

Abstract
Caveolin-1 (Cav-1) is a membrane scaffolding protein which functions to regulate intracellular compartmentalization of various signaling molecules. In the present studies, transgenic mice with a targeted disruption of the Cav-1 gene (Cav-1−/−) were used to assess the role of Cav-1 in acetaminophen-induced hepatotoxicity. Treatment of wild type mice with acetaminophen (300 mg/kg) resulted in centrilobular hepatic necrosis and increases in serum transaminases. This was correlated with decreased expression of Cav-1 in the liver. Acetaminophen-induced hepatotoxicity was significantly attenuated in Cav-1−/− mice, an effect that was independent of acetaminophen metabolism. Acetaminophen administration resulted in increased hepatic expression of the oxidative stress marker, lipocalin 24p3, as well as hemeoxygenase-1, but decreased glutathione and superoxide dismutase-1; no differences were noted between the genotypes suggesting that reduced toxicity in Cav-1−/− mice is not due to alterations in anti-oxidant defense. In wild type mice, acetaminophen increased mRNA expression of the pro-inflammatory cytokines, interleukin-1β and monocyte chemoattractant protein-1 (MCP-1), as well as cyclooxygenase-2, while 15-lipoxygenase (15-LOX), which generates anti-inflammatory lipoxins, decreased. Acetaminophen-induced changes in MCP-1 and 15-LOX expression were greater in Cav-1−/− mice. Although expression of tumor necrosis factor-α, a potent hepatocyte mitogen, was up-regulated in the liver of Cav-1−/− mice after acetaminophen, expression of proliferating cell nuclear antigen and survivin, markers of cellular proliferation, were delayed which may reflect the reduced need for tissue repair. Taken together, these data demonstrate that Cav-1 plays a role in promoting inflammation and toxicity during the pathogenesis of acetaminophen-induced injury.
Keywords: caveolin-1, liver, acetaminophen, antioxidants, cytokines, proliferation
Introduction
Acetaminophen is one of the most commonly used over the counter analgesics and the major cause of acute liver failure due to accidental and intentional overdose (Bower et al., 2007). Toxicity is characterized by centrilobular hepatic necrosis progressing to fulminant liver failure (Ramachandran and Kakar, 2009). Hepatotoxicity is initiated by N-acetyl-p-benzoquinone-imine (NAPQI), generated from acetaminophen via cytochrome P4502E1, which is predominantly localized in centrilobular regions of the liver (Lee et al., 1996). Other factors contributing to the hepatotoxicity of acetaminophen include inflammatory mediators, such as tumor necrosis factorα (TNFα), interleukin-1 (IL-1) and monocyte chemoattractant protein-1 (MCP-1), as well as reactive oxygen and nitrogen species (Gardner and Laskin, 2007; Laskin and Gardner, 2007; Laskin, 2009). Tissue repair following acetaminophen intoxication involves proliferation of remnant hepatocytes and the production of new extracellular matrix proteins (Chiu et al., 2003b; Gardner et al., 2003; Cozma et al., 2004; Bourdi et al., 2008). Accumulating evidence suggests that inflammatory mediators, including TNFα, are also involved in this process (Gardner and Laskin, 2007; Laskin and Gardner, 2007; Wullaert et al., 2007; Laskin, 2009). TNFα is a potent mitogen for hepatocytes (Yamada et al., 1997; Ledda-Columbano et al., 1998). It also stimulates extracellular matrix turnover and up-regulates antioxidants (Roderfeld et al., 2006; Han et al., 2009).
Caveolin-1 (Cav-1) is a membrane scaffolding protein that functions as an organizing center to concentrate various signaling molecules and negatively regulate their activity (Quest et al., 2004; Williams and Lisanti, 2004). Functional caveolin-binding motifs have been identified in a number of downstream cytokine and growth factor signaling molecules including tumor necrosis factor receptor-1 (TNFR1) and TNFR-associated factor-2, and in several proteins and serine/threonine and tyrosine kinases important in regulating cell cycle progression and cellular proliferation such as β-catenin, cyclin D1, and inhibitor of apoptosis proteins or survivin (Torres et al., 2006; Goetz et al., 2008; Parat, 2009). The role of Cav-1 in liver regeneration after injury is unknown. Under homeostatic conditions, Cav-1 is expressed in hepatocytes and sinusoidal cells, and in portal veins and hepatic arteries (Calvo et al., 2001; Shah et al., 2001; Malerod et al., 2002; Ogi et al., 2003). Following partial hepatectomy, Cav-1 has been reported to be up-regulated in hepatocytes (Pol et al., 2004; Fernandez et al., 2006; Mayoral et al., 2007). Moreover, defects in liver repair and reduced survival are observed in transgenic mice lacking the gene for Cav-1 (Fernandez et al., 2006) suggesting that Cav-1 may be important in stimulating wound repair processes following exposure to hepatotoxicants such as acetaminophen. To test this, we analyzed the effects of acetaminophen in Cav-1−/− mice. Unexpectedly, these mice were found to be less sensitive to the hepatotoxic effects of acetaminophen relative to wild-type mice. This was associated with decreased expression of the pro-inflammatory proteins, MCP-1 and IL-1, and increased expression of IL-10, which suppresses inflammation. These findings suggest that Cav-1 plays a role in promoting inflammation and tissue injury in this model of hepatotoxicity.
Materials and Methods
Animals and treatments
Male Cav-1−/− mice and wild-type B6J129SV F2 mice (7–9 weeks, 20–25 g) were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were maintained on food and water ad libitum and housed in microisolator cages. All animals received humane care in compliance with the institution’s guidelines, as outlined in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Mice were fasted overnight prior to administration of acetaminophen (300 mg/kg, I.P.) or phosphate-buffered saline (PBS) control. Blood samples were collected via cardiac puncture and analyzed for serum alanine and aspartate transaminase using diagnostic assay kits (ThermoElectron, Pittsburgh, PA).
Histology and immunohistochemistry
Livers were fixed over night at 4°C in 3% paraformaldehyde in PBS containing 2% sucrose, washed 3 times with 2% sucrose/PBS, transferred to 50% ethanol, and then paraffin embedded. Six micron tissue sections were prepared and stained with hematoxylin and eosin (Goode Histolabs, New Brunswick, NJ). For immunohistochemistry, sections were incubated overnight with rabbit antibody to hemeoxygenase-1 (HO-1, 1:1000; Stressgen/Assay Designs, Ann Arbor, MI), proliferating cell nuclear antigen (PCNA, 1:125, Abcam, Cambridge, MA), survivin (1:200; Abcam) or rabbit IgG control (Santa Cruz Biotechnology, Santa Cruz, CA). Antibody binding was visualized using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA). Three random sections of each liver were examined.
Western blot
Liver samples were homogenized in lysis buffer consisting of 20 mmol/L HEPES, 150 mmol/L NaCl, 10% glycerol, 1% Triton X-100, 1.5 mmol/L MgCl2, 1 mmol/L diethylene triamine pentacetic acid, 1 mmol/L phenylmethylsulfonylenediamine, 10 mmol/L sodium pyrophosphate, 50 mmol/L sodium fluoride, 2 mmol/L sodium orthovanadate and protease inhibitor cocktail (Sigma). Protein concentrations were assayed using a BCA Protein Kit (Pierce, Rockford, IL) with bovine serum albumin as the standard. Proteins were separated on 10% polyacrylamide gels and then transferred onto nitrocellulose membranes. Non-specific binding was blocked by incubation of the blots at room temperature with blocking buffer (5% nonfat dry milk, 10 mmol/L Tris-base, 200 nmol/L sodium chloride and 0.1% Tween 20) for 60 min. Membranes were then incubated overnight at 4oC with a monoclonal rabbit anti-Cav-1 antibody (1:1000, Cell Signaling Technology), or polyclonal rabbit anti-cyclooxygenase-2 (Cox-2) (1:250, Abcam). This was followed by incubation with goat anti-rabbit horse radish peroxidase antibody for 1 h at 20oC (1:10,000; BioRad, Carlsbad CA). Binding was detected using ECL Plus (GE Healthcare, Piscataway, NJ).
