

Abstract
Myxoma (MYXV) and vaccinia virus (VACV) have recently emerged as potential oncolytic agents that can infect and kill different human cancer cells. Although both are structurally similar, it is unknown whether the pathway(s) used by these poxviruses to enter and cause oncolysis in cancer cells are mechanistically similar. Here, we compared the entry of MYXV and VACV-WR into various human cancer cells and observed significant differences: 1- Low pH treatment accelerates fusion-mediated entry of VACV but not MYXV, 2- The tyrosine kinase inhibitor genistein inhibits entry of VACV, but not MYXV, 3- Knockdown of PAK1 revealed that it is required for a late stage downstream of MYXV entry into cancer cells, whereas PAK1 is required for VACV entry into the same target cells. These results suggest that VACV and MYXV exploit different mechanisms to enter into human cancer cells, thus providing some rationale for their divergent cancer cell tropisms.
Keywords: Myxoma, Vaccinia, Entry-fusion complex, Endocytosis, Macropinocytosis, PAK1, Genistein, Tyrosine kinase
Introduction
Poxviruses are large enveloped dsDNA viruses that replicate exclusively in the cytoplasm of infected cells (Moss, 2007). Historically, specific members of the different poxvirus genera have been more intensively studied than others because of their implications in human or veterinary diseases, or for use as vaccines, vectors or platforms for diverse therapies. For instance, variola virus (VARV), from the genus Orthopoxvirus, has been extensively studied as the causative agent of smallpox. VARV was eventually eradicated by vaccination with vaccinia virus (VACV), another related poxvirus of the genus Orthopoxvirus (Moss, 2007). VACV is one of the best studied and well characterized poxviruses, which has been used as a prototypical model to investigate poxvirus biology in general.
Recently, VACV has emerged as a potential oncolytic agent because of its rapid life cycle, strong target cell killing activity, inherent ability to preferentially replicate within tumor tissues (particularly for attenuated variants of VACV), large cloning capacity, well defined molecular biology, and its capacity to infect a variety of human cancer types (Kirn and Thorne, 2009; Thorne, 2008). A second poxvirus called Myxoma virus (MYXV), from the genus Leporipoxvirus, has also emerged as a potential oncolytic agent for treatment of human cancers (Stanford and McFadden, 2007). In contrast to VACV, which can productively infect a wide range of mammalian hosts, MYXV, can infect only lagomorphs. In European rabbits (Oryctolagus cuniculus), MYXV causes a lethal disease called myxomatosis (Fenner and Ratcliffe, 1965; Stanford et al., 2007). Although MYXV does not induce any known pathology in humans, or any other non-lagomorph host, this virus can efficiently replicate in vitro in a variety of transformed human cancer cells lines (Barrett et al., 2007a; Wang et al., 2006; Woo et al., 2008). The ability of MYXV to specifically infect and clear human cancer tissues in vivo has also been confirmed in immunodeficient mouse models using various xenografted human tumors (Lun et al., 2005; Lun et al., 2007; Wu et al., 2008). Because it has not been reported to induce any toxicity for humans, MYXV has also emerged as a promising candidate for virotherapy to treat a variety of human cancers, however unlike VACV, the molecular characteristics of MYXV infection have not been completely characterized.
One of the key tropism steps that can influence the ability of an oncolytic virus to distinguish normal cells from tumor cells is virus entry. Thus, elucidating and comparing the entry mechanism(s) used by MYXV and VACV to gain entry into human cancer cells is critical for advancing these two poxviruses as oncolytic agents. Recently, a significant number of studies have shed light on the mechanism(s) used by VACV to bind and enter into host mammalian cells, the majority of which have been performed with the intracellular mature virion (MV) form of VACV, since this is the most abundant and stable infectious form (Moss, 2006). Initial binding of VACV MVs to mammalian cells occurs by mechanisms that can be dependent on either cell surface glycosaminoglycans (GAGs) or other still-unidentified cellular moieties (Carter et al., 2005; Chiu et al., 2007; Chung et al., 1998 Chung et al., 2005; Foo et al., 2009; Hsiao et al., 1999). Studies using electron microscopy originally demonstrated that the VACV MVs from the Western Reserve (WR) and modified virus Ankara (MVA) strains enter cells by direct fusion with the plasma membrane (Carter et al., 2005). Later, it was confirmed that the fusion of VACV MV to the plasma membrane can occur at neutral pH, and is mediated by a multi-protein fusion complex carried by the virion (Moss, 2006). The number of viral proteins that comprise the poxviral entry-fusion complex (EFC) has continued to grow. For example, Sathehkumar and Moss provided recent evidence that VACV O3L (VACVWR069.5), a short open reading frame (ORF) of just 35 amino-acids, which possesses orthologs in MYXV (Cameron et al., 1999) and molluscum contagiosum virus (Senkevich, et al., 1997), is the newest identified integral component of the EFC, that is required for MV entry and membrane fusion (Satheshkumar and Moss, 2009).
Additionally, poxvirus MVs can also utilize a low-pH endosomal entry pathway (Townsley et al., 2006) but, interestingly, some strains of VACV cannot exploit this latter pathway (Bengali et al., 2009). A recent report suggests that VACV MV entry uses macropinocytosis, a transient growth factor-induced, and actin-dependent endocytic mechanism, which is utilized by large particles such as bacteria, apoptotic bodies, necrotic cells and viruses to penetrate into the cytoplasm of mammalian cells (Mercer and Helenius, 2008). The induction of macropinocytosis is preceded by the rapid activation of signaling pathways via tyrosine kinase receptors that ultimately trigger rearrangements of actin filaments at the plasma membrane, which favors the internalization of interacting particles (Mercer and Helenius, 2009).
