

Abstract
The versatility of polymers for tissue regeneration lies in the feasibility to modulate the physical and biological properties by varying the side groups grafted to the polymers. Biodegradable polyphosphazenes are high molecular weight polymers with alternating nitrogen and phosphorus atoms in the backbone. This study is the first of its kind to systematically investigate the effect of side group structure on the compressive strength of novel biodegradable polyphosphazene based polymers as potential materials for tissue regeneration. The alanine polyphosphazene based polymers, poly[bis(ethyl alanato) phosphazene] (PNEA), poly[(50% ethyl alanato) (50% methyl phenoxy) phosphazene] (PNEA50mPh50), poly[(50% ethyl alanato) (50% phenyl phenoxy) phosphazene] (PNEA50PhPh50) where investigated to demonstrate their mechanical properties and osteocompatibility.
Results of mechanical testing studies demonstrated the nature and the ratio of the pendent groups attached to the polymer backbone play a significant role in determining the mechanical properties of the resulting polymer. The compressive strength of PNEA50PhPh50 was significantly higher than poly(lactide-co-glycolide) (85:15 PLAGA) (p<0.05). Additional studies evaluated the cellular response and gene expression of primary rat osteoblast cells on PNEA, PNEA50mPh50 and PNEA50PhPh50 films as candidates for bone tissue engineering applications. Results of the in vitro osteocompatibility evaluation demonstrated that cells adhere, proliferate, and maintain their phenotype when seeded directly on the surface of PNEA, PNEA50mPh50, and PNEA50PhPh50 Moreover cells on the surface of the polymers expressed type I collagen, alkaline phosphatase, osteocalcin, osteopontin, and bone sialoprotein which are characteristic genes for osteoblast maturation, differentiation, and mineralization.
Keywords: Biodegradable polyphosphazenes, mechanical properties, osteocompatibility, gene expression
INTRODUCTION
Biodegradable polymers form an integral part of scaffold – based tissue engineering, and have been used as replacements for connective tissue in the past [1]. They are now currently used to assist or guide the regeneration of tissues or organs [2]. Biodegradable polymers used for tissue regeneration should be biocompatible and not elicit immunological or foreign body response, should be degradable with a controlled rate of degradation that matches the rate of new tissue formation, the degradation products should be non toxic and metabolized by the body, they should allow cellular attachment, proliferation, and differentiation and finally have suitable mechanical properties [2–4].
Natural polymers (such as collagen, fibrin, chitosan) and synthetic polymers (such as polyesters, polyanhydrides, polyphosphazenes) have been developed for various biomedical applications. Biodegradable polyphosphazenes are a novel class of polymers that have received a great deal of attention recently as potential biomaterials due to their synthetic flexibility, high biocompatibility, and predictable degradation rate [5, 6]. Polyphosphazenes are high molecular weight polymers with an alternating nitrogen and phosphorus backbone and with each phosphorus atom attached to two organic groups. The general structure of polyphosphazenes is shown in Figure 1 where R can be an alkoxy, aryloxy, or amino group.
Figure 1.

General structure of polyphosphazenes, where R can be either an alkoxy, or arloxy, or amino group.
The synthetic flexibility of polyphosphazenes allows a wide range of polymers to be synthesized using a highly reactive macromolecular intermediate, poly(dichlorophosphazene). Polyphosphazenes are synthesized via a two step reaction process (Figure 2). The first step involves the thermal ring opening polymerization of hexachlorocyclotriphosphazene (1) to form poly(dichlorophosphazene) (2) [7]. In the second step the reactive chlorine atoms in the macromolecular intermediate (2) are replaced by any of a broad range of organic side groups (3, 4).
Figure 2.

Macromolecular substitution reaction to form amino acid ester based polyphosphazenes.
The polyphosphazene platform of macromolecules constitutes more than a 700-member group of different polymers with a broad diversity of properties. The large number of polymers accessible in this system is a consequence of the wide variety of alcohols, phenols, and amines available to obtain polymers with different pendant groups and the fact that two or more different side groups can be incorporated into the same polymer molecule by simultaneous or sequential chlorine replacement reactions [7]. Due to the synthetic versatility of the polyphosphazene, the properties of the polymer can be tuned by an appropriate choice of side groups to suite a specific application. The chemical properties such as hydrophilic or hydrophobic character, solubility, biocompatibility, basicity or acidity, and susceptibility to hydrolysis [7] depend on the nature of the side group. Because the phosphorus – nitrogen backbone in these polymers is highly flexible, material properties such as glass – or melting temperatures and the mechanical properties of the polymer depend on the side groups attached to the phosphorus atom. Thus, these unique properties of polyphosphazenes have sparked great interest in their biomedical use [5, 8–11].
Amino acid substituted polyphosphazenes with various functional groups have been successfully synthesized [8–15]. The rate of hydrolysis for polyphosphazenes depends on the nature of the group attached to the α-carbon of the amino acid ester side group; bulkier side groups result in less hydrolytically sensitive polymers [16–20]. In the case of co-substituted polyphosphazenes, the nature of the co-substituent determines the rate of degradation.
