normal mucosa were isolated by incubating with 15 mM EDTA at 37°C for 20 minutes, as we described recently 18. The cell pellets were lysed with RIPA lysis buffer immediately or stored at −80°C for immunoblotting analysis. All animal studies were conducted in the University of Illinois at Chicago with an approval by the Animal Care and Use Committee at the University of Illinois at Chicago.
Cell lines and cell culture
Human colon cancer cell lines SW620 (wild-type β-catenin) and HCT116 (mutant β-catenin) were purchased from ATCC (American Type Culture Collection, Manassas, VA) and were maintained in MEM or McCoy's 5A medium, respectively. The medium was supplemented with 10% (v/v) fetal bovine serum (FBS), 1×antibiotic/ antimycotic (1×105 U/L streptomycin, 1×105 U/L penicillin, and 0.27 μmol/L amphotericin B). All cell lines were cultured in humidified incubator at 37 °C with 5% CO2. Sodium selenite (Sigma, St. Louis, MO) was dissolved in phosphate buffer solution (PBS) and stored at – 20°C at 1mol/L stock solution.
Cell proliferation assay
1 ×104 cells were seeded in each well of a 96-well plate and incubated overnight. The medium was removed. 1 × 10−4L of the medium with a final concentration of sodium selenite from physiological dosage (0, 25, 50, 100, 250, 500 and 1000 nmol/L) to pharmaceutical dosage (2.5, 5.0, 7.5, 10, 12.5 and 15 μmol/L) was added to each well, PBS was used as a control. Three independent experiments were performed. After 24, 48 and 72 h exposure to sodium selenite, cell proliferation was determined by MTS assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) according to the manufacturer's protocol (CellTiter 96 Non-Radioactive Cell Proliferation Assay Kit, Promega Corporation, Madison, WI).
Apoptosis analysis
6×105 cells were seeded in 6-well plates and incubated overnight until 40–50% confluency was reached. Cells were treated with different dosages of sodium selenite. PBS was used as a control. The cells were harvested at 24 ,48, and 72 h, washed with cold PBS, then fixed with 80% ethanol for 8 h at 4 °C, followed by staining with propidium iodide buffer (0.075 mmol/L propidium iodide, 0.1% sodium citrate, and 0.1%TritonX-100) for 3 h at 4 °C. 10,000 cells were analyzed for apoptosis using a Becton Dickinson FACScan (Becton Dickinson Immunocytometry Systems, San Jose, CA). The percentage of apoptotic cells was quantified using Cell Quest software.
Immunoblotting analysis
Sodium selenite treated cells were harvested at various time points. The cell pellets were lysed with RIPA (radio immunoprecipitation assay) lysis buffer (50 mmol/L Tris-HCl, pH 8.0, with 150 mmol/L sodium chloride, 1.0% Igepal CA-630 (NP-40), 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate). 30 μg of protein was loaded and separated in 12% SDS-PAGE gels and transferred to Immun-Blot PVDF membranes (Bio-Rad Laboratories). The following primary antibodies were used to probe the alterations of protein: JNK1, phosphorylated-JNK (p-JNK), β-catenin, cyclin D1 (Santa Cruz Biotechnology, Santa Cruz, CA), Caspase 3, cleaved caspase 3 (Cell Signaling Technology, Danvers, MA), CDK4 (BD PharMingen, San Diego, CA), cmyc (Sigma, St. Louis, MO), COX2 (Cayman Chemical, Ann Arbor, MI). All secondary antibodies against rabbit, goat or mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Signal was detected by enhanced chemoluminescence techniques (Amersham Life Science, Piscataway, NJ). β-actin (Sigma, St. Louis, MO) was used as loading control. Signal quantification was obtained using Quantity One software (Bio-Rad Laboratories, Hercules, CA) and normalized to β-actin. The ratio is presented.
Transfection and luciferase reporter assays
To examine the effect of sodium selenite on β-catenin/TCF transcriptional signaling, cells were plated in 24-well plates at a density of 2×105 cells/well and incubated overnight. The following day, cells were transiently transfected with β-catenin-TCF luciferase reporter construct TOPFlash which contains multiple optimal TCF/LEF (lymphocyte enhancing factor) binding sites that induce transcription of a luciferase reporter gene when activated by β-catenin, or FOPFlash which contains mutant and inactivated TCF/LEF binding sites, respectively (Upstate Biotechnology, Lake Placid, NY). To correct the differences caused by transfection efficiency, cells were co-transfected with a Renilla reporter plasmid using LipofectAMINE 2000 according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). 6 h after transfection, the medium was removed and the cells were treated with fresh medium supplemented with 5 μmol/L of sodium selenite for 24 , 48 and 72h, or treated with 0, 1, 2.5 ,5 μmol/L of sodium selenite for 48h. Cells were washed with cold PBS and lysed with passive lysis buffer (PLB) (Promega, Madison, WI). Luciferase activity was measured using the Dual Luciferase Report Assay System (Promega Corporation, Madison, WI). Lysate firefly luciferase values were normalized to Renilla luciferase activity. These experiments were triplicated.