In contrast to VACV, the mechanism(s) whereby MYXV enters mammalian cells have yet to be studied in detail, although the known virus-encoded members of the viral EFC identified in VACV are highly conserved in MYXV. With this in mind, we first sought to establish if there were any differences in the entry mechanisms between the MYXV Lausanne (MYXV-Lau) strain and the VACV Western Reserve (VACV-WR) strain, using several different human cancer cells as a model system. We report that substantial differences exist between these viruses with regard to the effects of low pH, and the requirement for endosomal acidification for entry. We used specific kinase inhibitors to demonstrate differences in the drug sensitivity to MV entry of both viruses. Specifically, we found that genistein, a tyrosine kinase inhibitor that blocks macropinocytosis, specifically inhibits the entry of MV particles from VACV, but not MYXV, into the same target cancer cells. These findings will further help our understanding of the various oncolytic mechanisms exploited by poxviruses and facilitate their development for oncolytic virotherapy.
Results
Replication of MYXV and VACV is cell-type dependent
Both MYXV and attenuated variants of VACV are potential oncolytic virus candidates for treatment of human cancers. In order to understand the mechanism of entry used by both viruses in various human cancer cells, we started this study comparing the infectivity of MYXV and VACV in several selected human cancer cell lines: A549 (lung carcinoma cells), HeLa (cervical carcinoma), Panc1 (pancreatic carcinoma) and BJAB (EBV-negative Burkitt B-cell lymphoma). We first used fluorescence microscopy to compare the ability of MYXV and VACV tagged with both EGFP (early/late expression) and Tomato Red (late expression only) to infect and spread within different cancer cell types. To do this, the indicator cells were infected with the recombinant vMyx-GFP-TrFP or VACV-GFP-TrFP at MOI of 0.1 and evaluated by fluorescence microscopy 48 hours post-infection (hpi). In each case, expression of GFP was a measure of early-late viral gene expression and TrFP was a measure of late gene expression only. We observed significant differences in infection progression by the viruses in some of the cancer cell types (Fig. 1). For instance, in A549 and HeLa cells, both MYXV (Fig. 1A) and VACV (Fig. 1B) appeared to initiate a permissive infection and underwent normal cell-to-cell spread. As well, the expected increase in the infectious progeny virus was observed for both viruses from A549 cells (Fig. 2A), which was comparable to titers in the control BGMK cells (Fig. 2B). In contrast, fluorescence microscopy analysis revealed the expression of both EGFP and TrFP from vMyx-GFP-TrFP and VACV-GFP-TrFP infection of BJAB (Fig.1A and 1B, respectively), however, single step growth curves revealed that infection of BJAB cells with MYXV did not produce new infectious virions while infection of these cells with VACV significantly fewer new virions then BGMK cells (Fig. 2C). The most notable differences between MYXV and VACV were observed with Panc1 cells, which supported productive replication for VACV but were completely nonpermissive for MYXV (Fig. 1A and 1B and Fig. 2D). Taken together, these results demonstrate that these viruses can differ significantly in terms of gene expression and progeny virus production in certain specific human cancer cells.
Fig. 1.

MYXV and VACV replication and spread is cell line-dependent. Selected human cancer cell lines were infected with purified MV of (A) vMyx-GFP-TrFP, or (B) VACV-GFP-TrFP MVs, at a multiplicity of 0.1 (MOI 0.1). At 48 hour after infection the formation of foci (for MYXV) or plaques (for VACV) was visualized by fluorescence microscopy.
Fig. 2.

One-step growth of MYXV and VACV is cell line-specific. Single growth curves were generated to evaluate the replication of MYXV and VACV in (A) A549, (B) BGMK (C) BJAB and (D) Panc1 cells. Cells were infected with either vMyx-GFP-TrFP or VACV-GFP-TrFP at a multiplicity of 5.0. Titers are expressed as log PFU/106 cells. The single growth curve for BGMK was used as a positive control, since this cell line is optimally permissive for both viruses.
MYXV entry into cancer cells, unlike VACV, is not stimulated by acidic pH
The entry mechanism(s) used by the mature virions (MVs) of VACV have been actively investigated in recent years. In this regard, Vanderplasschen and co-workers first reported that, while VACV MVs can enter cells by fusing with the plasma membrane at neutral pH, VACV EEVs use a low pH as the entry pathway (Vanderplasschen et al., 1998). Further studies conducted by Townsley and co-workers also demonstrated that VACV MVs can use the endocytic route via low-pH to enter to the cells, (Townsley et al., 2006). This latter conclusion is based on the evidence that a brief low-pH treatment (i.e. pH 5.0) accelerates VACV MV entry into host cells. In contrast, the role of pH has not been previously tested for MYXV. To determine whether MYXV entry also exploits this low-pH endosomal route, the rate of virus entry was quantified at neutral and acidic pH by measuring luciferase expression from either vMyx-GLuc (expressing Gaussia luciferase from a synthetic early/late promoter), or VACV-FLuc (expressing Firefly luciferase from the same promoter). This assay allowed us to compare the entry kinetics of each virus under different conditions.
In our initial studies, virus adsorption was synchronized by incubation of either, VACV-FLuc, or vMyx-GLuc to HeLa cells at 4°C for one hour. Then each virus was incubated at 37°C to allow the synthesis of the indicator luciferases. Although under these conditions, we were able to readily measure the VACV-FLuc luciferase activity (Supplemental Fig. S1C), we did not detect any vMyx-GLuc luciferase activity, (Supplemental Fig S1A). Next, we tested the ability of both viruses to enter the cells at room temperature (25°C), and observed similar results. Only when the adsorption of MYXV proceeded at 37°C we were able to measure any viral luciferase activity (Supplemental Fig S1A) indicating successful MYXV entry. To determine if this result was a peculiarity of HeLa cells, we next tested RK13 cells, a rabbit cell line permissive for MYXV and VACV. Virus entry was synchronized as described above. Surprisingly, we obtained the same results with RK13 cells (Supplemental Fig. S1, panels B and D) as with HeLa cells, suggesting that in contrast to VACV, MYXV has uniquely different temperature requirements for either binding and/or entry.