Our recent studies have been focused on modulating the properties of biodegradable polyphosphazenes by varying the side group chemistry as well as by blending polyphosphazenes with other degradable polymers such as poly(lactic acid-co-glycolic acid) (PLAGA) [21, 22]. By blending biodegradable polyphosphazenes with PLAGA, novel polymeric mixtures with varying glass transition temperatures and degradation profiles can be developed [21]. Another versatile process to modulate the properties of biodegradable polyphosphazenes is by varying the side groups. The effects of side groups on various properties of homo and co-polymers of ethyl alaninato substituted polyphosphazenes have been evaluated. The properties investigated include glass transition temperature, hydrolytic degradation, surface wettability, tensile strength and modulus. The glass transition temperature of the polymers was found to increase with increase in bulkiness of the side groups. The study demonstrated the feasibility of varying the properties of biodegradable polyphosphazenes by varying the bulkiness and hydrophobicity of the side groups [22].
Ethyl alanato derivates of polyphosphazenes have been used as membranes in the treatment of periodontal disease [23], as nerve guide conduits [24] and as scaffolds for orthopaedic applications [19, 25]. The objective of this study was to evaluate systematically the effect of side group chemistry on the compressive strength and osteocompatibility of biodegradable polyphosphazenes containing alanine. The mechanical properties and osteocompatibility of these alanine substituted polyphosphazenes, poly[bis(ethyl alanato) phosphazene] (PNEA), poly[(50% ethyl alanato) (50% methyl phenoxy) phosphazene] (PNEA50mPh50), poly[(50% ethyl alanato) (50% phenyl phenoxy) phosphazene] (PNEA50PhPh50) were evaluated. These are the first studies of their kind to fully examine these novel alanine based polyphosphazene polymers.
MATERIALS AND METHODS
The biodegradable polymers used for mechanical property evaluations in the present study were poly[bis(ethyl alanato) phosphazene] (PNEA), poly[(50% ethyl alanato) (50% methyl phenoxy) phosphazene] (PNEA50mPh50), poly[(50% ethyl alanato) (50% phenyl phenoxy) phosphazene] (PNEA50PhPh50). These polymers were synthesized by a two step process which involved the thermal ring opening polymerization of hexachlorocyclotriphosphazene (Espirix Technologies, USA) to form poly(dichlorophosphazene), followed by the sequential replacement of the chlorine atoms in the poly(dichlorophosphazene) by organic groups. All reactions were carried out in an argon atmosphere.
Synthesis of PNEA
Poly(dichlorophosphazene) (8.0 g; 0.069 mol) was dissolved in tetrahydrofuran (THF, Sigma, USA) and treated with L-alanine ethyl ester (Chem Impex, USA, 42.4 g; 0.276 mol) in the presence of triethylamine (Sigma, USA, 86.6 mL; 0.622 mol). The polymer (PNEA) was isolated and purified by successive precipitations from THF into hexanes (3X) followed by dialysis against a THF/methanol (Sigma, USA) mixture for 3 days [19, 25].
Synthesis of PNEA50 mPh50
Poly(dicholorophosphazene) (20.0 g; 0.173 mol) was treated with the sodium salt of p-methyl phenol (Sigma, USA, 20.53 g; 0.190 mol) in THF to obtain a partially substituted polymer. Substitution was then completed by replacing the remaining chlorine groups with excess of L-alanine ethyl ester (Chem Impex, USA, 79.54 g; 0.518 mol) in the presence of triethylamine (Sigma, USA, 144 mL; 1.04 mol). The reaction mixture was refluxed for two days to bring about complete substitution. The polymer was isolated by precipitation from THF into hexanes and was purified by repeated precipitations from THF into hexanes (2X) and ethanol (3X).
Synthesis of PNEA50PhPh50
Poly(dicholorophosphazene) (20.0 g; 0.173 mol) was treated with the sodium salt of p-phenylphenol (Sigma, USA, 32.31 g; 0.1898mol) in THF, to obtain a partially substituted polymer. Substitution was then completed by replacing the remaining chlorine groups with excess of L-alanine ethyl ester (Chem Impex, USA, 116.64 g, 0.759 mol) in the presence of triethylamine (Sigma, USA, 221 mL, 1.58mol). The reaction mixture was refluxed for two days to bring about complete substitution. The polymer was isolated by precipitation from THF into hexanes and purified by repeated precipitations from THF into hexanes and ethanol [19, 26, 27].
Physico-Chemical Analysis
The structures of the synthesized polymers were confirmed using multinuclear magnetic resonance spectroscopy (Bruker 360MHz Spectrometer, Solvent CDCl3). The glass transition temperatures of the polymers were measured using a differential scanning calorimeter (TA Instruments Q10 DSC, USA) at a heating rate of 10°C per minute under nitrogen atmosphere. Gel permeation chromatography (GPC, HP1090, Agilent Technologies, USA) was used to determine the molecular weights of the polymers.
Mechanical Property Evaluation
Cylindrical matrices with a diameter of 5mm and a height of 10mm were prepared as follows. Two hundred and fifty milligrams of the polymer were placed in a split dye and compressed at 1500 psi for 15 minutes. After applying the compressive force the mechanical properties of the polymers were measured using a uniaxial compressive testing machine (Instron, Canton MA) with a load cell of 500N at a rate of 10 mm/min until failure of the material. Cylindrical samples of 85:15 PLAGA were prepared as explained above and the mechanical properties were evaluated and compared to the values obtained for biodegradable polyphosphazenes.
Surface Analysis
The surfaces of PNEA, PNEA50mPh50, and PNEA50PhPh50 films were characterized using X-ray photoelectron spectroscopy [XPS, Physical Electronics 560, USA]. The XPS analysis was performed using magnesium X-rays at a pressure of 1.0 X 10−7 torr. The pass energy was 200 eV, and the aperture resolution was 1.6% which resulted in the energy resolution of 3.2 eV.