Immunocytochemical staining
Human osteosarcoma cells U2OS grown on a chamber slide (BD Biosciences, San Jose, CA) were treated with 5 μmol/L of sodium selenite or no sodium selenite MEM medium as a control. Forty-eight hours after treatment, cells were washed with cold PBS, fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min and rendered permeable by further treatment with 0.2% Triton X-100 for 5 min, blocked with 10% normal goat serum, and probed with goat anti-β-catenin polyclonal antibody (sc-1496; Santa Cruz Biotechnology, Santa Cruz, CA). Detection was with biotinylated anti-goat IgG, followed by incubation with avidin-biotin complex (Vector Laboratories, Burlingame, CA) and substrate 3’, 5’-diaminobenzidine, followed with hematoxylin counterstaining. These experiments were triplicated.
Small Interfering RNA Experiments
Targeted small interfering RNA for human JNK1 (si-JNK1)(Sigma, St. Louis, MO) (sequence of si-JNK1 was 5′-GGGAUUUGUUAUCCAAAAU-3′) was transfected into the human colon cancer cell line HCT116. 24 h after transfection, the cells were treated with 5 μmol/L of sodium selenite for 48 h. The small interfering RNA for human green fluorescence protein (si-GFP) (Sigma, St. Louis, MO) was used as control. Cell proliferation was determined by MTS, as described above.
Results
Selenium-enriched diet inhibited the added effects of western style diet on tumorigenesis of Muc2/p21 mice
To elucidate chemopreventive effects and the mechanisms of selenium in vivo, we fed the Muc2/p21 mice with either a western-style high-risk diet (high fat and low calcium and vitamin D) or the western-style diet supplemented with sodium selenite for 12 weeks or 24 weeks after weaning. After 12 weeks of feeding, the western style high-risk diet (high fat and low calcium and vitamin D) was additive to the genetic mutation of the Muc2 and p21 genes to augment intestinal tumor formation at an average of 2.1 tumors per mouse (2.1±0.7, mean ± SD, 9 mice) and tumor incidence of 100% (11/11) in Muc2−/−,p21+/− mice, compared to about 1 tumor per mouse (12 mice) and tumor incidence of 78% (7/9) in the Muc2−/−,p21+/− mice fed with AIN-76A control diet. However, intestinal tumorigenesis driven by the western-style diet was reversed by sodium selenite supplementation, reducing tumor numbers to 1.6 per mouse and tumor incidence to 83% (10/12) (Fig.1A).
Figure 1.

The Western-style diet (WD) accelerated mouse intestinal tumorigenesis, in comparison with the AIN-76A diet, but the Western-style diet supplemented with sodium selenite inhibited intestinal tumor formation in the Muc2−/−,p21+/− mice for 12 or 24 weeks (A), which occurred through activating JNK1 (JNK1 phosphorylation, p-JNK) and suppressing β-catenin and COX2 in intestinal epithelial cells from normal mucosa (B). β-actin was used as internal control. The image signal was quantified and normalized to β-actin. The ratio was presented. WD, western-style diet; WD+Sele, western-style diet supplemented with sodium selenite. N, number of animals studied in each group.
After 24 weeks of feeding, similar to our previous report 23, the Muc2−/−,p21+/− mice on AIN-76A control diet developed 1.9 intestinal tumors (1.9±0.2) per mouse, but the tumor number was increased to 4.3 (4.3±0.6) per mouse with feeding the Western-style high-risk diet (Fig.1A) (p=0.003). However, selenium-supplemented Western-style diet dramatically inhibited intestinal tumorigenesis, in terms of a significant reduction of tumor frequency from 4.3 to 1.9 per mouse (1.9±0.4) (p=0.008) (Fig. 1A).
To elucidate the underlying mechanisms of tumor inhibition activity of sodium selenite, we isolated mouse small intestinal epithelial cells from the mice that were on diet for 24 weeks, analyzed protein alterations by Western blotting, and found that tumor inhibition by sodium selenite was strongly associated with the activation of JNK1 (phosphorylated JNK1, p-JNK1) and suppression of β-catenin in mouse intestinal epithelial cells (Fig. 1B). Interestingly, inhibition of COX2 by sodium selenite was also seen in mouse intestinal epithelial cells (Fig. 1B).