In order to evaluate the effect of low-pH on MYXV entry, purified vMyx-GLuc MVs were adsorbed to the cells at 37°C and then were exposed to pH 5.0, at 37°C for 3 min. On the other hand, purified VACV-FLuc MVs were adsorbed to the cells at 4°C and then exposed to pH 5.0 for 3 min at 37°C. Consistent with previously reported results, a brief exposure to pH 5.0 resulted in a modest increase in the rate of VACV entry into both A549 (Fig. 3C), and HeLa (Fig. 3D) cells. In contrast, the same treatment actually decreases Gaussia luciferase expression from vMyx-GLuc in both A549 (Fig. 3A) and HeLa (Fig. 3B) cells. This difference was not due to adsorption of VACV at 4°C since adsorption of this virus at 37°C yielded comparable results (data not shown).
Fig. 3.

VACV and MYXV entry into cancer cells is differentially affected by brief low-pH exposure. A549 cells (panels A and C) and HeLa cells (panels B and D) were infected with vMyx-GLuc (panels A and B) or VACV-FLuc (panels C and D) at MOI of 5.0. After virus adsorption to the cells, unbound virus was removed and cells were washed with PBS followed by 3 min treatment in neutral pH (7.4) or low pH (5.0) at 37°C. Cells were washed in neutral pH media and the infection was allowed to proceed for different time points (2, 4, and 6 hours). At the given time point, cells were assayed for luciferase activity. Data represent the average ± standard error of samples in triplicate. Luciferase activity is reported in light units (LU).
Because the pH of the endosome varies from pH 6.0 for early endosomes to pH 5.0 for late endosomes, we tested the effect of a wide range of pHs starting from 5.0, 5.5, 6.0, 6.5, 7.0 and 7.4, to determine if MYXV entry could be activated at an acidic pH higher than 5.0. Interestingly, we found that pH 7.4 is the optimal pH for MYXV (Lausanne strain) to enter these cells, and that in contrast to VACV (Western Reserve strain), lower pHs have an inhibitory effect on MYXV entry, with pH 5.0 being the most inhibitory pH (results not shown). Because activation at low pHs can be time dependent, we measured luciferase activity at different time points starting at 1, 2, 3, 4, and 5 hours after virus adsorption. Interestingly, we always observed that a low pH had an inhibitory effect on MYXV entry (results not shown). Therefore, we conclude that the inhibitory effect on MYXV entry observed at acidic pHs is time independent.
Previous studies performed with VACV MVs revealed that the stimulation of virus entry through low-pH treatments pre-and post virus adsorption were not additive (Townsley and Moss, 2007). We found that two consecutive exposures of vMyx-GLuc to pH 5.0 (i.e. one prior virus adsorption, and the other after virus adsorption) exposure of at 37°C for 3 min did not increase Gaussia luciferase activity (Fig. S2A). On the other hand, treatment of VACV-FLuc MVs at pH 5.0 followed by adsorption at 4°C and then a second exposure to either, pH 7.4 or 5.0 resulted in similar Luc activities, although two consecutive treatments at pH 5.0 (pre-adsorption and post-adsorption) resulted in a slight increase in the VACV luciferase activity (Fig. S2B). Taken together these results suggest that MYXV and VACV binding and/or entry to at least some human cancer cells occurs by nonidentical mechanisms.
Finally, to directly address the potential for acid activation of the fusion machinery, we assessed the ability of MYXV to form syncytia at low pH. Our results demonstrate that MYXV(Lausanne strain), does not form syncytia at low pH (e.g. 4.6) whereas in contrast, VACV (Western Reserve strain), does form syncytia at the acidic pH 4.6, as expected (results not shown). Thus, we are confident that the entry differences we are reporting between VACV-WR and MYXV are biologically significant.
Inhibition of endosomal acidification affects both MYXV and VACV entry
Above, we have shown that in contrast to VACV entry, MYXV entry is not accelerated by low-pH treatment, (Fig. 3, Fig. S1, and Fig. S2). These data, however, do not rule out the possibility that MYXV uses an endocytic pathway as a means to enter cells. Previous studies have provided evidence for the uptake of VACV via the endocytic pathway (Townsley and Moss, 2007; Townsley et al., 2006). If MYXV uses the endocytic pathway, pharmacological inhibition of this pathway by inhibiting the relevant V-ATPases should also inhibit MYXV entry. To test this, we treated cells with bafilomycin-A1 and concanamycin-A, two specific inhibitors of V-ATPase (Bowman et al., 1988; Dröse and Altendorf, 1997),that is responsible for intracellular acidification (Klee et al., 1999), and determined the effects this treatment had on entry of MYXV and VACV (Fig. 4). Interestingly, at neutral pH, a decrease in the luciferase activity was observed for both viruses following treatment with either drug in a dose dependent manner, confirming previous observations performed with VACV (Townsley et al., 2006). To determine whether a low-pH treatment of bound MVs could bypass the inhibitory effect exerted by these drugs, both viruses were briefly exposed to pH 5.0 at 37°C for 3 min after binding. Significantly, we found that only the inhibition of VACV MV entry was alleviated under these conditions, while entry of MYXV remained constitutively blocked. These results not only confirm the ability of VACV to use the endocytic pathway for entry to cells, but also suggest the possibility that MYXV exploits the endocytic route to enter into cancer cells in a fashion that cannot be circumvented by transient low pH treatment.
Fig. 4.

Inhibitors of endosomal acidification decrease both VACV and MYXV entry. HeLa cells were pretreated with the indicated inhibitor for 1 hour at 37°C. While VACV-FLuc (panels C and D) was adsorbed to the cells at 4°C for 1 hour, vMyx-GLuc (panels A and B) was adsorbed to the cells at 37°C for 1 hour. Un-adsorbed viruses were removed and cells treated with either neutral pH (7.4), or acidic pH (5.0) in identical fashion as described elsewhere in this manuscript. Two hours after infection, cells were assayed for luciferase activity. Data represent the average of triplicates ± standard error.