Cell Isolation
Primary rat osteoblast (PRO) cells were isolated by the procedure explained by Schwartz [28]. Briefly, the calvaria of the rat pups was dissected, minced and rinsed in phosphate buffer saline solution. Sequential digestion of the calvarial explants was performed using collagenase and trypsin to produce separate lines of osteoblasts. The cells were cultured in Ham’s F-12 medium (Gibco, USA) supplemented with 12% fetal bovine serum (FBS, Gibco, USA) and 1% antibiotics (Penicillin–Streptomycin, Gibco, USA).
Polymer Film Preparation
The polymers, PNEA, PNEA50mPh50, and PNEA50PhPh50 were dissolved in dichloromethane to form a 7% (w/v) solution. The polymer solution was then poured into a petri dish coated with Bytac paper (Fisher Scientific, USA). The solvent was allowed to evaporate overnight and the polymer film was placed in a lyophilizer for 48 hours to completely remove any trace of organic solvents. Circular matrices, 10mm in diameter, were cut using a cork borer.
Cell Seeding
Cell proliferation, differentiation, and mineralization on the surface of poly[bis(ethyl alanato)phosphazene] [PNEA], poly[(50%ethyl alanato)(50%methyl phenoxy)phosphazene] [PNEA50mPh50] and poly[(50%ethyl alanato)(50%phenyl phenoxy)phosphazene] [PNEA50PhPh50] were evaluated to assess the osteocompatibility for bone regeneration. Each side of the polymer disk was exposed to ultraviolet light for 10 minutes to minimize bacterial contamination. The polymer disks were placed in a 48-well plate and washed with Ham’s F-12 media and 50,000 PRO cells were seeded on each scaffold to study the cell proliferation and alkaline phosphatase activity. To determine the gene expression of cells on the polymer surface 100,000 cells were seeded on each polymer matrix. The cells were cultured with Ham’s F-12 medium [Gibco, USA], 12% fetal bovine serum, and 1% antibiotics supplemented by 3mM of β-glycerolphosphate [Sigma, USA] and 10μg/ml of ascorbic acid [Fisher, USA]. The media was changed every other day.
Cell Adhesion and Proliferation
Cell adhesion and proliferation on the polymer scaffolds were evaluated quantitatively after 1, 3, and 7 days post seeding. The cells were washed with phosphate buffer saline solution and lysed with 1% triton X-100 [Biorad, USA]. The DNA concentration in cell lysate at each time point was determined using a Picogreen ds-DNA assay (Molecular Probes, OR, USA). The DNA concentration was measured as fluorescence using Tecan [Spectro Flour Plus, F129005, USA] at an emission and excitation wavelength of 485nm and 535nm respectively. The fluorescence was converted into cell number using a standard curve.
Alkaline Phosphatase Assay
The phenotypic marker of bone, alkaline phosphatase, was examined after 1, 3, and 7 days post seeding using an alkaline phosphatase substrate kit (Bio Rad, CA, USA). The cell lysate obtained from the DNA assay was used to evaluate the alkaline phosphatase activity. Briefly, 100μl of the cell lysate was added to 400μl of the substrate and solution was incubated for 30 minutes at 37°C. The reaction was stopped by the addition of 0.4M sodium hydroxide solution. The absorbance was measured using a Tecan [Spectro Flour Plus, F129005, USA] at 410nm. The absorbance was normalized based on the cell number.
Real Time Reverse Transcription PCR (RT-PCR)
The effect of the polymer surface on type I collagen (TIC), alkaline phosphatase (ALP), osteocalcin (OCN), osteopontin (OPN), and bone sialoprotein (BSP) expression were evaluated. After 7 days in culture, the polymer matrices were washed with PHOSPHATE BUFFER SALINE solution and the total RNA from the cells was isolated using Trizol (Gibco BRL, USA) following the procedure described by the manufacturer (Qiagen, 74106, USA). The RNA extracted was stabilized and centrifuged using a QIA Shredder Spin Column [Qiagen, USA] and dissolved in RNAse free water [Qiagen, USA]. The concentration of the RNA was measured using a spectrophotometer at 260nm.
The gene expression was determined using a real time RT-PCR [Applied Biosystems, ABI Prism, 7900 HT Sequence Detector System, USA]. The primers were designed on the basis of published gene sequences (NCBI and Pubmed) and shown in Table 1 [29].
Table 1.
Gene specific primers used in PCR Amplification.