Measurement of liver glutathione (GSH)
Livers were minced in ice cold 5% metaphosphoric acid (1:10), homogenized and then centrifuged at 3000g for 10 min at 4oC. Supernatants were filtered though a 0.2 μm syringe filter and reduced GSH quantified using a colorimetric assay kit (GSH-400, OxisResearch, Portland, OR). GSH was calculated based on the slope of a standard curve and expressed as μmol/g wet liver weight.
Quantitative real time PCR
Total RNA was extracted from liver samples (25 mg) using an RNeasy Miniprep kit (Qiagen Inc, Valencia, CA) and RNA reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) according to the manufacturers’ protocol. Standard curves were generated using serial dilutions from pooled randomly selected cDNA samples. Real time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on a 7900HT thermocycler using 96-well optical reaction plates according to manufacturer protocol. All PCR primer sequences were generated using Primer Express 2.0 (Applied Biosystems) and primers were synthesized by Integrated DNA Technologies (Coralville, IA). A minimum of three samples were analyzed for each experimental group, and all samples were run in duplicate. Primer sequences were: HO-1, CCTCACTGGCAGGAAATCATC; superoxide dismutase-1 (SOD-1), AGGCTGTACCAGTGCAGGAC; IL-1β, CCAAAAGATGAAGGGCTGCT; TNFα, AAATTCGAGTGACAAGCCGTA; MCP-1, GCCAGCTCTCTCTTCCTCCA; IL-10, GGTTGCCAAGCCTTATCGGA; survivin, TGAATCCTGCGTTTGAGTCG, 5-lipoxygenase (5-LOX), CAGGGAGAAGCTGTCCGAGT; 15-lipoxygenase (15-LOX), TCGGAGGCAGAATTCAAGGT, COX-2, CATTCTTTGCCCAGCACTTCAC; NADPH quinine oxidoreductase-1 (NQO1), ACGCCTGAGCCCAGATATTG; 24p3, GCCCAGGACTCAACTCAGAA; actin, TCACCCACACTGTGCCCATCTACGA; and glyceraldehyde 3-phophate dehydrogenase (GAPDH), TGAAGCAGGCATCTGAGGG.
Acetaminophen metabolism
Blood samples were collected by cardiac puncture and serum separated by centrifugation (3000g, 4oC, 10 min), frozen in liquid nitrogen and stored at −80°C Until analysis. Acetaminophen and conjugated metabolites were analyzed as previously described (Brunner and Bai, 1999). Briefly, samples were thawed and deproteinated with 6% perchloric acid containing 10 mg/ml theophylline as an internal standard. Precipitated proteins were sedimented by centrifugation (13,000g, 5 min, 4oC), supernatants collected and analyzed isocratically by HPLC (Shimadzu, Colombia, MD) fitted with a Luna 5 μm, C18 column, 250 mm × 2.0 mm (Phenomenex, Torrance, CA) with a guard column containing the same sorbent. Samples were analyzed at a constant flow rate of 0.2 ml/min, detected by UV absorbance at 254 nm with a mobile phase composed of 7% acetonitrile in 0.05 mmol/L sodium sulfate (pH 2.2).
Hepatic cytochrome P450-2E1 activity
To prepare microsomes, samples of whole liver (3 g) from control mice were homogenized in 2 volumes (w/v) of 10 mM Tris-base (pH 7.4) containing 1.5% KCl using a Teflon-glass homogenizer at 4oC. Homogenates were centrifuged at 1,000g (10 min, 4oC), supernatants collected and centrifuged at 11,000g (20 min, 4oC) to remove cellular debris, and then at 105,000g (1.5 h, 4oC) to isolate microsomes. Microsomes were resuspended in homogenization buffer containing 0.5 mM phenylmethanesulfonyl fluoride and centrifuged at 105,000g (90 min, 4oC). Pellets were resuspended in 0.25 M sucrose containing 10 mM Tris-base (pH 7.4) and stored at −80°C Until use (Cooper et al., 1993). Cytochrome P4502E1 was measured by metabolism of p-nitrophenol to p-nitrocatecol. To assay for cytochrome P450-2E1 activity, microsomes were incubated with 200 μM p-nitrophenol and 500 μM NADPH at 37oC for 30 min. Trichloroacetic acid (20%, w/v) was then added to stop the reaction. The mixture was then centrifuged (10,000g, 5 min, 4oC), supernatants collected, and mixed with 2 M NaOH. Changes in absorbance were measured spectrophotometrically at 535 nm. Concentrations of p-nitrocatechol in the samples were calculated based on a standard curve generated with authentic p-nitrocatechol (Koop, 1986).
Statistics
Data were analyzed using a Student’s t test or by one way ANOVA followed by Tukey’s post hoc analysis.
Results
Effects of acetaminophen on Cav-1 expression in the liver; role of Cav-1 in hepatotoxicity
In initial studies, we analyzed the effects of acetaminophen on expression of Cav-1 in the liver. Cav-1 protein was readily detected in livers of wild type mice treated with PBS control (Fig. 1). Administration of acetaminophen resulted in a rapid and time-dependent decrease in total liver Cav-1 which persisted for 72 h (Fig. 1). Subsequently, Cav-1 protein began to increase. To analyze the role of Cav-1 in acetaminophen-induced hepatotoxicity, we used transgenic mice with a targeted disruption of the gene for this protein. Consistent with previous studies (Drab et al., 2001; Le Lay and Kurzchalia, 2005), Cav-1 protein was not evident in livers of Cav-1−/− mice (Fig. 1). Treatment of these mice with acetaminophen had no effect on expression of this protein. In wild type mice, acetaminophen caused a rapid and time-related induction of hepatotoxicity. Thus, significant elevations in both alanine (ALT) and aspartate (AST) transaminases were noted within 3 h post-acetaminophen treatment; levels of these enzymes continued to increase for 18–24 h (Fig. 2). Subsequently, serum transaminase levels declined towards control. These changes were associated with histological alterations in the liver which were evident within 1–3 h and included hepatocellular cytoplasmic degeneration (Fig. 3 and not shown). This progressed to bridging necrosis by 6 h and severe congestion with moderate inflammation at 24 h (Fig. 3). At this time, greater than 75% of the liver lobules contained necrotic regions. Similar histopathology was observed at 48 h. By 72 h, however, liver regeneration was evident, characterized by the appearance of mitotic figures within hepatocytes adjacent to necrotic areas. Liver regeneration was nearly complete by 96 h (not shown). Acetaminophen-induced increases in serum transaminase levels were significantly reduced in Cav-1−/− mice, when compared to wild type mice (Fig. 2). In addition, in Cav-1−/− mice, histologic evidence of liver damage developed more slowly, and at 24 h included smaller areas (less than 50% lobular involvement) of the acinus (Fig. 3). By 48 h necrotic lesions began to coalesce. As observed in wild type mice, mitotic figures were also noted in livers of Cav-1−/− mice 72 h following acetaminophen, although at significantly lower levels (30.0 ± 9.2 in wild type mice versus 15.3 ± 0.9 in Cav-1−/− mice in 15 oil immersion fields). As noted in wild type mice, by 96 h, liver histology was similar to control (not shown).
Fig. 1.

Effects of acetaminophen on Cav-1 expression. Mice were treated with acetaminophen (APAP) or control (CTL). After 1–96 h in wild type mice or 18 h in Cav-1−/− mice, livers were collected and analyzed by Western blotting for Cav-1 expression. One representative section from 3 mice/treatment is shown.
Fig. 2.

Effects of acetaminophen treatment of mice on serum transaminases. Wild type and Cav-1−/− mice were treated with acetaminophen (APAP) or control (CTL). Serum was collected 1–96 h later and analyzed for alanine (ALT) and aspartate (AST) transanimase. Each bar represents the mean ± SE (n= 5–13 mice). aSignificantly different (p ≤ 0.05) from CTL; bSignificantly different (P ≤ 0.05) from wild type mice.
Fig. 3.