To further access whether MYXV uses the endocytic route as a means to enter the test cancer cells, the effect on viral entry was determined following treatment with pepstatin A, which inhibits the proteolysis of aspartic acid proteases, (Binkert et al., 2006). Interestingly, only a very modest decrease in the luciferase activity for MYXV-GLuc and VACV-FLuc was observed, suggesting that that MYXV infection does not require the activation of cellular acid-dependent endosomal proteases to enter cells and initiate early gene expression (results not shown). These results were somewhat unexpected,since it is well known that pepstatin A inhibits the proteolysis of aspartic acid proteases that in turn inhibit endosomal acidification (Zaidi et al., 2007), which should affect significantly VACV entry via the endocytic route. However, Okada and co-workers reported that pepstatin A also displays an alternative role inducing extracellular acidification, which is unrelated to its inhibition of aspartic proteases (Okada et al., 2003). We conlcude that the external acidification produced via pepstatin does not significantly affect MYXV and VACV entry into these cancer cells.
Genistein specifically compromises VACV, but not MYXV, entry into cancer cells
Recent studies have suggested that VACV MVs use macropinocytosis to enter host cells although the exact role of the virion phospholipids and “apoptosis mimicry” in this process remains controversial (Mercer and Helenius, 2008; Laliberte and Moss, 2009). Macropinocytosis, an actin-dependent endocytic process, is characterized by changes in the dynamics of actin filaments, which produce morphological changes of the plasma membrane. Macropinocytosis is induced by external stimuli that trigger the activation of receptor tyrosine kinases, depending on the size and nature of the stimulating ligands. In addition to this, re-arrangements of actin are regulated via the activation of phosphatidylinositol (PI) 3-kinase, and the Rho-family of small GTPases (Mercer and Helenius, 2009). The use of inhibitors that target either the kinases involved in macropinocytosis, or actin formation, has shed light into the mechanism(s) used by VACV MVs to enter host cells. To compare the effects of these inhibitors on MYXV and VACV entry, a select group of pharmacological inhibitors that act on a number of targets including tyrosine and serine-threonine kinases, disruption of actin polymerization, and inhibition of Na+/H+ exchange, were tested for their effects on viral entry (Table 1).
Table 1.
Drug inhibitors used for this study
| Inhibitor name and abbreviation | Description |
|---|---|
| K252a | An ATP analog that has been described as a serine/threonine kinase inhibitor and as a selective and potent inhibitor different members of the tyrosine kinase family (Morotti et al., 2002; Tapley et al., 1992). |
| Tyrphostin (AG17) | Tyrosine kinase inhibitor that decreases the levels of p21 in OCI-Ly8 immunoblastic lymphoma cell line. Induces apoptosis (Palumbo et al., 1997). |
| Wortmannin (WORT) | PI3-Kinase inhibitor (Chen and Wang, 2001). |
| Cytochalasin D (CTC-D) | An actin-disrupting drug that affects macropinocytosis (Mercer and Helenius, 2008; Stevenson and Begg, 1994). |
| Brefeldin A (BFA) | Fungal macrocyclic lactone that affects the activation of the small GTPase Arf1 (Damm et al., 2005). |
| 5-(N-Ethyl-N-isopropyl)-amiloride (EIPA) | Blocks Na+/H+ exchanger and a known inhibitor of macropinocytosis (Fretz et al., 2006; Nakase et al., 2004). |
| Staurosporin (STAU) | A protein kinase C (PKC) inhibitor and an inducer of apoptosis (Constantineseua et al., 1991; Wasilenko et al., 2001). |
| Genistein | Tyrosine kinase inhibitor (soy flavonoid) that blocks many different viruses at various stages (Andres et al., 2009). |
| Blebbistatin (BB) | A myosin II-depending inhibitor that inhibits VACV entry (Mercer and Helenius, 2008). |
HeLa or A549 cells were treated with 100 μM of inhibitor for 1 hour at 37°C, and then virus was adsorbed for 1 hour with the respective virus in the presence of the inhibitor. After virus adsorption, cells were washed and incubated at 37°C for 2 hours to allow the synthesis of luciferase and then assayed for luciferase activity. The results from this drug inhibitor screen revealed some intriguing differences between VACV and MYXV (Fig. 5). For example, we found that each virus responded differently to the tyrosine kinase inhibitor genistein in a cell line independent manner (Fig. 5, last columns in each panel). While VACV-FLuc entry into HeLa or A549 cells was dramatically inhibited by genistein (Fig. 5A and 5B, last columns marked with an asterisk, respectively), MYXV-GLuc entry was relatively unaffected by genistein (Fig. 5C, and 5D, last columns). To investigate if the divergent effects of genistein on the two viruses might be kinetic in nature, luciferase experiments were performed for A549 (Fig. 6A and 6B) and HeLa (Fig. 6C and 6D) cells at different time points. Consistent with the former results, genistein did not inhibit MYXV entry at any time point (Fig. 6A and 6C) whereas VACV entry was almost completely eliminated (Fig. 6B and 6D). These results clearly demonstrate that MYXV and VACV respond differently to gensitein, suggesting that the kinase targets of this drug are uniquely required for the entry and/or early viral gene expression of VACV, but not MYXV.
Fig. 5.

MYXV and VACV entry is affected differentially by drug inhibitors. Prior to infection, cells HeLa (panels A and C) and A549 (panels B and D) were treated with each indicated inhibitor at the appropriate concentration during 1 hour at 37°C. Either vMyx-GLuc (panels C and D), or VACV-FLuc (panels A and B) was adsorbed to the cells at 37°C for 1 hour in the presence of inhibitor. After adsorption, cells were washed and then incubated with media supplemented with the appropriate inhibitor for 1 hour. Cells were subsequently assayed for luciferase activity. Cycloheximide (CHX) was added to the cells to prevent the early viral protein synthesis and serves as an internal control to quantify newly synthesized luciferase. Data represent the average ± standard error of samples in triplicate, and are expressed in light units (LU).
Fig.6.

Genistein differentially affects MYXV and VACV entry in a time-independent manner. Kinetic experiments were performed for A549 (panels A and B) and HeLa (panels C and D). Cells pre-treated with genistein were infected with vMyx-GLuc (panels A and C) or VACV-FLuc (panels B and D) for 1 hour, at 37°C. At the given time points post-infection, luciferase activity is expressed in light units, (LU).