Sodium selenite inhibited cell proliferation and promoted apoptosis in colon cancer cells
To elucidate the anti-cancer activities of sodium selenite and the underlying mechanism in vitro, we treated human colon cancer cell lines HCT116 and SW620 with physiological dosages (0, 25, 50, 100, 250, 500 and 1000 nmol/L) and pharmaceutical dosages (2.5, 5, 7.5, 10, 12.5 and 15 μmol/L) of sodium selenite for 48h, respectively, and found that the physiological range of sodium selenite did not affect colon cancer cell proliferation (data not shown), however, the pharmaceutical dosage of sodium selenite dramatically inhibited cell growth, and cell growth inhibition was in a dose-dependent manner in both cell lines (Fig. 2A, C). We therefore decided to use the pharmaceutical dosage of sodium selenite for the rest of our study. To investigate duration effect, cells were treated with 5 μmol/L of sodium selenite for 12, 24, 48, 72, 96 and 120 h, respectively. As shown in Fig. 2B and 2D, sodium selenite inhibited cell proliferation in a time-dependent manner and its anticancer ability can be extended to more than five days, indicating that a low dosage and a long term intake of sodium selenite supplemental may be used as an approach for the chemoprevention and chemotherapy of colon cancer.
Figure 2.
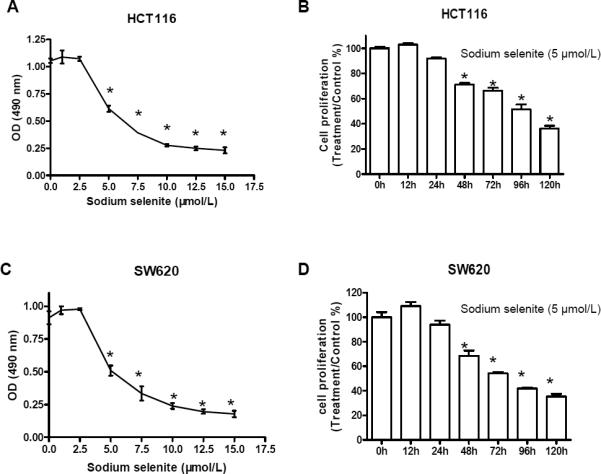
Sodium selenite inhibited cell proliferation in human colon cancer lines HCT116 and SW620. A and C, cell proliferation inhibition by sodium selenite occurred in a dosage dependent manner (48h); B and D, inhibition of cell proliferation by sodium selenite was time-dependent (5 μmol/L). * p<0.01, compared to the control group (0 μmol/L, or 0 h, respectively). These experiments were triplicated independently.
We also found that sodium selenite caused cell cycle arrest – S/G2 phase increased about 1.2 fold in both HCT116 and SW620 cell lines after 24 h and about 1.8 fold in both cell lines after 48 h treatment of 5 μmol/L of sodium selenite, respectively, which is consistent with previous reports 27-30.
To determine whether sodium selenite has the ability to induce apoptosis in HCT116 and SW620 cells, we used P.I. staining cooperated with flow cytometry scanning to analyze apoptosis in both cell lines with treatment of sodium selenite at the final concentration of 2.5, 5 and 7.5 μmol/L for 48 h. As shown in Fig. 3A and 3D, a lower dosage of sodium selenite (2.5 μmol/L) could induce apoptosis, and a higher dosage from 5 μmol/L significantly induced apoptosis in both colon cancer cell lines, indicating that the induction of apoptosis by sodium selenite was dosage-dependent. We then treated the HCT116 and SW620 cells with 5 μmol/L of sodium selenite for 24, 48 and 72 h, respectively, and found that sodium selenite-induced apoptosis in both cell lines was in a time-dependent manner (Fig. 3B and 3E). The longer both cell lines were treated with sodium selenite, the more cells underwent apoptosis. To further confirm the induction of apoptosis, western-blot was performed and an increased cleavage of Caspase 3 was seen in both colon cancer cell lines after 48 and 72 h treatment of 5 μmol/L of sodium selenite (Fig. 3C and 3F).
Figure 3.

Sodium selenite promoted apoptosis in colon cancer lines HCT116 and SW620. A and D, apoptosis induction by sodium selenite was in a dosage dependent manner (48h); B and E, apoptosis induction by sodium selenite was time-dependent (5μmol/L); C and F, increase of cleaved Caspase 3, a biomarker of apoptosis, was observed in both colon cancer cell lines after 48 h and 72 h treatment of 5 μmol/L of sodium selenite, while the total caspases 3 was slightly reduced. “C” referred as “control”, and “T” referred as “sodium selenite treatment”. β-actin was used as loading control. The image signal was quantified and normalized to β- actin. The ratio was presented. These experiments were triplicated independently. (* p<0.05, ** p<0.01, compared to control, respectively).
Sodium selenite inhibited β-catenin signaling in colon cancer cells
β-catenin signaling pathway plays critical roles in carcinogenesis and is a promising target for colorectal cancer prevention and chemotherapy. Thus, we determined whether sodium selenite-mediated inhibition of cell proliferation and induction of apoptosis was associated with β-catenin signaling. As shown in figure 4, β-catenin was indeed inhibited by sodium selenite in both HCT116 and SW620 cell lines, and the inhibition of β-catenin occurred in a dose- and time- dependent manner. Downstream targets of β-catenin, such as cyclin D1, CDK4 and c-myc, were also suppressed by sodium selenite (Fig. 4A-4D).
Figure 4.