| Genes | Sequences | Length | |
|---|---|---|---|
| GAPDH | Forward | 5′-TGCATCCTGCACCACCAA-3′ | 18 |
| Reverse | 5′-CCTTCCACAATGCCAAAGTTG-3′ | 21 | |
| Type I Collagen(TIC) | Forward | 5′-ATGGTGCTCCTGGTGCCA-3′ | 18 |
| Reverse | 5′-TGTATTCGATGACTGTCTTG-3′ | 20 | |
| Alkaline Phosphatase (ALP) | Forward | 5′-AACCTCGAGCAGGAACACAAG-3′ | 21 |
| Reverse | 5′-CAACGGCAGCAGGAATCC-3′ | 18 | |
| Osteocalcin (OCN) | Forward | 5′-ATGAGGACCCTCTCTCTGCTCACT-3′ | 24 |
| Reverse | 5′-CCCAGCATCATGAGGGCCTGGATCTT-3′ | 26 | |
| Osteopontin (OPN) | Forward | 5′-CAACCATGAGACTGGCAGTGGTTTGC-3′ | 26 |
| Reverse | 5′-GCCTCTTCTTTAATTGACCTCAGAAG-3′ | 26 | |
| Bone Sialoprotein (BSP) | Forward | 5′-AGTTGCCAGATTTACCAAACATGA-3′ | 24 |
| Reverse | 5′-TGTATTAAAAACCAGCCATTGTTCA-3′ | 26 | |
Statistics
Analysis of variance (one-way ANOVA) was used to determine the level of significance for the mechanical properties of the different polymers (n=6). Analysis of variance (two-way ANOVA) was used to evaluate the significance between the polymer at different time points (n=4 for cell proliferation and alkaline phosphatase). In both cases the statistical significance was evaluated at p< 0.05.
RESULTS
The structures of the polymers were confirmed using 31P and 1H NMR (Table 2). The molecular weights of the, PNEA, PNEA50mPh50 and PNEA50PhPh50 were 85,000 g/mol, 408,000 g/mol, 4,600,000 g/mol and 4,350,000 g/mol respectively (Table 2). The surfaces of the polymers were analyzed using an X-ray photoelectron spectroscopy. The results showed that there was a higher percentage of carbon on the surface of PNEA50PhPh50 when compared to PNEA50mPh50 and PNEA (Table 2). The glass transition temperatures of PNEA, PNEA50mPh50, and PNEA50PhPh50 were −3°C, −6°C, and 35.2°C respectively (Table 2).
Table 2.
Characterization, and thermal properties of, poly[bis(ethyl alanato)phosphazene] [PNEA], poly[(50%ethyl alanato) (50%methylphenoxy)phosphazene] [PNEA50mPh50], and poly[(50%ethyl alanato) (50%phenylphenoxy)phosphazene] [PNEA50PhPh50] and poly[(85%lactic acid)(15%glycolic acid)] [85:15 PLAGA].
| Polymer | Structure | NMR (ppm) | Mw (g/mol) | Tg (°C) | XPS (%) | ||
|---|---|---|---|---|---|---|---|
| 31P | 1H | ||||||
| PNEA | 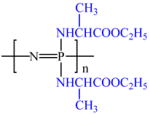 |
−1.10 | 4.4 (1H) | 408,000 | −3.0 | C | 56.1 |
| 4.1 (2H) | O | 26.2 | |||||
| 1.6 (3H) | N | 12.4 | |||||
| 1.3 (3H) | P | 5.3 | |||||
| PNEA50mPh50 | 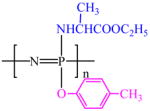 |
−5.07 | 6.8 (4H ) | 4,608,000 | −6.0 | C | 63.2 |
| 3.9 (3H) | O | 22.3 | |||||
| 2.2 (3H) | N | 9.2 | |||||
| 1.8 (3H) | P | 5.2 | |||||
| PNEA50PhPh50 | 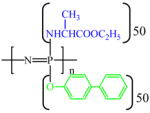 |
1.78 | 7.4 (9H) | 4,350,000 | 35.2 | C | 67.0 |
| −5.12 | 4.4 (1H) | O | 26.8 | ||||
| −14.58 | 4.1 (2H) | N | 4.1 | ||||
| 1.4 (3H) 1.2 (3H) |
P | 2.0 | |||||
| PLAGA | 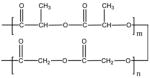 |
– | – | 1,35,000 | 65.0 | – | |
The mechanical properties of the PNEA, PNEA50mPh50 and PNEA50PhPh50 were evaluated and compared to 85:15 PLAGA (Figure 3). The presence of alanine side groups in the polymer was found to significantly increase the compressive strength of PNEA. With the linkage of bulkier side groups to the polymer chain as in the case of PNEA50mPh50 and PNEA50PhPh50 the compressive strength was 25.0±11.3MPa and 59.24±25.59MPa respectively (Figure 3). The compressive strength of 85:15 PLAGA under the current fabrication technique was 34.9±5.7MPa.
Figure 3.

Compressive strength of amino acid ester based polyphosphazenes compared with 85:15 PLAGA. Statistical significance at p<0.05, n=6.
Cell adhesion and proliferation of primary rat osteoblast cells on the surface of PNEA, PNEA50mPh50, and PNEA50PhPh50 was evaluated using a ds-DNA quantification kit and the cell number on the surface of the polymers was calculated using a standard curve. The cell number on the polymer films were comparable after 3 days in culture (p<0.05) (Figure 4). At day 7, the cell number on the surface of PNEA50PhPh50 was significantly higher than PNEA (p<0.05) (Figure 4).
Figure 4.

Number of primary rat osteoblast cells on poly[bis(ethyl alanato) phosphazene] [PNEA], poly[(50% ethyl alanato) (50% methyl phenoxy) phosphazene] [PNEA50mPh50], poly[(50% ethyl alanato) (50% phenyl phenoxy) phosphazene] [PNEA50PhPh50], and tissue culture polystyrene [TCPS] over 7 days. Statistical significance at p<0.05, n=4.
The phenotypic marker of bone, alkaline phosphatase (ALP) activity of the cells on the surface of the polymers was evaluated. Figure 5 shows the ALP activity per cell on the surfaces of the matrices. Cells on the polymer surfaces expressed early ALP activity (at day 1) and continued to express ALP activity up to day 7.