Low-pH exposure decreases MYXV entry even in the presence of genistein
Recently, it was demonstrated that the inhibition produced by genistein on VACV MV entry could be bypassed by transient low pH exposure (Mercer and Helenius, 2008) suggesting that, at least for VACV, this inhibitor blocks virus entry. Here, we have shown that MYXV entry is instead inhibited by low-pH exposure (Fig. 3A and 3C) and that genistein does not significantly affect MYXV entry (Fig. 5).
Effect of genistein on MYXV replication at late times
Our luciferase reporter results suggest that genistein specifically affects VACV entry but not that of MYXV. To determine if genistein affects later stages of MYXV replication and progeny virus production, we first analyzed the spread of virus infection in cancer cells in the presence of genistein. To do this, HeLa cells were infected with vMyx-GFP-TrFP or VACV-GFP-TrFP at a MOI of 0.1. At 48 hpi, the synthesis of EGFP and TrFP gene was examined by fluorescence microscopy. As expected, genistein dramatically inhibited VACV early (EGFP) and late (TrFP) gene expression, whereas MYXV was not affected (Fig. 7A). We also measured the effect of genistein on the percent of EGFP-positive cells of both viruses using flow cytometry following infection with each virus at a MOI of 0.1. The results demonstrate that although genistein does not affect the extent of the number of MYXV GFP+ cells, it dramatically reduced the percent of vaccinia GFP+ cells (Fig. 7B).
Fig. 7.

Genistein differentially affects MYXV and VACV. (A) HeLa cells were treated with genistein for 1 hour and then infected with vMyx-GFP-TrFP or VACV-GFP-TrFP at a MOI of 1.0. After 1 hour of virus adsorption, cells were incubated in a media containing genistein. Virus propagation was assessed by fluorescence microscopy 72 hours post-infection. (B) To measure cell-cell spread, the percentage of GFP+ cells in untreated (no genistein) and genistein-treated samples was determined by flow cytometry. Cells were infected with each virus at a MOI of 1.0, trypsinized and fixed in formaldehyde before analysis. (C) and (D): To investigate the effect of genistein on MYXV and VACV virus titers, cells were harvested at the given time points and then lysed by repeated freeze-thawing. Virus titers were determined as described in Materials and Methods.
Next, we investigated the effect of genistein on the replication of both viruses in a single step growth cycle in HeLa cells infected with each virus at a MOI of 5.0. Cells were collected at the indicated times points after infection and the infectious progeny virus titrated on indicator BSC-40 cells. Although both GFP and TrFP were synthesized (Fig. 7A), MYXV progeny virus synthesis was essentially blocked by genistein (Fig. 7C). As expected, VACV progeny were not synthesized in the presence of gensitein (Fig. 7D). These results suggest that, unlike VACV, the entry of MYXV is not blocked by genistein but this inhibitor does cause a post-replicative late block to MYXV progeny virus assembly. In fact, this was demonstrated by two means: (i) the expression kinetics for representative early and late MYXV and VACV genes was analyzed by real-time PCR (Supplemental Fig. S3, panels A and B, respectively), and (ii) Western blot analysis (Supplemental Fig. S4, panels A and B, respectively), which revealed that genistein differentially affected early and late MYXV and VACV protein synthesis. Taken together, our results demonstrate that genistein inhibits MYXV not at entry, but rather at a post-entry stage, whereas in contrast VACV is blocked by this drug at the early stage of virus entry.
MYXV, but not VACV, entry into cancer cells is not dependent on PAK1
PAK1 is reported as an essential cell kinase factor for entry of VACV MV (Mercer and Helenius, 2008). To determine the requirement for PAK1 in MYXV entry, we used small interfering RNA (siRNA) to knock down PAK1 in HeLa cells, which was assessed at the protein level by Western blot analysis (Fig. 8A, lane 3). Both non-targeting siRNA (NT siRNA) (Fig. 8A, lane 2), and untreated cells [Mock (-)], (Fig. 8A, lane 1), were used as controls. The effect of knocking down PAK1 significantly reduced VACV early and late gene expression (Fig. 8C) much more extensively than for MYXV (Fig. 8B) The results obtained for MYXV in HeLa cells are consistent with previous studies performed with mouse NIH3T3 murine fibroblasts, which revealed that PAK1 is required for a late stage of MYXV replication downsteam of binding and entry (Johnston et al., 2003). To further confirm that PAK1 is not required for MYXV entry into cancer cells, we measured luciferase activity for vMyx-GLuc (Fig. 8D) and VACV-FLuc (Fig. 8E). At 72 hours after PAK1 siRNA transfection, HeLa cells were infected with either vMyx-GLuc or VACV-FLuc at an MOI of 5.0. Two hours post-infection, cells were assayed for luciferase activity (Fig. 8D and 8E). Knockdown of PAK1 caused only modest reduction in MYXV entry that did not reach significance (Fig. 8D), in contrast, VACV entry was significantly decreased (Fig. 8E). In general, these results demonstrate that PAK1 is required for VACV entry but does not play a critical role in MYXV entry, although it is required for a late stage in MYXV replication, which confirm previous findings of our group (Johnston et al., 2003). The specific requirement of PAK1 for VACV entry into cancer cells but not that for MYXV, constitutes another difference between the entry of these viruses.
Fig. 8.

Entry of MYXV and VACV into cancer cells is differentially affected by knockdown of PAK1. (A) Representative Western blots confirming PAK1 knock down. HeLa cells were transfected for 72 hours with 50 nM siRNA directed against PAK1 (lane 3), or against a non-targeting siRNA, (NT siRNA), (lane 2). Untransfected cells are referred as mock (-) (lane 1). Equal sample loading was confirmed by detection of the housekeeping protein actin. Replication of vMyx-GFP-TrFP (panel B) and VACV-GFP-TrFP (panel C) in HeLa cells was evaluated by fluorescence microscopy. 72 hours after transfection, cells were infected with the recombinant fluorescent viruses at a MOI of 1.0. Formation of MYXV foci, or VACV plaques was evaluated 48 hpi. Entry of MYXV-GLuc (D) or VACV-FLuc (E) into cells was assessed 72 hours post-transfection by measuring luciferase activity after 2 hours infection with vMyx-GLuc or VACV-FLuc at MOI of 5.0.