Sodium selenite activated JNK1 (increase of phosphorylated JNK1, p-JNK1) and inhibited β-catenin signaling in dose- and time-dependent manner in colon cancer cells HCT116 (A and B) and SW620 (C and D), respectively. “C” referred as “control”, and “T” referred as “sodium selenite treatment”. β-actin was used as loading control. The image signal of p-JNK1 and β-catenin was quantified and normalized to β-actin. The ratio was presented. These experiments were triplicated independently.
To further determine whether the suppression of β-catenin expression by sodium selenite corresponded to the reduction of β-catenin/TCF transcriptional activity, the two colon cancer cell lines were transiently co-transfected with the TCF/LEF luciferase reporter construct (TOPFlash) or mutant TCF/LEF luciferase reporter construct (FOPFlash) and Renilla reporter plasmid, respectively. Sodium selenite significantly reduced β-catenin/TCF/LEF luciferase activity (TOPFlash) in both cell lines, compared to FopFlash control (Fig. 5). The reduction of β-catenin transcriptional activities occurred furthermore in a dosage-dependent manner (Fig. 5A, 5C). Additional, sodium selenite-mediated reduction of β-catenin/TCF/LEF transcriptional activity occurred in a time-dependent manner (Fig. 5B, 5D). The reduction of β-catenin/TCF transcriptional activity could be affected by the reduction of β-catenin expression, which was consistent with the reduced expression of β-catenin, c-myc, cyclin D1 and CDK4 as shown in figure 4.
Figure 5.
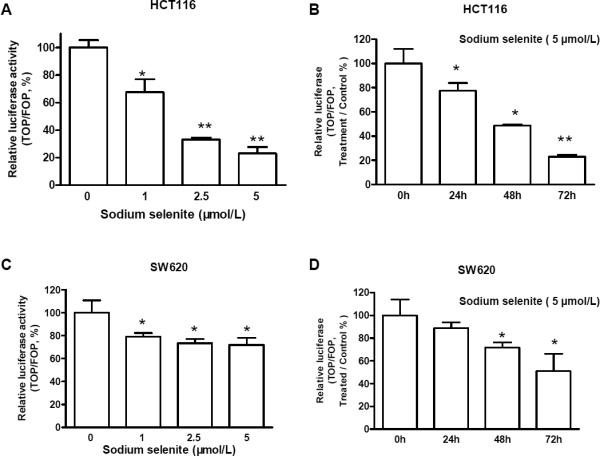
Sodium selenite inhibited β-catenin/TCF4 transcriptional activities in dose- and time-dependent manner in colon cancer cells HCT116 (A and B) and SW620 (C and D). As described in “Materials and Methods”, colon cancer cells were co-transfected with TOPFlash (TOP) or FOPFlash (FOP) and with Renilla reporter plasmid using LipofectAMINE 2000. 6 h after transfection, the medium was removed and the cells were treated with fresh medium supplemented with 5 μmol/L of sodium selenite for 24 , 48 h and 72h, or treated with 0, 1, 2.5 ,5 μmol/L of sodium selenite for 48h. PBS was used as control. The presented data were the results from three independent luciferase activity assays (*p<0.05, **p<0.01, compared to control, respectively.)
β-catenin activation, i.e., translocation into the nuclei, is an important event in promoting cell growth and carcinogenesis. To determine whether selenium inhibits of β-catenin nuclear translocation, in a pilot experiment, we treated human osteosarcoma cell line U2OS with 5 μmol/L of sodium selenite for 48 hours. Immunocytochemical staining showed reduction of nuclear β-catenin expression as well as cytoplasmic expression by sodium selenite, compared to the control (Fig. 6), indicating selenium could cause β-catenin translocation from nuclei into cytoplasm in which β-catenin was degraded via other mechanisms, such as activation JNK1, as we recently reported that activated JNK1 interacts with and inhibits β-catenin expression 21.
Figure 6.
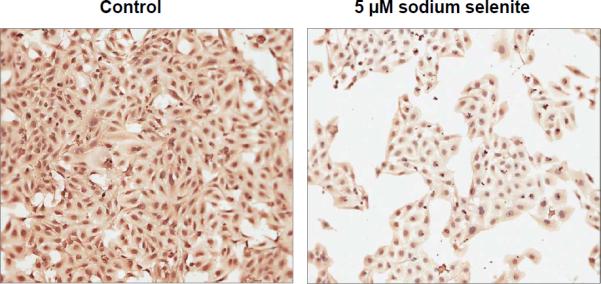
Sodium selenite inhibited nuclear β-catenin expression as well as cytoplasmic expression in human osteosarcoma cells after 48 h treatment of 5 μmol/L of sodium selenite.