Figure 5.

Alkaline phosphatase activity expressed by the cells on poly[bis(ethyl alanato) phosphazene] [PNEA], poly[(50% ethyl alanato) (50% methyl phenoxy) phosphazene] [PNEA50mPh50], poly[(50% ethyl alanato) (50% phenyl phenoxy) phosphazene] [PNEA50PhPh50], and tissue culture polystyrene [TCPS] over 7 days. Statistical significance at p<0.05, n=4.
Figure 6 shows the real time RT-PCR of the genes, type I collagen (TIC), alkaline phosphatase (ALP), osteocalcin (OCN), osteopontin (OPN), and bone sialoprotein (BSP) on the surface of the polymers after 7 days in culture. TIC gene expression of the cells on the polymer surfaces of culture was comparable to each other (p<0.05) (Figure 6). The cells on the surface of the polymers expressed ALP (Figure 6). The OCN expression on the surface of PNEA50mPh50, and PNEA50PhPh50 was significantly higher than PNEA (p<0.05) (Figure 6). OPN expression on PNEA was significantly higher than PNEA50mPh50 and, PNEA50PhPh50 (p<0.05) (Figure 6). The BSP expression of cells on PNEA, PNEA50mPh50, and PNEA50PhPh50 was comparable to each other (p<0.05) (Figure 6).
Figure 6.

Gene expression of type I collagen (TIC), alkaline phosphatase (ALP), osteocalcin (OCN), osteopontin (OPN), and bone sialoprotein (BSP) by the cells on the surface of poly[bis(ethyl alanato) phosphazene] [PNEA], poly[(50% ethyl alanato) (50% methyl phenoxy) phosphazene] [PNEA50mPh50], poly[(50% ethyl alanato) (50% phenyl phenoxy) phosphazene] [PNEA50PhPh50], and tissue culture polystyrene [TCPS] after 7 days using a real time RT-PCR. Quantitative values were determined by the Delta-Delta method and normalized with the house keeping gene, GAPDH.
DISCUSSION
Biodegradable polymers are currently used for various medical applications [1, 30]. The polymers used for these applications should be biocompatible, biodegradable, the degradation products should be non–toxic and they should be easily removed from the body [2–4]. In our earlier studies we have established the high osteocompatibility of glycine substituted polyphosphazenes [8, 31–34]. We have synthesized and evaluated in vitro and in vivo alanine substituted polyphosphazenes that could potentially be used for various biomedical applications [19, 22, 25–27].
The elemental analysis of the surfaces of the polymer films were measured using an X-ray photoelectron spectroscopy (XPS) (Table 2). The percentage of the carbon atoms on the surface of PNEA50PhPh50 was higher than PNEA suggesting that the bulky aromatic sides groups in PNEA50PhPh50 are oriented towards the surface. This confirms the hydrophobic nature of phenyl phenoxy substituted polyphosphazene when compared to the PNEA and PNEA50mPh50. The hydrophobic or hydrophilic nature of the polymer also affects cellular adhesion and proliferation.
The compressive strength of PNEA50PhPh50 was found to be 59.24±25.59 MPa and the compressive strength of PNEA50mPh50 24.98±11.26 MPa was found to be comparable to PNEA (46.61±17.56 MPa). The compressive strength of high strength polyphosphazene, PNEA50PhPh50 was significantly higher than 85:15 PLAGA (34.9 ± 5.7MPa) (Figure 3). Also the compressive strength of PNEA, and PNEA50mPh50 was comparable to 85:15 PLAGA (Figure 3). This suggests that the presence of aromatic groups in the side chain increases the steric hinderance and reduces the torsion of the polymer backbone resulting in increased stiffness and polymer rigidity [22]. These results demonstrate that the mechincal property of polyphosphazenes can be modulated via side chain chemistry.
The proliferation, differentiation, and mineralization of primary rat osteoblast (PRO) cells were examined on PNEA, PNEA50mPh50, and PNEA50PhPh50. The proliferation of cells on biomaterials can be divided into three stages – an initial lag phase during which the cells adapt to the new environment and replace the extra cellular matrix (ECM) that has been degraded during trypsination since the polymer surface does not contain ECM; a log phase, where the cell number increases exponentially where the cells are still in an undifferentiated state; and finally a plateau phase during which the cells express phenotypic markers [35, 36]. In this study PRO cells were able to adhere, grow, and express osteoblastic genes when cultured on these surfaces demonstrating the alanine based polymers (PNEA, PNEA50mPh50, and PNEA50PhPh50) osteocompatibility.
Alkaline phosphatase (ALP) is one of the earliest phenotypic markers expressed by osteoblasts cells [37]. The cells on the surface of PNEA, PNEA50mPh50, and PNEA50PhPh50 expressed ALP throughout the study (Figure 5). The increased expression of ALP suggests that the PRO cells have shifted to a more differentiated stage.