Discussion
Binding and entry into a cell are key steps for successful viral replication and progression of infection. It has been reported that most chordopoxviruses can bind and enter a much wider variety of mammalian cells in vitro than is predicted based on their host tropisms in vivo, and to date no specific cell surface receptor for cell entry by poxviruses has been reliably reported (McFadden, 2005). However, differences in poxvirus binding and entry have been noted even between similar strains of VACV into the same cells (Bengali et al., 2009), suggesting that the binding and entry stage might indeed play a role in mediating cellular tropism by poxviruses. This issue is particularly important for viruses that are being exploited for oncolytic virotherapy, and may play a discriminatory role in how such viruses distinguish normal vs cancerous cells. We initiated this study to establish if differences might exist between MYXV and VACV in their modes to infect and replicate in a select test group of human cancer cells. In this regard, we found that infection with MYXV is selectively restrictive to some cancer cells, such as pancreatic cancer cells (Panc1), whereas VACV can infect a larger selection of human cancer cell types. These initial results prompted us to focus on the early stages of infection, in particular virion entry, with the goal to establish any differences between these viruses.
The poxviruses MYXV (Lausanne strain), and VACV (WR strain) exploit nonidentical mechanisms to enter into the same human cancer cells. Using early promoter driven luciferase expression assays, we found that entry of MYXV in cancer cells is uniquely inhibited at low pH. In contrast, low pH treatment significantly accelerated VACV entry into the same cells. Interestingly, the use of inhibitors of endosomal acidification, like bafilomycin A1, and concanamycin-A, two inhibitors of the vacuolar H+-ATPase (Dröse and Altendorf, 1997), reduced both VACV and MYXV entry in a dose-dependent manner, suggesting that both viruses utilize an endocytic route of entry. However, they differ significantly in their ability to be stimulated by low pH. VACV, under low pH treatment, can overcome the inhibitory effects mediated by these inhibitors of the endosomal acidification, whereas MYXV is unable to do so.
During poxvirus infection of mammalian cells, rapid cell signaling events are induced that participate in virus entry, including the activation of a wide range of tyrosine and serine-threonine kinases (like PAK1), which were found to play pivotal roles in VACV entry (Mercer and Helenius, 2008). In this regard, we confirmed that genistein, a soy isoflavonoid and a tyrosine kinase inhibitor, blocks VACV entry as reported previously (Mercer and Helenius, 2008) but here we demonstrate that genistein does not inhibit MYXV entry into the same cancer cells. Nevertheless, genistein is still inhibitory to MYXV replication, but blocks the virus growth cycle at a later post-entry stage. The fact that genistein selectively affects VACV but not MYXV entry and alters MYXV replication only at late stage(s), suggests that this inhibitor may target intracellular molecules that are differentially required for VACV and MYXV replication. Mercer and Helenius reported evidence supporting the effect of genistein on reducing the phosphorylation of PAK1 and the concomitant reduction of VACV MV entry (Mercer and Helenius, 2008). This seemed to be in contrast to a previous publication by our lab (Johnston et al., 2003) showing that PAK1 was required for MYXV replication in mouse cells only at later (i.e. post-entry) stages of virus replication. In this study, we show that blocking PAK1 expression by siRNA was indeed selectively inhibitory to the entry of VACV, but not MYXV, into the same human cancer cells.
Recent reports have claimed that VACV uses macropinocytosis as an efficient entry mechanism that allows the virus to subvert the innate cellular apoptotic response pathway to gain entry into mammalian cells (Mercer and Helenius, 2008) but the exact role of externalized phosphatidyl serine on the input virions has been questioned (Laliberte and Moss, 2009). Since macropinocytosis is an actin- and tyrosine kinase-dependent mechanism, we tested whether various inhibitors could selectively block cellular pathways required for VACV, but not MYXV, entry into human cancer cells. Our data support a role for genistein in inhibiting VACV at the entry level, while for MYXV the effect of this inhibitor is not at the entry step but rather at a much later post-entry level. This result allows us to make a functional differentiation between these two poxviruses at the level of entry into cancer cells. The role of isoflavones like genistein at inhibiting the replication of many distinct viruses has been well documented, ranging from virus binding, (Andres et al., 2007; Hayashi et al., 1997) and entry (Kubo et al., 2003; Li et al., 2000; Sharma-Walia et al., 2004) to virus replication (Evers et al., 2005; Kim et al., 2006; Robin et al., 2001; Yura et al., 1993), viral protein translation, as well as the formation of viral glycoproteins complexes (Andres et al., 2007). Here we show that the kinase targets for genistein are required at very different levels for VACV (i.e. entry) and MYXV (i.e. late virus assembly).
Regardless of the mechanism, the results presented here uncover new insights with regard to entry of poxviruses to cancer cells. This is of considerable interest because both MYXV and attenuated variants of VACV are currently being developed for oncolytic virotherapy, and these two viruses exhibit very distinct oncolytic properties (Kirn and Thorne, 2009; Stanford and McFadden, 2007). Establishing the mechanism(s) by which MYXV and VACV can differentially enter and initiate infections of human cancer cells and thereby exert oncolysis in different models of cancer needs to be addressed in future studies.
Materials and Methods
Cell culture and infections
All cells used in this study, were grown at 37°C in a humidified 5% CO2 atmosphere. Unless otherwise mentioned, all media used in this study were always supplemented with 100 μg/mL penicillin-streptomycin, and 2 mM L-glutamine. CV-1 cells (ATCC# CCL70) were cultured in Minimum Essential Medium (MEM; Gibco BRL, Grand Island, NY) supplemented with 1X MEM nonessential amino acids (Cellgro, Mediatech, Herndon, VA), 1 mM sodium pyruvate and 5% fetal bovine serum (FBS); (Gibco BRL). The cell lines HeLa (ATCC# CCL-2), A549 (ATCC# CCL-185), RK13 (ATCC CCL-37), BSC-40 (a generous gift of Dr. Richard Condit), and BGMK (Wang et al., 2008) were maintained in Dulbecco's modified Eagle's medium, (DMEM) supplemented with 10% FBS. BJAB (a generous gift of Dr. Sankar Swaminathan) and Panc1 (ATCC# CRL-1469) cells were maintained in Iscove's Modified Dulbecco's Medium (IMDM) (Invitrogen) supplemented 20% FBS).