JNK1 played an important role in sodium selenite-mediated inhibition of cell proliferation and suppression of β-catenin signaling
Previous studies have shown that JNK1 plays a critical role in intestinal cell maturation, including apoptosis, differentiation and proliferation 18. Our recent data also showed that the JNK1 interacts with β-catenin and downregulates β-catenin signaling in vivo and in vitro 21. Therefore, we elucidated whether the JNK1 was involved in selenium-mediated promotion of apoptosis and suppression of cell proliferation and β-catenin signaling in colon cancer cells. In both colon cancer cell lines HCT116 and SW620, phosphorylation of JNK1 (an active form of JNK1) was increased by 3.2 or 1.5 fold, respectively, corresponding with a 20% to 60% of inhibition of β-catenin and its targets (i.e. c-myc, cyclin D1 and CDK4) (Fig. 4). These effects of sodium selenite were in dose- and time-dependent manners, indicating that JNK1 was functioning in the effects of sodium selenite. Taken above, one could explain that selenium activates JNK1, triggering apoptosis (Fig.3), then the activated JNK1 inhibited β-catenin, resulting in cell growth inhibition (Fig.2).
To determine whether JNK1 was required for sodium selenite-mediated inhibition of cell proliferation and suppression of β-catenin signaling, we used targeted human JNK1 small interfering RNA (siRNA) to knockdown JNK1 expression and treated HCT116 cells with 5 μmol/L sodium selenite for 48 h. Our previous study demonstrated that this siRNA (si-JNK1) was efficient to knockdown JNK expression and abrogated butyrate-induced apoptosis 18. We found that cell proliferation was significantly inhibited (Fig. 7A) and the β-catenin protein levels were dramatically suppressed (Fig. 7B) in GFP-siRNA control transfected cells. However, knockdown of JNK1 by siRNA attenuated selenium-mediated inhibition of cell proliferation (Fig. 7A), because the activation of JNK1 was not induced as much as in the control cells (Fig.7B). Consequently, the suppression of β-catenin, c-myc, CDK4 and cylin-D1 expression in JNK1 siRNA transfected cells by sodium selenite was much less than those in the cells without si-JNK1 transfection (Fig.7B).
Figure 7.

Cell growth inhibition by sodium selenite was JNK1 dependent in colon cancer cells HCT116. Knockdown of JNK1 by siRNA abrogated sodium selenite-mediated inhibition of cell growth (A) and suppression of the expression of β-catenin signaling pathway (B) (48 h). si-GFP was a small interfering RNA targeting human green fluorescence protein (GFP) and was used as mock control; si-JNK1 was a small interfering RNA targeting human JNK1. The knockdown efficiency was confirmed by western blotting. These experiments were triplicated independently. The image signal was quantified and normalized to β-actin. The ratio was presented right below the image. (* p<0.01, compared to the “0”)
Discussion
Several randomized controlled trials have demonstrated significant cancer prevention of selenium in colorectal cancer 3, 31-33. The milestone was cancer prevention by selenium supplementation conducted by Clark and his colleagues 3, although the SELECT trial was failed 8, in part due to the formulation of selenium (L-selenomethoinine in SELECT trial v.s. selenized yeast in Clark's trial) and others addressed earlier. In some other studies and our current in vitro and in vivo studies, selenium showed effective inhibition of colorectal carcinogenesis. Moreover, we also found that sodium selenite alone or in combination with sulindac were effective in preventing intestinal tumor formation in the Apc/p21 mice (Bi and Yang, unpublished data). Therefore, selenium is still a promising chemopreventive agent. However, some issues should be considered for human use of selenium for cancer prevention, such as selection of population with determination of selenium status and glutathione peroxidase 1 genotypes, different dosage and formulation, etc.
It has been reported that anti-cancer activity of selenium was associated with multiple mechanisms, such as anti-oxidation 34, 35, anti-inflammation and/or suppression of β-catenin by 1,4-phenylene bis(methylene) selenocyanate 7, 36, 37, and increasing phosphorylation of mitogen-active protein kinase (MAPK) in prostate cancer cells by methylseleninic acid 38, 39. But we found that tumor inhibition by sodium selenite in colorectal cancer was linked to the phosphorylation of JNK1 and consequent inhibition of β-catenin and its transcriptional targets c-myc, cyclin D1 and CDK4, leading to apoptosis induction and cell proliferation inhibition.
It has been known that JNK1 plays important roles in the regulation of cell proliferation, differentiation and apoptosis in response to cellular stress (e.g. cytokines and ultraviolet irradiation) and chemopreventive agents (e.g. sulindac). Our data strongly suggest that JNK1 plays a critical role in selenium-mediated chemoprevention of colorectal cancer – the activation of JNK1 might be one of the key factors through which selenium inhibited cell proliferation and induced apoptosis in colorectal cancer cells and prevented intestinal cancer formation in Muc2/p21 mouse model of colorectal cancer. However, lack of JNK1 abrogated inhibition of cell proliferation by selenium, which proves the necessity of JNK1 in selenium-mediated tumor inhibition. Therefore, this report is the first to reveal a novel mechanism of selenium in cancer chemoprevention.