Effects of acetaminophen on liver histology. Wild type and Cav-1−/− mice were treated with acetaminophen (APAP) or control (CTL). Livers were collected 3–96 h later and sections stained with hematoxylin and eosin (original magnification X200; insets, X400). One representative section from 3 mice/treatment is shown.
We next examined expression of lipocalin 24p3, an acute phase protein and a marker of oxidative stress (Roudkenar et al., 2007; Roudkenar et al., 2008). Low levels of 24p3 mRNA were evident in the livers of both wild type and Cav-1−/− mice (Fig. 4, upper panel). Acetaminophen treatment of the mice resulted in a time-dependent increase in 24p3 mRNA levels which reached a maximum at 24 h in both genotypes. Loss of Cav-1 had no significant effect on hepatic expression of 24p3.
Fig. 4.

Effects of acetaminophen on 24p3 and NQO1 mRNA expression. Livers were collected 1–96 h after treatment of wild type (open bars) or Cav-1−/− mice (closed bars) with control (CTL) or acetaminophen (APAP). Samples were analyzed by real time PCR. Each bar represents the mean ± SE (n = 3–8 mice). aSignificantly different (p ≤ 0.05) from CTL.
To exclude the possibility that differences in hepatotoxicity between wild type and Cav-1−/− mice were due to alterations in acetaminophen metabolism, we measured serum levels of acetaminophen conjugates and hepatic cytochrome P4502E1 activity. HPLC analysis of serum samples collected 30 min after acetaminophen administration revealed the presence of unconjuguated acetaminophen (266.3 ± 4.0 and 230.5 ± 6.1 μg/ml, n = 3 mice), acetaminophen-glucuronide (386.8 ± 22.9 and 417.4 ± 47.5 μg/ml, n = 3 mice), and acetaminophen-sulfate, which was below the level of quantification, in wild type and Cav-1−/− mice, respectively (Fig. 5). No significant differences were observed between the genotypes. We also noted generally similar rates of cytochrome P4502E1 mediated hydroxylation of p-nitrophenol in microsomal fractions of livers from wild type and Cav-1−/− mice (0.54 ± 0.04 and 0.46 ± 0.06 nmol/min/mg, respectively). NQO1, a phase II enzyme important in detoxification of the acetaminophen metabolite, NAPQI (Vasiliou et al., 2006) was also analyzed. Consistent with previous studies (Aleksunes et al., 2008), NQO1 mRNA levels were found to increase in the livers of wild type mice following administration of acetaminophen (Fig. 4). This was first observed 6 h post exposure (Fig. 4). Similar increases in NQO1 mRNA expression were noted in Cav-1−/− mice.
Fig. 5.

Effects of loss of Cav-1 on acetaminophen metabolism. Serum was collected 30 min after administration of acetaminophen to wild type and Cav-1−/− mice and analyzed by HPLC for the presence of APAP, APAP-glucuronide (-gluc), and APAP-sulfate (-sulf) metabolites, with retention times of 15, 9.1 and 8 min respectively; IS, internal standard. One representative chromatogram from 3 mice/strain is shown.
Effects of loss of Cav-1 on hepatic antioxidants
In further studies, we determined if reduced sensitivity of Cav-1−/− mice to acetaminophen was associated with alterations in levels of GSH and antioxidants important in protecting against liver injury (Nakae et al., 1990; Ferret et al., 2001; Chiu et al., 2003a; Fujimoto et al., 2009; Mladenovic et al., 2009; Yoshikawa et al., 2009). GSH was detected in livers of wild type and Cav-1−/− mice; however levels of this antioxidant were significantly lower in Cav-1−/− mice (Table 1). In both mouse strains, acetaminophen administration caused a rapid (within 1 h) decrease in GSH which returned to control levels after 6 h (Table 1). SOD-1 mRNA was also identified in livers of both wild type and Cav-1−/− mice (Fig. 6); a similar time-dependent decrease in SOD expression was observed in both mouse strains which reached a maximum 24 h after acetaminophen (Fig. 6). In contrast to SOD-1, a rapid and transient increase in HO-1 mRNA was noted in the livers of both wild type and Cav-1−/− mice after acetaminophen administration which reached a maximum within 3 h and returned to control levels by 18–24 h. Increases in HO-1 expression were also evident in histologic sections of liver 24 h post-treatment (Fig. 6 and not shown). Interestingly, HO-1 was predominantly localized in macrophages within the hepatic sinusoids. No significant differences were noted in acetaminophen-induced HO-1 mRNA or protein expression between the genotypes.
Table 1.
Effects of acetaminophen treatment of mice on hepatic GSH levels.
| Time after Acetaminophen (h) | Wild Type | Cav-1−/− |
|---|---|---|
| Control | 7.2 ± 0.9 | 4.5 ± 0.8a |
| 1 | 0.6 ± 0.1b | 0.8 ± 0.1b |
| 3 | 0.7 ± 0.1b | 2.0 ± 0.9b |
| 6 | 4.6 ± 2.1 | 4.8 ± 1.0 |
| 18 | 4.7 ± 0.5 | 7.4 ± 1.5 |
| 24 | 5.8 ± 0.1 | 5.7 ± 1.7 |
Wild type and Cav-1−/− mice were treated with acetaminophen or control. Livers were collected 1–24 h later and analyzed for GSH content as described in the Materials and Methods. Data are presented as μmol GSH/g liver, mean ± SE (n = 3–5 mice).
Significantly different (P ≤ 0.05) from wild type mice.
Significantly different (p ≤ 0.05) from control.
Fig. 6.

Effects of acetaminophen on SOD-1 and HO-1 expression. Livers were collected 1–24 h after treatment of wild type or Cav-1−/− mice with control (CTL) or acetaminophen (APAP). Left panel: Liver samples from wild type (open bars) and Cav-1−/− (closed bars) mice were analyzed by real time PCR. Each bar represents the mean ± SE (n = 3–15 mice). aSignificantly different (p ≤ 0.05) from CTL; bSignificantly different (p≤0.05) from wild type mice. Right panel: Liver samples from wild type (Panels A, C, and E) and Cav-1−/− mice (Panels B, D, and F) were collected 24 h after treatment with CTL (Panels A and B) or APAP (Panels C-F) and stained with antibody to HO-1 (Panels A-D) or rabbit IgG (Panels E and F). Antibody binding was visualized using a Vectastain Elite ABC kit (original magnification, X1000). One representative section from 3 mice/treatment is shown.
Effects of loss of Cav-1 on acetaminophen-induced alterations in expression of inflammatory mediators
In our next series of studies, we analyzed the effects of loss of Cav-1 on acetaminophen-induced expression of pro- and anti-inflammatory mediators. Consistent with previous studies (Gardner et al., 2003; Dambach et al., 2006), acetaminophen treatment of wild type mice resulted in a time-dependent increase in expression of mRNA for the pro-inflammatory cytokines IL-1β and MCP-1 (Fig. 7). Whereas, significant increases in IL-1β were detectable at 3 h, increases in MCP-1 were delayed for 18 h. In Cav-1−/− mice, acetaminophen-induced increases in expression of MCP-1 mRNA occurred earlier (within 3 h) and were significantly greater when compared to wild type mice, while changes in IL-1β were generally similar in the two mouse strains. In both wild type and Cav-1−/− mice, expression of these mediators returned to control levels by 48 h (not shown). We also analyzed the effects of loss of Cav-1 on expression of TNFα, which exerts both pro- and anti-inflammatory actions in the liver (Rutherford and Chung, 2008; Han et al., 2009). Acetaminophen treatment of wild type mice had no significant effect on TNFα mRNA expression in the liver. In contrast, a significant increase in expression of this cytokine was observed within 3 h in Cav-1−/− mice which persisted for at least 24 h (Fig. 7). Acetaminophen administration was also associated with increased expression of the anti-inflammatory cytokine, IL-10 in Cav-1−/− mice, but not in wild type mice (Fig. 7). This response was rapid, appearing within 1 h and persisting for 6 h.
Fig. 7.