Pharmacological Drugs
Unless otherwise noted in the text, each pharmacological inhibitor was added to the cells 1 hour before virus adsorption and maintained throughout the course of the experiment. The inhibitors blebbistatin (BB), staurosporine (STAU), wortmannin (WORT), cytochalasin-D (CTC-D), AG17, K252a, ethylisopropyl amiloride (EIPA), brefeldin A (BFA), genistein, bafilomycin A1, concanamycin A, , cycloheximide (CHX) and cytosine arabinoside (AraC) were obtained from Sigma-Aldrich. Pepstatin A was obtained from Fisher.
Viruses
All experiments were performed with MYXV, Lausanne strain, and VACV, Western Reserve (WR) strain. Recombinant vMyx-GFP-TrFP expressing both enhanced green fluorescent protein (EGFP) driven by a synthetic early/late poxvirus promoter and Tomato red fluorescent protein (TrFP) driven by poxvirus P11 late promoter has been described previously (Bartee et al., 2009). Recombinant VACV expressing both EGFP (under synthetic E/L promoter) and TrFP (under P11 late promoter) was created by an intergenic insertion of the TrFP. Recombinant vaccinia virus (WR) expressing the firefly luciferase under a synthetic poxvirus early-late promoter (VACV-FLuc) was generated by Dr. Peter Turner (Turner and Moyer, 2008) and was a kind gift from Dr. Dick Moyer (Department of Molecular Genetics and Microbiology, University of Florida). Recombinant myxoma virus expressing the Gaussia luciferase driven by a poxvirus synthesis early/late promoter (vMyx-GLuc) was constructed in the following manner. The coding regions for monomeric Cherry red fluorescent protein (mCherry, from R. Tsien) and Guassia luciferase (Nanolight) were separated by a “translational slip” sequence (2A) from foot and mouth disease virus (FMDV) such that the single transcript encoded two independent proteins. This RFP/luciferase cassette was amplified from the expression plasmid pT-REx RFP2AGLuc (gift from Dr. Stojdl, Children's Hospital of Eastern Ontario) using primers JB08.05. GCGCTGCAGAAAAATTGAAATTTTATTTTTTTTTTTTGGAATATAAATAATGGTGTCCAAGGGGGAGGAGGAC (Pst1 italics and vaccinia virus synthetic early/late promoter is underlined) and JB07.05 GCGCTGCAGTTAGTCACCCCCGGCTCCCTTAATC (Pst1 site italicized). The amplified product was cloned into the Pst1 site of the plasmid pBS:63KO-GFP which has been described previously (Barrett et al., 2007b). This new transfer vector, pBS:63KO E/L Cherry2AGLuc, was transfected into BGMK cells that had been infected with vMyx-Lau one hour earlier. Infected/transfected cells were collected 48 h post transfection and recombinant virus, expressing mCherry and GLuc were purified through successive rounds of plaque purification on BGMK cells. For the present study, all virus stocks were grown in CV-1 cells and purified by centrifugation through a sucrose cushion and two successive sucrose gradient sedimentations as described previously (Earl et al., 1998).
Luciferase assays
Luciferase assays were performed using either MYXV gaussia luciferase virus (vMyx-GLuc) or vaccinia firefly luciferase (VACV-FLuc). A total of 2.5 × 104 cells of the indicated cell line were seeded in each well of white bottom 96-well plate (Fisher). Cells were infected with each virus at a MOI of 5.0 for 1 hour at 37°C. Cells were subsequently washed with 1X PBS and incubated with fresh media. At the indicated time point, equal amount of either, Firefly substrate BriteLite™ plus assay kit, (PerkinElmer), or coelenterazine, the substrate for Gaussia luciferase, (New England BioLabs Inc) mixed with lysis buffer was added to the cells and incubated for 1 min or 10 sec. Bioluminescence signal intensity was quantified on a Thermo Appliskan, 100-240V luminometer (Thermo Electron corp.). Each experiment was performed at least three times in triplicate. Luciferase activity is reported in light units (LU).
Fluorescence microscopy and flow cytometry
A total of 2.5 × 104 cells of the respective cancer cell line were plated in each well of a 96-well plate. The following day, cells were infected with either vMyx-GFP-TrFP, or VACV-GFP-TrFP at MOI of 0.1 and 1.0. At 24, 48, and 72 hours after infection the sizes and shapes of GFP, and TrFP foci for MYXV and plaques for VACV were observed using a Leica DMI 6000B microscope. For FACS analysis, 72 hours post-infection, cells were harvested using 1% trypsin and fixed in 2% para-formaldehyde. Percent of GFP+ cells was quantified by using flow cytometry on a BD FACSCalibur.
Real-Time PCR
Real-time PCR experiments were performed as described previously (Bartee et al., 2009). Briefly, 2 μL of total RNA was used to prepare cDNA. Genomic DNA was removed from total RNA, using the DNA free kit (Ambion) according to the manufacturer's recommendations. Subsequently, 1 μL of the deoxynucleoside triphosphates (100 mM) and 1 μL of random hexamer primers (50 μg/mL) were added, and the mixture was incubated for 5 min at 65°C. After this incubation, the tube was allowed to cool at room temperature, and 6 μL of 5X reaction buffer, 3 μL dithiothreitol (0.1 M), 1 μL of RNasin (Promega), and 1 μL of Superscript III reverse transcriptase (Invitrogen) were added. The resulting mixture was incubated for 1 hour at 42°C and then for 15 min at 72°C. The final reaction was diluted 1:10 with sterile water and used for Sybr green-based real time PCR. Then, 4 μL of diluted cDNA was added to 21 μL of PCR mix containing 0.5 U of Taq polymerase (NEB), 1X Thermo Pol buffer, 0.1X Sybr green dye (Molecular Probes), 0.5X Rox reference dye (Invitrogen), a 100 μL concentration of deoxynucleoside triphosphates (Invitrogen), 4 mM MgCl2, (Invitrogen), 4 ng of forward primer and 4 ng of reverse primer. The resulting 25 μg reaction was run on an ABL 7300 real-time PCR machine under the following conditions: 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Primers used in real-time PCR are listed in table 2.