Increasing evidence shows that there is crosstalk between MAPK and Wnt signaling through other pathways, such as Akt, p38 and p53. But most recently we found that the MAPK JNK1 interacts with Wnt/β-catenin signaling and negatively regulates β-catenin signaling through GSK3β and the proteasome pathway 21. We found that activation of JNK1 dramatically inhibited β-catenin expression and transcriptional activities, but loss of JNK1 upregulated β-catenin signaling in HEK293T cells and human colon cancer cells. Most impressively, mice with JNK1 deficiency spontaneously developed intestinal tumors, which were linked to upregulation of β-catenin, resulting in increase of cell proliferation and decrease of cell differentiation and apoptosis in intestinal mucosa and tumor tissues 18. Here we show that phosphorylation of JNK1 stimulated by sodium selenite correlates with downregulation of β-catenin in translational and transcriptional level, further supporting our observation that JNK1 suppresses β-catenin.
Activation and nuclear accumulation of β-catenin is known as a key event of transformation and carcinogenesis in colon cancer, therefore, targeting β-catenin has been a promising strategy for cancer prevention and therapy. Herein again, we found that sodium selenite inhibited β-catenin expression and transcription, recued nuclear expression. It will be very interesting to investigate whether it was a direct target of selenium or a subsequence of activation of JNK1 by sodium selenite or both. Studying this will improve our understanding of the chemopreventive mechanisms by selenium.
We recently reported that intestinal tumorigenesis in the Muc2−/− was associated with chronic intestinal inflammation, in terms of elevation of several cytokines and COX2 levels in Muc2−/− mice 40. These results led us to determine whether sodium selenite functions in inhibiting inflammation since sodium selenite significantly prevented intestinal tumor formation (Fig. 1A). Interestingly, we found that COX2 expression was almost completely eliminated in sodium selenite-fed Muc2−/− mouse intestinal epithelial cells (Fig. 1B). However, whether sodium selenite affected other cytokines and/or MAPK JNK1 regulates COX2 as seen in the β-catenin signaling cascade is still under investigation.
Studies from our group and others have suggested several potential regulations among JNK1, β-catenin and COX2, such as regulation of COX2 by JNK1 in MEFs 41, 42, regulation of COX2 by β-catenin in Apc+/− mice, regulation of β-catenin by COX2 in Apc/Muc2 mice 40, and regulation of β-catenin by PGE2 in vitro 43, 44. However, none of these regulators is well defined and it is important to be elucidated, which will provide important evidence of the regulations among JNK1, β-catenin and COX2, providing a rational for developing chemopreventive strategies of colorectal cancer.
In summary, our data demonstrate that sodium selenite is effective to promote apoptosis and inhibit proliferation, and to eventually inhibit intestinal carcinogenesis by targeting JNK1 and β-catenin signaling pathway, which provides more evidence of the anti-cancer bioactivity of selenium and improved our understanding of the mechanism of selenium-mediated chemoprevention and chemotherapy in colorectal cancer.
Acknowledgement
We would like to thank Drs. Anna Velcich and Leonard Augenlicht (Albert Einstein Cancer Center, Bronx, NY) for providing Muc2−/− mice and support of experimental design and data interpretation, thank Dr. Philip Leder (Howard Hughes Medical Institute, Harvard University Medical School, Boston, MA) for providing p21−/− mice, and thanks Dr.Deming Gou (University of Illinois at Chicago, Chicago, IL) for providing reagents. Thanks to Dr.Nicole M Pohl (University of Illinois at Chicago, Chicago, IL) for critical reading and editing this manuscript and thanks to Lindsay Gallagher (University of Illinois, Chicago, IL) for technical assistance.
Grant support: This work was supported in part by the National Cancer Institute grant (CA112081 to W.Y.) and the American Institute for Cancer Research grant (05A121 to W.Y.). Fang W is a recipient of the State Scholarship Fund of China (2007-3020).