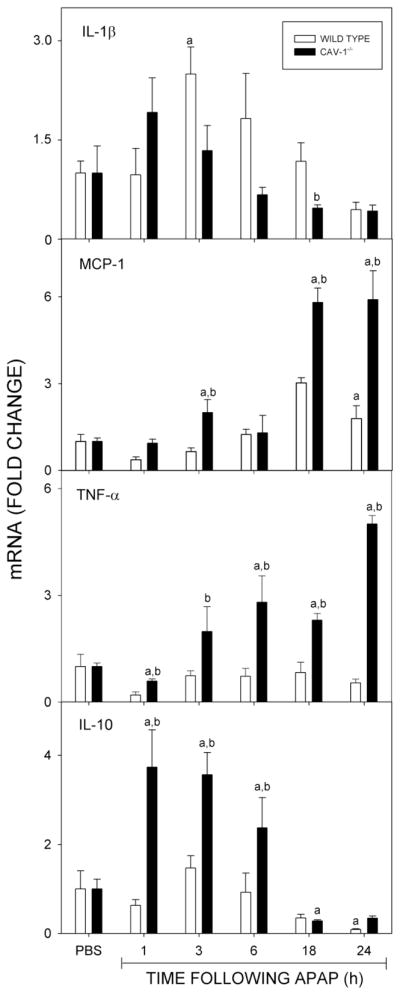
Effects of acetaminophen on mRNA expression of pro- and anti-inflammatory cytokines. Livers, collected 1–24 h after treatment of wild type (open bars) or Cav-1−/− (closed bars) mice with control (CTL) or acetaminophen (APAP) were analyzed by real time PCR. Each bar represents the mean ± SE (n = 3–15 mice). aSignificantly different (p ≤ 0.05) from CTL; bSignificantly different (p ≤ 0.05) from wild type mice.
We also analyzed expression of enzymes mediating the production of eicosaniods, including COX-2, 5-LOX and 15-LOX, which are involved in both initiation and resolution of inflammation (Harizi et al., 2008). Treatment of wild type mice with acetaminophen resulted in a marked increase in expression of COX-2 mRNA which reached a maximum at 3 h and persisted at these levels for 24 h (Fig. 8, upper panel). Subsequently, COX-2 mRNA levels declined toward control. This correlated with increases in expression of COX-2 protein (Fig. 8, lower panel). COX-2 protein also increased in Cav-1−/− mice peaking at 24 h, but this was attenuated relative to wild type mice. In contrast, acetaminophen had no significant effect on COX-2 mRNA expression in Cav-1−/− mice. Acetaminophen also had no significant effect on 5-LOX expression in either wild type or Cav-1−/− mice, while 15-LOX mRNA decreased in both mouse stains (Fig. 9). These effects occurred more rapidly in Cav-1−/− mice, when compared to wild type mice, and persisted for at least 96 h.
Fig. 8.

Effects of acetaminophen on COX-2 expression. Livers were collected 1–96 h after treatment of wild type (open bars) or Cav-1−/− (closed bars) mice with control (CTL) or acetaminophen (APAP). Upper panel: Livers were analyzed by real time PCR. Each bar represents the mean ± SE (n = 3–8 mice). aSignificantly different (p ≤ 0.05) from CTL; bSignificantly different (p ≤ 0.05) from wild type mice. Lower panel: Liver samples collected 6–96 after APAP administration to wild type (W) or Cav-1−/− (C) mice were analyzed by Western blotting. One representative blot from 3 mice/treatment is shown.
Fig. 9.

Effects of acetaminophen on expression of 5-LOX and 15-LOX. Livers were collected 1–96 h after treatment of wild type (open bars) or Cav-1−/− (closed bars) mice with control (CTL) or acetaminophen (APAP) and analyzed by real time PCR. Each bar represents the mean ± SE (n = 3–8 mice). aSignificantly different (p ≤ 0.05) from CTL; bSignificantly different (p ≤ 0.05) from wild type mice.
Effects of loss of Cav-1 on hepatocyte proliferation
We next determined if hepatotoxicity of acetaminophen in Cav-1−/− mice was associated with increased proliferation of remnant hepatocytes, a key step in tissue repair. In initial experiments, we analyzed expression of PCNA, a nuclear protein that is up-regulated in proliferating cells (Mendieta-Condado et al., 2009). In wild type mice acetaminophen caused in a time dependent increase in PCNA staining in hepatocyte nuclei in periportal regions of the liver, which was evident within 3 h and remained elevated for at least 96 h (Fig. 10). The appearance of PCNA positive hepatocytes was delayed until 18 h in Cav-1−/− mice. However, by 24 h, significantly greater numbers of PCNA labeled hepatocytes were observed in Cav-1−/− mice when compared to wild type mice. PCNA positive macrophages were also noted in livers of both wild type and Cav-1−/− mice after acetaminophen administration. Whereas at early times (3–6 h), these cells were scattered throughout the liver lobule, at later times (48–96 h), PCNA positive macrophages were more abundant in centrilobular regions of the liver. Greater numbers of these cells were noted in wild type when compared to Cav-1−/− mice (113.7 ± 32.7 cells/10 high power fields versus 10.3 ± 3.3/10 high power fields; n = 3 mice at 3 h post exposure).
Fig. 10.


Effects of acetaminophen on PCNA expression in the liver. Sections were prepared 1–96 h after treatment of wild type (WT) or Cav-1−/− mice with CTL or acetaminophen (APAP). Upper panel: The number of PCNA-positive hepatocytes was counted in 5 randomly selected microscopic fields (400x), centered on the portal triad. A minimum of 500 hepatocytes were analyzed in each liver section. Data are presented as % PCNA-positive cells. Each bar represents the mean ± SE (n = 6 sections from 3 mice/treatment). aSignificantly different (p ≤ 0.05) from CTL; bSignificantly different (p ≤ 0.05) from wild type mice. Lower panel: Liver samples from wild type and Cav-1−/− mice were stained with antibody to PCNA. Antibody binding was visualized by using a Vectastain Elite ABC kit (original magnification, X400). Arrows, PCNA labeled hepatocyte nuclei; arrowheads, PCNA labeled macrophages. One representative section from 3 mice/treatment is shown.
We also examined expression of survivin, a 16.5 kDa protein implicated in the regulation of cell-cycle progression (Li et al., 1998; Reed, 2001). Following acetaminophen administration to wild type mice, survivin protein expression increased in hepatocyte nuclei located adjacent to portal veins and necrotic zones in the livers. This was evident 3 h post exposure and persisted for at least 72 h (Fig. 11 and not shown). Survivin was also up-regulated in the livers of Cav-1−/− mice after acetaminophen, however, the kinetics were distinct. Thus, survivin expression was delayed until 24 h post exposure in Cav-1−/− mice; subsequent expression of this protein decreased towards control levels (Fig. 11). Greater expression of survivin was noted in wild type mice relative to Cav-1−/− mice at all time points examined.
Fig. 11.

Effects of acetaminophen on expression of survivin. Liver samples from wild type and Cav-1−/− were collected 3–72 h after treatment with control (CTL) or acetaminophen and stained with antibody to survivin. Antibody binding was visualized by using a Vectastain Elite ABC kit (original magnification, X400). Arrows indicate survivin stained hepatocyte nuclei. One representative section from 3 mice/treatment is shown.
Discussion
Cav-1 is an integral membrane protein important in negatively regulating cell signaling and proliferation (Williams and Lisanti, 2004; Shatz and Liscovitch, 2008). The present studies show that acetaminophen administration decreases expression of Cav-1 protein in the livers of wild type mice. Similar down-regulation of Cav-1 protein has been described in the lung in response to toxicants such as bleomycin and ozone (Koslowski et al., 2004; Odajima et al., 2007; Fakhrzadeh et al., 2008). In contrast, partial hepatectomy results in increased Cav-1 expression (Fernandez et al., 2006; Mayoral et al., 2007). This suggests that the regulatory molecules controlling liver injury and repair after acetaminophen and partial hepatectomy are distinct.