The gene expression of type I collagen (TIC), alkaline phosphatase (ALP), osteocalcin (OCN), osteopontin (OPN), and bone sialoprotein (BSP) was evaluated using a real time RT-PCR after 7 days. During the differentiation of osteoblast cells, the extra cellular matrix which contains TIC is up regulated [38]. Type I collagen is expressed by osteoblastic tissues and binds to the collagenous matrix protein, which in turn initiates and controls mineralization [39–43]. The presence of TIC indicates the deposition of extra cellular matrix of the protein at the gene level. The expression of ALP gene marks the beginning of osteoblast differentiation [37]. OCN is produced during the late phases of differentiation by mature osteoblast cells and is found in mineralized matrix [38, 43, 44]. The presence of ALP, and OCN demonstrates the presence of mature osteoblast cells on the surface of the polymer. The expression of OPN is usually higher during proliferation and then decreases. It later peaks during the mineralization front and may control mineralization [44–46]. BSP plays a crucial role in matrix mineralization, hydroxyapatite nucleation, cell attachment, calcium binding, cell signaling, and collagen binding [47, 48]. In this study we observed that the PRO cells on the polymer surfaces expressed all the genes that are characteristic of osteoblast phenotype, differentiation, maturation, and mineralization, indicating their osteocompatibility. Our results correspond to the trend reported for cellular differences dependent on side chain and composition of the polymer. For example, previous studies in our laboratory have demonstrated by adding an ethyline glycinato group to polyphosphazene resulted in increased cellular growth and phenotype expression compared to other subgroups added [32]. We feel that this is a similar trend when looking at the level of PNEA in comparison to PNEA50mPh50, and PNEA50PhPh50. This may help substantiate why we have observed a difference in protein and gene expression between the various groups. Future studies will help determine this theory and will be investigated thoroughly.
CONCLUSION
The present study evaluated the effect of side group chemistry on the compressive strength of biodegradable polyphosphazenes. The incorporation of aromatic side groups significantly affected the mechanical properties of the polymers depending on the nature of the side groups present. This is highly advantageous as it allows different polymers to be synthesized with varying properties depending upon the nature of application. The compressive strength of PNEA50PhPh50 was significantly higher when compared to 85:15 PLAGA, which is a FDA approved polymer for certain biomedical applications. Osteocompatability of PRO cells on PNEA, PNEA50mPh50, and PNEA50PhPh50 films were evaluated. PRO cells adhered, and proliferated on the polymer surface and the cells expressed alkaline phosphatase activity. Cells on the surface of the polymers expressed genes such as type I collagen, alkaline phosphatase, osteocalcin, osteopontin, and bone sialoprotein which are characteristic genes expressed by osteoblast cells during differentiation, and mineralization and maybe dependent on side group substitution and polymer composition. We feel these novel based alanine polyphosphazene materials are ideal candidates for tissue and organ regeneration and very promising. Future studies will further evaluate their osteocompatibility at the gene level, expand their in vivo applications, and enhance their mechanical properties to be suitable for weight bearing applications.
Acknowledgments
The authors acknowledge the financial support from NIH grant #AR 46560.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nair LS, Laurencin CT. Biodegradable polymers as biomaterials. Prog Polym Sci. 2007;32:762–798. [Google Scholar]
- 2.Hutmacher D, Kirsch A, Ackermann KL, Huerzeler MB. Matrix and carrier materials for bone growth factors: state of the art and future perspectives. In: Stark GB, Horch R, Tancos E, editors. Biological matrices and tissue reconstruction. Berlin: Springer; 1998. pp. 197–206. [Google Scholar]
- 3.Hutmacher DW. Scaffolds in tissue engineering of bone and cartilage. Biomaterials. 2000;21:2529–2543. doi: 10.1016/s0142-9612(00)00121-6. [DOI] [PubMed] [Google Scholar]
- 4.Seal BL, Otero TC, Panitch A. Polymeric biomaterials for tissue engineering and organ regeneration. Mat Sci Engg R. 2001;R34:147–230. [Google Scholar]
- 5.Ibim SM, Ambrosio A, Larrier D, Allcock HR, Laurencin CT. Controlled macromolecule release from poly(phosphazene) matrices. J Control Release. 1996;40:31–39. [Google Scholar]