Abbreviations
- JNK1
c-Jun NH2-terminal kinase 1
- APC
adenomatous polyposis coli
- MAPK
mitogen-activated protein kinase
- TCF
T cell factor
- LEF
lymphoid enhancer factor
- COX2
cyclooxygenase 2
- siRNA
small interfering RNA
References
- 1.Wang L, Bonorden MJ, Li GX, Lee HJ, Hu H, Zhang Y, Liao JD, Cleary MP, Lu J. Methyl-selenium compounds inhibit prostate carcinogenesis in the transgenic adenocarcinoma of mouse prostate model with survival benefit. Cancer Prev Res (Phila Pa) 2009;2:484–95. doi: 10.1158/1940-6207.CAPR-08-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Facompre N, El-Bayoumy K. Potential stages for prostate cancer prevention with selenium: implications for cancer survivors. Cancer Res. 2009;69:2699–703. doi: 10.1158/0008-5472.CAN-08-4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark LC, Combs GF, Jr., Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, Krongrad A, Lesher JL, Jr., et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. Jama. 1996;276:1957–63. [PubMed] [Google Scholar]
- 4.Greenwald P, Anderson D, Nelson SA, Taylor PR. Clinical trials of vitamin and mineral supplements for cancer prevention. Am J Clin Nutr. 2007;85:314S–7S. doi: 10.1093/ajcn/85.1.314S. [DOI] [PubMed] [Google Scholar]
- 5.Hawk ET, Levin B. Colorectal cancer prevention. J Clin Oncol. 2005;23:378–91. doi: 10.1200/JCO.2005.08.097. [DOI] [PubMed] [Google Scholar]
- 6.Ip C. Lessons from basic research in selenium and cancer prevention. J Nutr. 1998;128:1845–54. doi: 10.1093/jn/128.11.1845. [DOI] [PubMed] [Google Scholar]
- 7.Peters U, Takata Y. Selenium and the prevention of prostate and colorectal cancer. Mol Nutr Food Res. 2008;52:1261–72. doi: 10.1002/mnfr.200800103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA, Parsons JK, Bearden JD, 3rd, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Jama. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 10.Gavert N, Ben-Ze'ev A. β-Catenin signaling in biological control and cancer. J Cell Biochem. 2007;102:820–8. doi: 10.1002/jcb.21505. [DOI] [PubMed] [Google Scholar]
- 11.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–98. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 12.Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327–35. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- 13.Nathke I. Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nat Rev Cancer. 2006;6:967–74. doi: 10.1038/nrc2010. [DOI] [PubMed] [Google Scholar]
- 14.Herbst A, Kolligs FT. Wnt signaling as a therapeutic target for cancer. Methods Mol Biol. 2007;361:63–91. doi: 10.1385/1-59745-208-4:63. [DOI] [PubMed] [Google Scholar]
- 15.Luo J, Chen J, Deng ZL, Luo X, Song WX, Sharff KA, Tang N, Haydon RC, Luu HH, He TC. Wnt signaling and human diseases: what are the therapeutic implications? Lab Invest. 2007;87:97–103. doi: 10.1038/labinvest.3700509. [DOI] [PubMed] [Google Scholar]
- 16.Han A, Song Z, Tong C, Hu D, Bi X, Augenlicht LH, Yang W. Sulindac suppresses β-catenin expression in human cancer cells. Eur J Pharmacol. 2008;583:26–31. doi: 10.1016/j.ejphar.2007.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song Z, Tong C, Liang J, Dockendorff A, Huang C, Augenlicht LH, Yang W. JNK1 is required for sulindac-mediated inhibition of cell proliferation and induction of apoptosis in vitro and in vivo. Eur J Pharmacol. 2007;560:95–100. doi: 10.1016/j.ejphar.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tong C, Yin Z, Song Z, Dockendorff A, Huang C, Mariadason J, Flavell RA, Davis RJ, Augenlicht LH, Yang W. c-Jun NH2-terminal kinase 1 plays a critical role in intestinal homeostasis and tumor suppression. Am J Pathol. 2007;171:297–303. doi: 10.2353/ajpath.2007.061036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 20.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 21.Hu D, Fang W, Han A, Gallagher L, Davis RJ, Xiong B, Yang W. c-Jun N-terminal kinase 1 interacts with and negatively regulates Wnt/β-catenin signaling through GSK3β pathway. Carcinogenesis. 2008;29:2317–24. doi: 10.1093/carcin/bgn239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, Kucherlapati R, Lipkin M, Yang K, Augenlicht L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 2002;295:1726–9. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 23.Yang W, Velcich A, Lozonschi I, Liang J, Nicholas C, Zhuang M, Bancroft L, Augenlicht LH. Inactivation of p21WAF1/cip1 Enhances Intestinal Tumor Formation in Muc2-/- Mice. Am J Pathol. 2005;166:1239–46. doi: 10.1016/S0002-9440(10)62342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–84. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 25.Yang K, Lamprecht SA, Shinozaki H, Fan K, Yang W, Newmark HL, Kopelovich L, Edelmann W, Jin B, Gravaghi C, Augenlicht L, Kucherlapati R, et al. Dietary calcium and cholecalciferol modulate cyclin D1 expression, apoptosis, and tumorigenesis in intestine of adenomatous polyposis coli1638N/+ mice. J Nutr. 2008;138:1658–63. doi: 10.1093/jn/138.9.1658. [DOI] [PubMed] [Google Scholar]