To investigate the role of Cav-1 in acetaminophen-induced hepatotoxicity, Cav-1−/− mice were used. Unexpectedly, these mice were found to be less sensitive to the hepatotoxic effects of acetaminophen than wild type mice. Analysis of plasma levels of acetaminophen-glucuronide and -sulfate metabolites after acetaminophen administration, as well as hepatic cytochrome P4502E1 activity, indicated that there were no major differences in acetaminophen metabolism or the capacity to metabolize acetaminophen between Cav-1−/− mice and wild type mice. This is supported by findings that levels of NQO1, which is important in detoxification of NAPQI, were generally similar in the two mouse strains.
Antioxidants play a key role in protecting against acetaminophen-induced injury (Nakae et al., 1990; Ferret et al., 2001; Chiu et al., 2003a; Fujimoto et al., 2009; Mladenovic et al., 2009; Yoshikawa et al., 2009). Consistent with previous studies (Gardner et al., 2002; Chiu et al., 2003a), we observed a significant reduction in whole liver GSH levels in wild type mice following acetaminophen administration. Similar results were observed in Cav-1−/− mice. GSH is known to be present at high levels in hepatocytes (Yuan and Kaplowitz, 2009). In addition to scavenging reactive oxygen species, it plays a key role in detoxification of the cytotoxic acetaminophen metabolite, NAPQI (Jaeschke, 1990). Interestingly, baseline levels of GSH were significantly lower in livers of Cav-1−/− mice, when compared to wild type mice. Our findings that Cav-1−/− mice are less sensitive to acetaminophen suggest that reduced levels of GSH are not key to the pathogenesis of hepatotoxicity in these mice. As observed with GSH, no significant differences in expression of HO-1, SOD-1 or lipocalin 24p3 were noted between the genotypes following acetaminophen administration. Thus, it appears that protection of Cav-1−/− mice against the hepatotoxic effects of acetaminophen is not due to alterations in oxidative stress or antioxidant defense.
Acetaminophen administration to both wild type and Cav-1−/− mice was associated with up-regulation of pro- and anti-inflammatory mediators which have been implicated in the pathogenesis of liver injury and repair, including IL-1β, MCP-1, IL-10, and TNFα (Liu and Kaplowitz, 2006; Laskin, 2009). Of particular note are IL-10 and MCP-1 which play important roles in mediating the resolution of inflammation and initiating tissue repair following acetaminophen intoxication, in part, by recruiting and activating M2 repair macrophages (Hogaboam et al., 2000; Dambach et al., 2002; Holt et al., 2008). In Cav-1−/− mice, expression of both IL-10 and MCP-1 increased more rapidly when compared to wild type mice; moreover, expression levels were greater. Early production of these mediators may contribute to hepatoprotection in Cav-1−/− mice against acetaminophen. We also found that TNFα mRNA expression increased in response to acetaminophen in Cav-1−/− mice, with no effect in wild type mice. Accumulating evidence suggests that TNFα is a key mediator of hepatocyte proliferation following injury (Bruccoleri et al., 1997; Chiu et al., 2003b; Okamoto et al., 2009; Shimizu et al., 2009). TNFα also up-regulates hepatic antioxidant defense, and induces matrix remodeling and tissue repair in various models of liver injury (Haruyama et al., 2000; Gharaee-Kermani and Phan, 2001; Chiu et al., 2003a). TNFα may play a similar hepatoprotective role in Cav-1−/− mice after acetaminophen-intoxication. Previously, studies have reported increases in TNFα mRNA levels in wild type mice following acetaminophen intoxication (Gardner et al., 2002; Dambach et al., 2006). Differences between our findings and earlier reports are likely due to genetic distinctions between the mouse strains used (Mayoral et al., 2007). The present studies also show that acetaminophen-induced changes in IL-1β mRNA levels were similar in wild type and Cav-1−/− mice. This indicates that Cav-1 is not required for hepatic IL-1β expression in this model.
Arachidonic acid metabolites, including prostaglandins, leukotrienes and lipoxins are important mediators of the responses to tissue injury, contributing to both pro- and anti-inflammatory activity (Harizi et al., 2008; Serhan, 2008). Prostaglandins are generated from arachidonic acid via the actions of COX-2. The present studies demonstrate that COX-2 mRNA and protein are upregulated in livers of wild type mice after acetaminophen administration. Although COX-2 protein was also up-regulated in Cav-1−/− mice following acetaminophen, levels were reduced when compared to wild type mice. Moreover, there were no significant changes in COX-2 mRNA. These data indicate that the proinflammatory activity of COX-2 may play a more prominent role in hepatotoxicity in this model. This is consistent with previous reports demonstrating that COX-2−/− mice are protected from acetaminophen-induced hepatotoxicity (Reilly et al., 2001).
Leukotrienes and lipoxins are produced via the sequential activation of 5-LOX and 15-LOX which are expressed in leukocytes and endothelium at sites of tissue damage (Romano, 2006; Kronke et al., 2009). Whereas, products of 5-LOX are generally proinflammatory, 15-LOX generates anti-inflammatory lipoxins (Serhan, 2008). Although no significant changes were noted in 5-LOX expression after acetaminophen administration in either wild type or Cav-1−/− mice, 15-LOX decreased in both genotypes. In Cav-1−/− mice, this decrease occurred more rapidly and was persistent, while in wild type mice it was delayed and transient. The rapid and persistent decrease in Cav-1−/− mice may reflect reduced need for anti-inflammatory eicosanoids due to decreased hepatotoxicity.
The outcome of the response to hepatotoxicants is determined, in part, by the ability of remnant hepatocytes to proliferate, a major step in tissue repair (Mehendale, 2005; Fausto et al., 2006). Cav-1 has been reported to regulate multiple pathways important for tissue regeneration, including TNFα signaling (Gassmann and Werner, 2000; Lopez-Rivera et al., 2005; Canbay et al., 2007). Although TNFα was rapidly up-regulated in Cav-1−/− mice after acetaminophen administration, hepatocyte proliferation, assessed by PCNA expression, was delayed when compared to wild type mice, suggesting that alternative signaling molecules control hepatocyte proliferation in Cav-1−/− mice. Of note is our observation that PCNA expression was significantly greater in Cav-1−/− mice relative to wild type mice 24 hr post acetaminophen. This may contribute to the hepatoprotective effects of loss of Cav-1. Interestingly, macrophages located within the sinusoids in the midzonal and portal regions and necrotic areas of the liver were found to express PCNA, indicating that these cells rapidly proliferate in response to acetaminophen-induced injury. Macrophage proliferation in the liver has been described after partial body irradiation, carbon tetrachloride intoxication, or depletion by liposome-encapsulated dichloromethylene diphosphate, suggesting that this may be a generic response to tissue injury (Bouwens et al., 1986; Yamamoto et al., 1996; Orfila et al., 1999). The finding that there are greater numbers of proliferating macrophages in acetaminophen-treated wild type mice may be indicative of more severe injury in the liver when compared to Cav-1−/− mice. The phenotype of the proliferating macrophages in the liver is unknown and is likely to influence the ultimate pathogenic response to acetaminophen (Laskin, 2009). Thus, whereas M1 macrophages contribute to tissue injury, M2 cells are involved in repair. It may be that early proliferation of M1 macrophages in wild type mice plays a role in tissue injury, while delayed proliferation of M2 macrophages initiates repair and is important in hepatoprotection in Cav-1−/− mice. Delayed proliferation of mitogen producing M2 macrophages in Cav-1−/− mice is consistent with the delay in the initiation of hepatocyte proliferation observed after acetaminophen, as well as the decreased need for tissue repair.
Survivin is a member of the family of inhibitor of apoptosis proteins and functions as a regulator of mitosis and apoptosis (Mita et al., 2008). Expression of survivin is associated with inhibition of cell death and is required for cell division (Altieri, 2008). Survivin has recently been reported to be important in liver regeneration following partial hepatectomy (Baba et al., 2009). Within 3 h of acetaminophen administration to wild type mice, we found that survivin protein expression was up-regulated in hepatocyte nuclei located adjacent to portal veins and spreading to necrotic zones in the livers by 24 h. Evidence suggests that expression of survivin is controlled by Cav-1 via the β-catenin-Tcf/Lef-dependent signaling pathway and it has been suggested that the anti-proliferative and pro-apoptotic properties of Cav-1 are due to reduced survivin expression (Torres et al., 2006). Our observation that acetaminophen-induced expression of survivin in livers of Cav-1−/− mice was attenuated, relative to wild type mice, and hepatocyte proliferation increased 24 h post acetaminophen are consistent with this idea.