- 6.Allcock HR, Pucher SR, Scopelianos AG. Poly[(amino acid ester)phosphazene] as substrates for the controlled release of small molecules. Biomaterials. 1994;15:563–569. doi: 10.1016/0142-9612(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 7.Allcock HR. Chemistry and applications of polyphosphazenes. New Jersey: Wiley Interscience; 2002. [Google Scholar]
- 8.Laurencin CT, Koh HJ, Neenan TX, Allcock HR, Langer RS. Controlled release using a new biodegradable polyphosphazene matrix system. J Biomed Mater Res. 1987;21:1231–1246. doi: 10.1002/jbm.820211006. [DOI] [PubMed] [Google Scholar]
- 9.Allcock HR, Kwon S. Glyceryl polyphosphazenes: synthesis, properties, and hydrolysis. Macromolecules. 1988;21:1980–1985. [Google Scholar]
- 10.Allcock HR. Polyphosphazenes as new biomedical and bioactive materials. In: Langer RS, Chasen M, editors. Biodegradable polymers as drug delivery systems. New York: Marcel Dekker; 1990. pp. 163–193. [Google Scholar]
- 11.Andrianov AK, Payne LG. Protein release from polyphosphazene matrices. Adv Drug Del Rev. 1998;31:185–196. doi: 10.1016/s0169-409x(97)00122-1. [DOI] [PubMed] [Google Scholar]
- 12.Allcock HR, Fuller TJ, Matsumura K. Hydrolysis pathways for aminophosphazenes. Inorg Chem. 1982;21:515–521. [Google Scholar]
- 13.Allcock HR, Fuller TJ, Mack DP, Matsumura K, Smeltz KM. Synthesis of poly[(amino acid alkyl ester)phosphazenes] Macromolecules. 1977;10:824–830. [Google Scholar]
- 14.Tanigami T, Ohta H, Orii R, Yamaura K, Matsuzawa S. Degradation of poly[bis(ethylamino)phosphazene] in aqueous solution. J Inorg Org Polymers. 1995;5:135–153. [Google Scholar]
- 15.Allcock HR, Cheng JY. Poly(organophosphazenes) with oligopeptides as side groups: prospective biomaterials. Macromolecules. 1991;24:993–999. [Google Scholar]
- 16.Ruiz EM, Ramirez CA, Aponte MA, Barbosa-Canovas GV. Degradation of poly[(bis glycine ethyl ester)phosphazene] in aqueous media. Biomaterials. 1993;14:491–496. doi: 10.1016/0142-9612(93)90235-t. [DOI] [PubMed] [Google Scholar]
- 17.Crommen J, Vandorpe J, Schacht E. Degradable polyphosphazenes for biomedical applications. J Control Release. 1993;24:167–180. [Google Scholar]
- 18.Allcock HR, Pucher SR, Scopelianos AG. Poly[(amino acid ester) phosphazene]: synthesis, crystallinity, and hydrolytic sensitivity in solution and the solid state. Macromolecules. 1994;27:1071–1075. [Google Scholar]
- 19.Sethuraman S, Nair LS, El-Amin S, Farrar R, Nguyen MT, Singh A, Allcock HR, et al. In vivo biodegradability and biocompatibility evaluation of novel alanine ester based polyphosphazenes in a rat model. J Biomed Mater Res A. 2006;77:679–687. doi: 10.1002/jbm.a.30620. [DOI] [PubMed] [Google Scholar]
- 20.Nair LS, Lee DA, Bender JD, Barrett EW, Greish YE, Brown PW, Allcock HR, et al. Synthesis, characterization and osteocompatibility evaluation of novel alanine-based polyphosphazenes. J Biomed Mater Res A. 2006;76:206–213. doi: 10.1002/jbm.a.30532. [DOI] [PubMed] [Google Scholar]
- 21.Krogman NR, Singh A, Nair LS, Laurencin CT, Allcock HR. The miscibility of hydrolytically unstable polyphosphazene/poly(lactide-co-glycolide) blends. Biomacromolecules. 2007;8:1306–1312. doi: 10.1021/bm061064q. [DOI] [PubMed] [Google Scholar]
- 22.Singh A, Krogman NR, Sethuraman S, Nair LS, Sturgeon JL, Brown PW, Laurencin CT, et al. Effect of side group chemistry on the properties of biodegradable L-Alanine co-substituted polyphosphazenes. Biomacromolecules. 2006;7:914–918. doi: 10.1021/bm050752r. [DOI] [PubMed] [Google Scholar]
- 23.Veronese FM, Marsilio F, Lora S, Caliceti P, Passi P, Orsolini P. Polyphosphazene membranes and microspheres in periodontal diseases and implant surgery. Biomaterials. 1999;20:91–8. doi: 10.1016/s0142-9612(97)00104-x. [DOI] [PubMed] [Google Scholar]
- 24.Langone F, Lora S, Veronese FM, Caliceti P, Parnigotto PP, Valenti F, Palma G. Peripheral nerve repair using a poly(organo)phosphazene tubular prosthesis. Biomaterials. 1995;16:347–53. doi: 10.1016/0142-9612(95)93851-4. [DOI] [PubMed] [Google Scholar]
- 25.Nair LS, Bender JD, Singh A, Sethuraman S, Greish YE, Brown PW, Allcock HR, et al. Biodegradable poly[bis(ethyl alanato)phosphazene]-poly(lactide-co-glycolide) blends: miscibility and osteocompatibility evaluations. Mater Res Soc Symp Proc. 2005;844:Y9.7.1–Y9.7.7. [Google Scholar]
- 26.Sethuraman S, Lakshmi S, Singh A, Bender JD, Greish YE, Brown PW, Allcock HR, et al. Synthesis & Evaluation of Novel Amino Acid Eater Phenyl Phenoxy Phosphazene for Bone Tissue Engineering. Combined Orthopaedic Research Society Meeting; Banff, Canada. 2004. [Google Scholar]
- 27.Sethuraman S, Lakshmi S, Singh A, Greish YE, Brown PW, Allcock HR, Laurencin CT. Novel biomaterials based on phenyl phenoxy substituted biodegradable polyphosphazenes. (Patent Pending) [Google Scholar]
- 28.Schwartz E. Bone. In: Freshney I, editor. Culture of animal cells. New York: Wiley-Liss; 1994. pp. 332–333. [Google Scholar]