- 26.Yang K, Yang W, Mariadason J, Velcich A, Lipkin M, Augenlicht L. Dietary components modify gene expression: implications for carcinogenesis. J Nutr. 2005;135:2710–4. doi: 10.1093/jn/135.11.2710. [DOI] [PubMed] [Google Scholar]
- 27.Jiang C, Li H, Tripp CP. Infrared method for in situ studies of polymer/surfactant adsorption on silica powders from aqueous solution. Applied spectroscopy. 2003;57:1419–24. doi: 10.1366/000370203322554590. [DOI] [PubMed] [Google Scholar]
- 28.Lu J, Pei H, Ip C, Lisk DJ, Ganther H, Thompson HJ. Effect on an aqueous extract of selenium-enriched garlic on in vitro markers and in vivo efficacy in cancer prevention. Carcinogenesis. 1996;17:1903–7. doi: 10.1093/carcin/17.9.1903. [DOI] [PubMed] [Google Scholar]
- 29.Sinha R, Said TK, Medina D. Organic and inorganic selenium compounds inhibit mouse mammary cell growth in vitro by different cellular pathways. Cancer Lett. 1996;107:277–84. doi: 10.1016/0304-3835(96)04373-x. [DOI] [PubMed] [Google Scholar]
- 30.Zeng H, Davis CD, Finley JW. Effect of selenium-enriched broccoli diet on differential gene expression in min mouse liver(1,2). The Journal of nutritional biochemistry. 2003;14:227–31. doi: 10.1016/s0955-2863(03)00005-6. [DOI] [PubMed] [Google Scholar]
- 31.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, Hess TM, Woloj GM, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–84. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 32.Flossmann E, Rothwell PM. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet. 2007;369:1603–13. doi: 10.1016/S0140-6736(07)60747-8. [DOI] [PubMed] [Google Scholar]
- 33.Meyskens FL, Jr., McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, Kelloff G, Lawson MJ, Kidao J, McCracken J, Albers CG, Ahnen DJ, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila Pa) 2008;1:32–8. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu J, Jiang C. Selenium and cancer chemoprevention: hypotheses integrating the actions of selenoproteins and selenium metabolites in epithelial and non-epithelial target cells. Antioxid Redox Signal. 2005;7:1715–27. doi: 10.1089/ars.2005.7.1715. [DOI] [PubMed] [Google Scholar]
- 35.Diwadkar-Navsariwala V, Diamond AM. The link between selenium and chemoprevention: a case for selenoproteins. J Nutr. 2004;134:2899–902. doi: 10.1093/jn/134.11.2899. [DOI] [PubMed] [Google Scholar]
- 36.Narayanan BA, Narayanan NK, Desai D, Pittman B, Reddy BS. Effects of a combination of docosahexaenoic acid and 1,4-phenylene bis(methylene) selenocyanate on cyclooxygenase 2, inducible nitric oxide synthase and β-catenin pathways in colon cancer cells. Carcinogenesis. 2004;25:2443–9. doi: 10.1093/carcin/bgh252. [DOI] [PubMed] [Google Scholar]
- 37.Rao CV, Cooma I, Rodriguez JG, Simi B, El-Bayoumy K, Reddy BS. Chemoprevention of familial adenomatous polyposis development in the APC(min) mouse model by 1,4-phenylene bis(methylene)selenocyanate. Carcinogenesis. 2000;21:617–21. doi: 10.1093/carcin/21.4.617. [DOI] [PubMed] [Google Scholar]
- 38.Hu H, Jiang C, Ip C, Rustum YM, Lu J. Methylseleninic acid potentiates apoptosis induced by chemotherapeutic drugs in androgen-independent prostate cancer cells. Clin Cancer Res. 2005;11:2379–88. doi: 10.1158/1078-0432.CCR-04-2084. [DOI] [PubMed] [Google Scholar]
- 39.Jiang C, Wang Z, Ganther H, Lu J. Distinct effects of methylseleninic acid versus selenite on apoptosis, cell cycle, and protein kinase pathways in DU145 human prostate cancer cells. Mol Cancer Ther. 2002;1:1059–66. [PubMed] [Google Scholar]
- 40.Yang K, Popova NV, Yang WC, Lozonschi I, Tadesse S, Kent S, Bancroft L, Matise I, Cormier RT, Scherer SJ, Edelmann W, Lipkin M, et al. Interaction of Muc2 and Apc on Wnt signaling and in intestinal tumorigenesis: potential role of chronic inflammation. Cancer Res. 2008;68:7313–22. doi: 10.1158/0008-5472.CAN-08-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang D, Li J, Song L, Ouyang W, Gao J, Huang C. A JNK1/AP-1-dependent, COX-2 induction is implicated in 12-O-tetradecanoylphorbol-13-acetate-induced cell transformation through regulating cell cycle progression. Mol Cancer Res. 2008;6:165–74. doi: 10.1158/1541-7786.MCR-07-0181. [DOI] [PubMed] [Google Scholar]
- 42.Zhang D, Li J, Wu K, Ouyang W, Ding J, Liu ZG, Costa M, Huang C. JNK1, but not JNK2, is required for COX-2 induction by nickel compounds. Carcinogenesis. 2007;28:883–91. doi: 10.1093/carcin/bgl186. [DOI] [PubMed] [Google Scholar]
- 43.Buchanan FG, DuBois RN. Connecting COX-2 and Wnt in cancer. Cancer Cell. 2006;9:6–8. doi: 10.1016/j.ccr.2005.12.029. [DOI] [PubMed] [Google Scholar]
- 44.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-β-catenin signaling axis. Science. 2005;310:1504–10. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]