In summary, the present studies demonstrate that protection of Cav-1−/− mice from acetaminophen-induced liver injury is associated with increased expression of cytokines implicated in down-regulation of inflammation and initiation of tissue repair including IL-10, MCP-1 and TNFα. This is correlated with reduced expression of COX-2, an enzyme important in the generation of proinflammatory eicosaniods. Taken together, these data suggest that Cav-1 plays a role in promoting acetaminophen-induced hepatotoxicity. This is surprising since Cav-1 has been shown to play an important role in regulating signaling molecules involved in cell replication, a key step in tissue repair in other models of tissue injury (Quest et al., 2008). Further studies are necessary to determine if the pro-inflammatory activity of Cav-1 is unique to the pathogenesis of hepatotoxicity induced by acetaminophen.
Acknowledgments
This work was supported by NIH grants GM034310, CA132634, AR055073, ES004738, and ES005022.
Abbreviations
- Cav-1
caveolin-1
- IL
interleukin
- MCP-1
monocyte chemoattractant protein-1
- LOX
lipoxygenase
- NAPQI
N-acetyl-p-benzoquinone-imine
- TNFα
tumor necrosis factor-α
- TNFR
tumor necrosis factor receptor
- HO-1
hemeoxygenase-1
- PCNA
proliferating cell nuclear antigen
- COX-2
cyclooxygenase-2
- GSH
glutathione
- SOD-1
superoxide dismutase-1
- NQO1
NADPH quinine oxidoreductase-1
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- ALT
alanine transaminase
- AST
aspartate transaminase
Footnotes
Conflict of interest statement: The authors declare that is are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aleksunes LM, Slitt AL, Maher JM, Augustine LM, Goedken MJ, Chan JY, Cherrington NJ, Klaassen CD, Manautou JE. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol Appl Pharmacol. 2008;226:74–83. doi: 10.1016/j.taap.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri DC. New wirings in the survivin networks. Oncogene. 2008;27:6276–6284. doi: 10.1038/onc.2008.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba HA, Wohlschlaeger J, Schmitz KJ, Nadalin S, Lang H, Benesch A, Gu Y, Biglarnia AR, Sotiropoulos GC, Takeda A, Takeda N, von Wnuck Lipinski K, Levkau B. Survivin is upregulated during liver regeneration in rats and humans and is associated with hepatocyte proliferation. Liver Int. 2009;29:585–592. doi: 10.1111/j.1478-3231.2008.01911.x. [DOI] [PubMed] [Google Scholar]
- Bourdi M, Korrapati MC, Chakraborty M, Yee SB, Pohl LR. Protective role of c-Jun N-terminal kinase 2 in acetaminophen-induced liver injury. Biochem Biophys Res Commun. 2008;374:6–10. doi: 10.1016/j.bbrc.2008.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwens L, Knook DL, Wisse E. Local proliferation and extrahepatic recruitment of liver macrophages (Kupffer cells) in partial-body irradiated rats. J Leukoc Biol. 1986;39:687–697. doi: 10.1002/jlb.39.6.687. [DOI] [PubMed] [Google Scholar]
- Bower WA, Johns M, Margolis HS, Williams IT, Bell BP. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102:2459–2463. doi: 10.1111/j.1572-0241.2007.01388.x. [DOI] [PubMed] [Google Scholar]
- Bruccoleri A, Gallucci R, Germolec DR, Blackshear P, Simeonova P, Thurman RG, Luster MI. Induction of early-immediate genes by tumor necrosis factorα contribute to liver repair following chemical-induced hepatotoxicity. Hepatology. 1997;25:133–141. doi: 10.1002/hep.510250125. [DOI] [PubMed] [Google Scholar]
- Brunner LJ, Bai S. Simple and rapid assay for acetaminophen and conjugated metabolites in low-volume serum samples. J Chromatogr B Biomed Sci Appl. 1999;732:323–329. doi: 10.1016/s0378-4347(99)00290-x. [DOI] [PubMed] [Google Scholar]
- Calvo M, Tebar F, Lopez-Iglesias C, Enrich C. Morphologic and functional characterization of caveolae in rat liver hepatocytes. Hepatology. 2001;33:1259–1269. doi: 10.1053/jhep.2001.23937. [DOI] [PubMed] [Google Scholar]
- Canbay A, Bechmann L, Gerken G. Lipid metabolism in the liver. Z Gastroenterol. 2007;45:35–41. doi: 10.1055/s-2006-927368. [DOI] [PubMed] [Google Scholar]
- Chiu H, Gardner CR, Dambach DM, Brittingham JA, Durham SK, Laskin JD, Laskin DL. Role of p55 tumor necrosis factor receptor 1 in acetaminophen-induced antioxidant defense. Am J Physiol Gastrointest Liver Physiol. 2003a;285:G959–G966. doi: 10.1152/ajpgi.00219.2003. [DOI] [PubMed] [Google Scholar]
- Chiu H, Gardner CR, Dambach DM, Durham SK, Brittingham JA, Laskin JD, Laskin DL. Role of tumor necrosis factor receptor 1 (p55) in hepatocyte proliferation during acetaminophen-induced toxicity in mice. Toxicol Appl Pharmacol. 2003b;193:218–227. doi: 10.1016/j.taap.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Cooper KO, Reik LM, Jayyosi Z, Bandiera S, Kelley M, Ryan DE, Daniel R, McCluskey SA, Levin W, Thomas PE. Regulation of two members of the steroid-inducible cytochrome P450 subfamily (3A) in rats. Arch Biochem Biophys. 1993;301:345–354. doi: 10.1006/abbi.1993.1154. [DOI] [PubMed] [Google Scholar]
- Cozma LG, Alexa ID, Dobrescu G. Transcriptional and electron microscopic analysis of extracellular matrix proteoglycans in acute acetaminophen intoxication. Rev Med Chir Soc Med Nat Iasi. 2004;108:452–457. [PubMed] [Google Scholar]
- Dambach DM, Durham SK, Laskin JD, Laskin DL. Distinct roles of NF-κB p50 in the regulation of acetaminophen-induced inflammatory mediator production and hepatotoxicity. Toxicol Appl Pharmacol. 2006;211:157–165. doi: 10.1016/j.taap.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Dambach DM, Watson LM, Gray KR, Durham SK, Laskin DL. Role of CCR2 in macrophage migration into the liver during acetaminophen-induced hepatotoxicity in the mouse. Hepatology. 2002;35:1093–1103. doi: 10.1053/jhep.2002.33162. [DOI] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Fakhrzadeh L, Laskin JD, Laskin DL. Regulation of caveolin-1 expression, nitric oxide production and tissue injury by tumor necrosis factor-alpha following ozone inhalation. Toxicol Appl Pharmacol. 2008;227:380–389. doi: 10.1016/j.taap.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- Fernandez MA, Albor C, Ingelmo-Torres M, Nixon SJ, Ferguson C, Kurzchalia T, Tebar F, Enrich C, Parton RG, Pol A. Caveolin-1 is essential for liver regeneration. Science. 2006;313:1628–1632. doi: 10.1126/science.1130773. [DOI] [PubMed] [Google Scholar]
- Ferret PJ, Hammoud R, Tulliez M, Tran A, Trebeden H, Jaffray P, Malassagne B, Calmus Y, Weill B, Batteux F. Detoxification of reactive oxygen species by a nonpeptidyl mimic of superoxide dismutase cures acetaminophen-induced acute liver failure in the mouse. Hepatology. 2001;33:1173–1180. doi: 10.1053/jhep.2001.24267. [DOI] [PubMed] [Google Scholar]
- Fujimoto K, Kumagai K, Ito K, Arakawa S, Ando Y, Oda S, Yamoto T, Manabe S. Sensitivity of liver injury in heterozygous Sod2 knockout mice treated with troglitazone or acetaminophen. Toxicol Pathol. 2009;37:193–200. doi: 10.1177/0192623308329282. [DOI] [PubMed] [Google Scholar]