- 29.Sun JS, Chang WHS, Chen LT, Huang YC, Juang LW, Lin FH. The influence on gene-expression profiling of osteoblasts behavior following treatment with the ionic products of sintered β-dicalcium pyrophosphate dissolution. Biomaterials. 2004;25:607–616. doi: 10.1016/s0142-9612(03)00567-2. [DOI] [PubMed] [Google Scholar]
- 30.Chandra R, Rustgi R. Biodegradable polymers. Prog Polym Sci. 1998;23:1273–1335. [Google Scholar]
- 31.Laurencin CT, Morris CD, Jacques HP, Schwartz ER, Keaton AR, Zou L. Osteoblast culture on bioerodible polymers: studies of initial cell adhesion and spread. Polym Adv Tech. 1992;3:359–364. [Google Scholar]
- 32.Laurencin CT, Norman ME, Elgendy HM, El-Amin SF, Allcock HR, Pucher SR, Ambrosio AA. Use of polyphosphazenes for skeletal tissue regeneration. J Biomed Mater Res. 1993;27:963–973. doi: 10.1002/jbm.820270716. [DOI] [PubMed] [Google Scholar]
- 33.Ambrosio AA, Allcock HR, Katti DS, Laurencin CT. Degradable polyphosphazene/poly(α-hroxyester) blends: degradation studies. Biomaterials. 2002;23:1667–1672. doi: 10.1016/s0142-9612(01)00293-9. [DOI] [PubMed] [Google Scholar]
- 34.Lakshmi S, Katti DS, Laurencin CT. Biodegradable polyphosphazenes for drug delivery applications. Adv Drug Del Rev. 2003;55:467–482. doi: 10.1016/s0169-409x(03)00039-5. [DOI] [PubMed] [Google Scholar]
- 35.Termine JD, Kleinman HK, Whitson SW, Conn KM, McGarvey ML, Martin GR. Osteonectin, a bone-specific protein linking mineral to collagen. Cell. 1981;26:99–105. doi: 10.1016/0092-8674(81)90037-4. [DOI] [PubMed] [Google Scholar]
- 36.Stein GS, Lian JB. Molecular mechanisms mediating proliferation/differentiation interrelationships during progressive development of the osteoblast phenotype. Endocr Rev. 1993;1:424–442. doi: 10.1210/edrv-14-4-424. [DOI] [PubMed] [Google Scholar]
- 37.Lian JB, Stein GS. Concepts of osteoblast growth and differentiation: basis for modulation of bone cell development and tissue formation. Crit Rev Oral Biol Med. 1992;3:269–305. doi: 10.1177/10454411920030030501. [DOI] [PubMed] [Google Scholar]
- 38.Xie J, Baumann MJ, McCabe LR. Osteoblasts respond to hydroxyapatite surfaces with immediate changes in gene expression. J Biomed Mater Res. 2004;71A:108–117. doi: 10.1002/jbm.a.30140. [DOI] [PubMed] [Google Scholar]
- 39.Bronzert DA, Bates SE, Sheridan JP, Lindsey R, Valverius EM, Stampfer MR, et al. Transforming growth factor-beta induces platelet-derived growth factor (PDGF) messenger RNA and PDGF secretion while inhibiting growth in normal human mammary epithelial cells. Mol Endocrinol. 1990;4:981–999. doi: 10.1210/mend-4-7-981. [DOI] [PubMed] [Google Scholar]
- 40.Ohsawa K, Neo M, Matsuoka H, Akiyama H, Ito H, Kohna H, et al. The expression of bone matrix protein mRNAs around ®-TCP particles implanted into bone. J Biomed Mater Res. 2000;52:460–466. doi: 10.1002/1097-4636(20001205)52:3<460::aid-jbm3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 41.Franceschi RT, Iyer BS. Relationship between collagen synthesis and expression of the osteoblast phenotype in MC3T3-E1 cells. J Bone Miner Res. 1992;7:235–246. doi: 10.1002/jbmr.5650070216. [DOI] [PubMed] [Google Scholar]
- 42.Genovese C, Rowe D, Kream B. Construction of DNA sequences complementary to rat alpha 1 and alpha 2 collagen mRNA and their use in studying the regulation of type I collagen synthesis by 1, 25- dihydroxy vitamin D. Biochemistry. 1984;23:6210–6216. doi: 10.1021/bi00320a049. [DOI] [PubMed] [Google Scholar]
- 43.Cowles EA, DeRome ME, Pastizzo G, Brailey LL, Gronowicz GA. Mineralization and the expression of matrix proteins during in vivo bone development. Calcif Tissue Int. 1998;62:74–82. doi: 10.1007/s002239900397. [DOI] [PubMed] [Google Scholar]
- 44.Sato M, Yasui N, Nakase T, Kawahata H, Sugimoto M, Hirota S, Kitamura Y, et al. Expression of bone matrix proteins mRNA during distraction osteogenesis. J Bone Miner Res. 1998;13:1221–1231. doi: 10.1359/jbmr.1998.13.8.1221. [DOI] [PubMed] [Google Scholar]
- 45.Dodds RA, Connor JR, James IE, Rykaczewski EL, Appelbaum E, Dul E, Gowen M. Human osteoclasts, not osteoblasts, deposit osteopontin onto resorption surface: an in vitro and ex vivo study of remodeling bone. J Bone Miner Res. 1995;10:1666–1680. doi: 10.1002/jbmr.5650101109. [DOI] [PubMed] [Google Scholar]
- 46.Dehardt DT, Noda M. Osteopontin expression and function: role in bone remodeling. J Cell Biochem Suppl. 1998;31:92–102. [PubMed] [Google Scholar]
- 47.Robey PG. Bone matrix proteoglycans and glycoprotein. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principle of bone biology. New York: Academic Press; 1996. pp. 155–166. [Google Scholar]
- 48.Wang C, Duan Y, Markovic B, Barbara J, Howlett CR, Zhang X, Zreiqat H. Proliferation and bone-related gene expression of osteoblasts grown on hydroxyapatite ceramics sintered at different temperatures. Biomaterials. 2004;25:2949–2956. doi: 10.1016/j.biomaterials.2003.09.088. [DOI] [PubMed] [Google Scholar]