- Gardner CR, Laskin DL. Sinusoidal cells in liver injury and repair. In: Sahu S, editor. Hepatotoxicity: from Genomics to In Vitro and In Vivo Models. John Wiley & Sons, Ltd; West Sussex: 2007. pp. 341–370. [Google Scholar]
- Gardner CR, Laskin JD, Dambach DM, Chiu H, Durham SK, Zhou P, Bruno M, Gerecke DR, Gordon MK, Laskin DL. Exaggerated hepatotoxicity of acetaminophen in mice lacking tumor necrosis factor receptor-1. Potential role of inflammatory mediators. Toxicol Appl Pharmacol. 2003;192:119–130. doi: 10.1016/s0041-008x(03)00273-4. [DOI] [PubMed] [Google Scholar]
- Gardner CR, Laskin JD, Dambach DM, Sacco M, Durham SK, Bruno MK, Cohen SD, Gordon MK, Gerecke DR, Zhou P, Laskin DL. Reduced hepatotoxicity of acetaminophen in mice lacking inducible nitric oxide synthase: potential role of tumor necrosis factor-α and interleukin-10. Toxicol Appl Pharmacol. 2002;184:27–36. [PubMed] [Google Scholar]
- Gassmann MG, Werner S. Caveolin-1 and -2 expression is differentially regulated in cultured keratinocytes and within the regenerating epidermis of cutaneous wounds. Exp Cell Res. 2000;258:23–32. doi: 10.1006/excr.2000.4904. [DOI] [PubMed] [Google Scholar]
- Gharaee-Kermani M, Phan SH. Role of cytokines and cytokine therapy in wound healing and fibrotic diseases. Curr Pharm Des. 2001;7:1083–1103. doi: 10.2174/1381612013397573. [DOI] [PubMed] [Google Scholar]
- Goetz JG, Lajoie P, Wiseman SM, Nabi IR. Caveolin-1 in tumor progression: the good, the bad and the ugly. Cancer Metastasis Rev. 2008;27:715–735. doi: 10.1007/s10555-008-9160-9. [DOI] [PubMed] [Google Scholar]
- Han D, Ybanez MD, Ahmadi S, Yeh K, Kaplowitz N. Redox regulation of tumor necrosis factor signaling. Antioxid Redox Signal. 2009;11:2245–2263. doi: 10.1089/ars.2009.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med. 2008;14:461–469. doi: 10.1016/j.molmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Haruyama T, Ajioka I, Akaike T, Watanabe Y. Regulation and significance of hepatocyte-derived matrix metalloproteinases in liver remodeling. Biochem Biophys Res Commun. 2000;272:681–686. doi: 10.1006/bbrc.2000.2837. [DOI] [PubMed] [Google Scholar]
- Hogaboam CM, Bone-Larson CL, Steinhauser ML, Matsukawa A, Gosling J, Boring L, Charo IF, Simpson KJ, Lukacs NW, Kunkel SL. Exaggerated hepatic injury due to acetaminophen challenge in mice lacking C-C chemokine receptor 2. Am J Pathol. 2000;156:1245–1252. doi: 10.1016/S0002-9440(10)64995-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol. 2008;84:1410–1421. doi: 10.1189/jlb.0308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke H. Glutathione disulfide formation and oxidant stress during acetaminophen-induced hepatotoxicity in mice in vivo: the protective effect of allopurinol. J Pharmacol Exp Ther. 1990;255:935–941. [PubMed] [Google Scholar]
- Koop DR. Hydroxylation of p-nitrophenol by rabbit ethanol-inducible cytochrome P-450 isozyme 3a. Mol Pharmacol. 1986;29:399–404. [PubMed] [Google Scholar]
- Koslowski R, Barth K, Augstein A, Tschernig T, Bargsten G, Aufderheide M, Kasper M. A new rat type I-like alveolar epithelial cell line R3/1: bleomycin effects on caveolin expression. Histochem Cell Biol. 2004;121:509–519. doi: 10.1007/s00418-004-0662-4. [DOI] [PubMed] [Google Scholar]
- Kronke G, Katzenbeisser J, Uderhardt S, Zaiss MM, Scholtysek C, Schabbauer G, Zarbock A, Koenders MI, Axmann R, Zwerina J, Baenckler HW, van den Berg W, Voll RE, Kuhn H, Joosten LA, Schett G. 12/15-lipoxygenase counteracts inflammation and tissue damage in arthritis. J Immunol. 2009;183:3383–3389. doi: 10.4049/jimmunol.0900327. [DOI] [PubMed] [Google Scholar]
- Laskin DL. Macrophages and inflammatory mediators in chemical toxicity: A battle of forces. Chem Res Toxicol. 2009;22:1376–1385. doi: 10.1021/tx900086v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskin DL, Gardner CR. Nonparenchymal cells, inflammatory macrophages, and hepatotoxicity. In: Kaplowitz N, DeLeve LD, editors. Drug-Induced Liver Disease. Informa Heatlthcare; New York: 2007. pp. 159–184. [Google Scholar]
- Le Lay S, Kurzchalia TV. Getting rid of caveolins: phenotypes of caveolin-deficient animals. Biochim Biophys Acta. 2005;1746:322–333. doi: 10.1016/j.bbamcr.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Ledda-Columbano GM, Curto M, Piga R, Zedda AI, Menegazzi M, Sartori C, Shinozuka H, Bluethmann H, Poli V, Ciliberto G, Columbano A. In vivo hepatocyte proliferation is inducible through a TNF and IL-6-independent pathway. Oncogene. 1998;17:1039–1044. doi: 10.1038/sj.onc.1202018. [DOI] [PubMed] [Google Scholar]
- Lee SS, Buters JT, Pineau T, Fernandez-Salguero P, Gonzalez FJ. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem. 1996;271:12063–12067. doi: 10.1074/jbc.271.20.12063. [DOI] [PubMed] [Google Scholar]
- Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- Liu ZX, Kaplowitz N. Role of innate immunity in acetaminophen-induced hepatotoxicity. Expert Opin Drug Metab Toxicol. 2006;2:493–503. doi: 10.1517/17425255.2.4.493. [DOI] [PubMed] [Google Scholar]
- Lopez-Rivera E, Lizarbe TR, Martinez-Moreno M, Lopez-Novoa JM, Rodriguez-Barbero A, Rodrigo J, Fernandez AP, Alvarez-Barrientos A, Lamas S, Zaragoza C. Matrix metalloproteinase 13 mediates nitric oxide activation of endothelial cell migration. Proc Natl Acad Sci USA. 2005;102:3685–3690. doi: 10.1073/pnas.0408217102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malerod L, Juvet K, Gjoen T, Berg T. The expression of scavenger receptor class B, type I (SR-BI) and caveolin-1 in parenchymal and nonparenchymal liver cells. Cell Tissue Res. 2002;307:173–180. doi: 10.1007/s00441-001-0476-9. [DOI] [PubMed] [Google Scholar]
- Mayoral R, Fernandez-Martinez A, Roy R, Bosca L, Martin-Sanz P. Dispensability and dynamics of caveolin-1 during liver regeneration and in isolated hepatic cells. Hepatology. 2007;46:813–822. doi: 10.1002/hep.21746. [DOI] [PubMed] [Google Scholar]
- Mehendale HM. Tissue repair: an important determinant of final outcome of toxicant-induced injury. Toxicol Pathol. 2005;33:41–51. doi: 10.1080/01926230590881808. [DOI] [PubMed] [Google Scholar]
- Mendieta-Condado E, Pichardo-Olvera M, Sanchez-Sevilla L, Chagoya de Sanchez V, Hernandez-Munoz R. Adenosine administration accelerates progression of the cell cycle during rat liver regeneration induced by one-third hepatectomy. J Pharmacol Exp Ther. 2009;331:122–132. doi: 10.1124/jpet.109.156620. [DOI] [PubMed] [Google Scholar]
- Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000–5005. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- Mladenovic D, Radosavljevic T, Ninkovic M, Vucevic D, Jesic-Vukicevic R, Todorovic V. Liver antioxidant capacity in the early phase of acute paracetamol-induced liver injury in mice. Food Chem Toxicol. 2009;47:866–870. doi: 10.1016/j.fct.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Nakae D, Yamamoto K, Yoshiji H, Kinugasa T, Maruyama H, Farber JL, Konishi Y. Liposome-encapsulated superoxide dismutase prevents liver necrosis induced by acetaminophen. Am J Pathol. 1990;136:787–795. [PMC free article] [PubMed] [Google Scholar]
- Odajima N, Betsuyaku T, Nasuhara Y, Nishimura M. Loss of caveolin-1 in bronchiolization in lung fibrosis. J Histochem Cytochem. 2007;55:899–909. doi: 10.1369/jhc.7A7203.2007. [DOI] [PubMed] [Google Scholar]
- Ogi M, Yokomori H, Oda M, Yoshimura K, Nomura M, Ohshima S, Akita M, Toda K, Ishii H. Distribution and localization of caveolin-1 in sinusoidal cells in rat liver. Med Electron Microsc. 2003;36:33–40. doi: 10.1007/s007950300004. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Kimura M, Watanabe N, Ogihara M. Tumor necrosis factor (TNF) receptor-2-mediated DNA synthesis and proliferation in primary cultures of adult rat hepatocytes: the involvement of endogenous transforming growth factor-α. Eur J Pharmacol. 2009;604:12–19. doi: 10.1016/j.ejphar.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Orfila C, Lepert JC, Alric L, Carrera G, Beraud M, Vinel JP, Pipy B. Expression of TNF-alpha and immunohistochemical distribution of hepatic macrophage surface markers in carbon tetrachloride-induced chronic liver injury in rats. Histochem J. 1999;31:677–685. doi: 10.1023/a:1003851821487. [DOI] [PubMed] [Google Scholar]
- Parat MO. The biology of caveolae: achievements and perspectives. Int Rev Cell Mol Biol. 2009;273:117–162. doi: 10.1016/S1937-6448(08)01804-2. [DOI] [PubMed] [Google Scholar]
- Pol A, Martin S, Fernandez MA, Ferguson C, Carozzi A, Luetterforst R, Enrich C, Parton RG. Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Mol Biol Cell. 2004;15:99–110. doi: 10.1091/mbc.E03-06-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quest AF, Gutierrez-Pajares JL, Torres VA. Caveolin-1: an ambiguous partner in cell signalling and cancer. J Cell Mol Med. 2008;12:1130–1150. doi: 10.1111/j.1582-4934.2008.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quest AF, Leyton L, Parraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem Cell Biol. 2004;82:129–144. doi: 10.1139/o03-071. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Kakar S. Histological patterns in drug-induced liver disease. J Clin Pathol. 2009;62:481–492. doi: 10.1136/jcp.2008.058248. [DOI] [PubMed] [Google Scholar]
- Reed JC. The survivin saga goes in vivo. J Clin Invest. 2001;108:965–969. doi: 10.1172/JCI14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly TP, Brady JN, Marchick MR, Bourdi M, George JW, Radonovich MF, Pise-Masison CA, Pohl LR. A protective role for cyclooxygenase-2 in drug-induced liver injury in mice. Chem Res Toxicol. 2001;14:1620–1628. doi: 10.1021/tx0155505. [DOI] [PubMed] [Google Scholar]
- Roderfeld M, Geier A, Dietrich CG, Siewert E, Jansen B, Gartung C, Roeb E. Cytokine blockade inhibits hepatic tissue inhibitor of metalloproteinase-1 expression and up-regulates matrix metalloproteinase-9 in toxic liver injury. Liver Int. 2006;26:579–586. doi: 10.1111/j.1478-3231.2006.01271.x. [DOI] [PubMed] [Google Scholar]
- Romano M. Lipid mediators: lipoxin and aspirin-triggered 15-epi-lipoxins. Inflamm Allergy Drug Targets. 2006;5:81–90. doi: 10.2174/187152806776383152. [DOI] [PubMed] [Google Scholar]
- Roudkenar MH, Halabian R, Ghasemipour Z, Roushandeh AM, Rouhbakhsh M, Nekogoftar M, Kuwahara Y, Fukumoto M, Shokrgozar MA. Neutrophil gelatinase-associated lipocalin acts as a protective factor against H2O2 toxicity. Arch Med Res. 2008;39:560–566. doi: 10.1016/j.arcmed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Roudkenar MH, Kuwahara Y, Baba T, Roushandeh AM, Ebishima S, Abe S, Ohkubo Y, Fukumoto M. Oxidative stress induced lipocalin 2 gene expression: addressing its expression under the harmful conditions. J Radiat Res (Tokyo) 2007;48:39–44. doi: 10.1269/jrr.06057. [DOI] [PubMed] [Google Scholar]
- Rutherford A, Chung RT. Acute liver failure: mechanisms of hepatocyte injury and regeneration. Semin Liver Dis. 2008;28:167–174. doi: 10.1055/s-2008-1073116. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Controlling the resolution of acute inflammation: a new genus of dual anti-inflammatory and proresolving mediators. J Periodontol. 2008;79:1520–1526. doi: 10.1902/jop.2008.080231. [DOI] [PubMed] [Google Scholar]
- Shah V, Cao S, Hendrickson H, Yao J, Katusic ZS. Regulation of hepatic eNOS by caveolin and calmodulin after bile duct ligation in rats. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1209–G1216. doi: 10.1152/ajpgi.2001.280.6.G1209. [DOI] [PubMed] [Google Scholar]
- Shatz M, Liscovitch M. Caveolin-1: a tumor-promoting role in human cancer. Int J Radiat Biol. 2008;84:177–189. doi: 10.1080/09553000701745293. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Togo S, Kumamoto T, Makino H, Morita T, Tanaka K, Kubota T, Ichikawa Y, Nagasima Y, Okazaki Y, Hayashizaki Y, Shimada H. Gene expression during liver regeneration after partial hepatectomy in mice lacking type 1 tumor necrosis factor receptor. J Surg Res. 2009;152:178–188. doi: 10.1016/j.jss.2007.12.785. [DOI] [PubMed] [Google Scholar]
- Torres VA, Tapia JC, Rodriguez DA, Parraga M, Lisboa P, Montoya M, Leyton L, Quest AF. Caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. J Cell Sci. 2006;119:1812–1823. doi: 10.1242/jcs.02894. [DOI] [PubMed] [Google Scholar]
- Vasiliou V, Ross D, Nebert DW. Update of the NAD(P)H:quinone oxidoreductase (NQO) gene family. Hum Genomics. 2006;2:329–335. doi: 10.1186/1479-7364-2-5-329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TM, Lisanti MP. The caveolin genes: from cell biology to medicine. Ann Med. 2004;36:584–595. doi: 10.1080/07853890410018899. [DOI] [PubMed] [Google Scholar]
- Wullaert A, van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor-κB: effects on liver homeostasis and beyond. Endocr Rev. 2007;28:365–386. doi: 10.1210/er.2006-0031. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Kirillova I, Peschon JJ, Fausto N. Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc Natl Acad Sci USA. 1997;94:1441–1446. doi: 10.1073/pnas.94.4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Naito M, Moriyama H, Umezu H, Matsuo H, Kiwada H, Arakawa M. Repopulation of murine Kupffer cells after intravenous administration of liposome-encapsulated dichloromethylene diphosphonate. Am J Pathol. 1996;149:1271–1286. [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa Y, Morita M, Hosomi H, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. Knockdown of superoxide dismutase 2 enhances acetaminophen-induced hepatotoxicity in rat. Toxicology. 2009;264:89–95. doi: 10.1016/j.tox.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med. 2009;30:29–41. doi: 10.1016/j.mam.2008.08.003. [DOI] [PubMed] [Google Scholar]