Table 2.
Primers used for real-time PCR
| Primer | ||
|---|---|---|
| Gene |
|
|
| Direction* | Sequence (5'-3') | |
| MYXV M-T7 | For | CGTGGATCAATGTGTGTGAA |
| MYXV M-T7 | Rev | CAAGACACGACGTCCAAATC |
| MYXV SERP-1 | For | CGTGACGTTTAACTCGGAGA |
| MYXV SERP-1 | Rev | CTCGTCTTCATACGAACGGA, |
| VACV F11L | For | ACAGGATTCGTCATTCCAGA |
| VACV F11L | Rev | CAATTCCAATTGTTGCCTGT |
| VACV F17R | For | TGCATCTGCTCATACTCCGT |
| VACV F17R | Rev | GGGCGATGAGGGTTTATCTA |
For, forward; Rev, reverse
Virus titration
MYXV and VACV viruses were propagated and titrated by focus and plaque formation, respectively on BSC-40 cells as described previously (Opgenorth et al., 1992).Briefly, each cell line was seeded in 6-well dish during one overnight. Cells were then infected with either, vMyx-GFP-TrFP, or VACV-GFP-TrFP at an MOI of 5.0. At the given time points post-infection [e.g. 0 hpi (1 hour after virus adsorption), 4, 8, 12, 24, and 48 hpi], cells were harvested and frozen. To release the virus, cells were lysed three times via freeze-thawing and then sonicated. Virus titers were determined by using the limiting dilution method on BSC-40 cells.
Virus infection at low pH
Cells were mock treated or treated with gensitein at the optimal concentration (100 μM) for 1 hour at 37°C. Subsequently, vMyx-GLuc, or VACV-FLuc was adsorbed to the cells. Cells were then washed with cold PBS, followed by 5 min incubation at 37°C and 7.4 and 5.0. Subsequently, cells were washed in neutral pH media (e.g. 7.4), and infections were allowed to proceed for the indicated time with or without inhibitor. Cells were then processed for luciferase activity. When used, cycloheximide (CHX), (100 μg/mL) was added as a control for the early protein synthesis.
Inhibition of endosomal acidification
Cells (2×104 per well) seeded in 96 well plates were pretreated for 1 hour at 37°C with different concentrations of bafilomycin A1 (BFA1), concanamycin A, in DMEM or mock treated (no inhibitor). VACV MV was adsorbed to the cells at MOI of 5.0 at 4°C for 1 hour. MYXV was adsorbed to the cells at MOI of 5.0 for 1 hour, at 37°C. After virus adsorption, cells were washed with PBS and then incubated during 5 min in neutral pH (7.4) or acidic pH (5.0), at 37°C. Cells incubated at 37°C and pH 7.4 in the presence of the respective inhibitor. Luciferase assays were performed 2 hours after virus adsorption.
siRNA transfection
A validated siRNA against PAK1 (TCCACTGATTGCTGCAGCTAA); and a non-targeting siRNA (NT-siRNA) were obtained from Qiagen. Shortly before transfection, a total of 2×105 HeLa cells were plated in each well of six well plates (2.3 mL volume). Cells were transfected according to manufacturer's specifications, by mixing 50 nM of each siRNA, with Hiperfect transfection reagent from Qiagen. 72 hours after transfection, cells were infected with recombinant, double label fluorescent viruses at an MOI of 1.0 to access virus spread. To investigate virus entry to the cells, recombinant vMyx-GLuc, or VACV-FLuc viruses were used at an MOI of 5.0. For fluorescence experiments the foci were evaluated 48 hpi. Luciferase activity was measured 2 hpi.
Western blot analysis
Cells (e.g. untreated, or treated with different inhibitors) were infected with either vMyx-GFP-TrFP, or VACV-GFP-TrFP at a MOI of 5 for 1 hour at 37°C, and harvested at 2, 4, and 24 hpi to monitor viral protein expression. Harvested cells were suspended in RIPA buffer [20 mM Tris-HCl pH 7.5, 137 mM NaCl, 15% glycerol, 20 mM NaF, 1% NP40, 10 mM NaPPi, 1% NaV3O4, 1% phenylmethylsulphonyl fluoride, 25 mM β-glycerolphosphate, 0.25% sodium deoxycholate, inhibitor cocktail (Sigma-Aldrich), a complete Mini, EDTA-free tablet (Roche) and nanopure water]. Suspensions were sonicated to solubilize proteins. The levels of protein were quantified using the Bradford assay (Bio Rad), and 25 μg of each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Separated proteins were transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad) and blocked for 1 hour at room temperature with 5% non-fat dry milk in TBST (25 mM Tris-buffered saline with 0.1% Tween 20). Primary antibodies against each viral protein were diluted in 5% milk-TBST and incubated with the membranes for 2 hours at room temperature. After washing, membranes were incubated for 1 hour at room temperature with horseradish peroxidase-conjugated secondary antibodies diluted 1/5,000 in 5% milk-TBST. To monitor the expression of PAK1, membranes were incubated overnight at 4°C with the primary antibody rabbit polyclonal against PAK1 (Cell Signaling Technologies), diluted 1/1,000 in 5% bovine serum albumin-TBST. Membranes were then incubated for 1 hour with the respective secondary antibody in 5% milk-TBST. Western blots were analyzed using the chemiluminescence reagent (Millipore). Loading of equal amounts of protein from each sample was confirmed by detection of β Actin. To control early and late viral protein expression, 1.0 mg/mL of cycloheximide (CHX) and 50 μg/mL cytosine arabinoside (AraC) were used, respectively.
Supplementary Material
Fig. S1. Efficient adsorption MYXV to human and rabbit cell lines requires 37°C. HeLa cells (panels A and C) and rabbit RK13 cells (panels B and D) were incubated with vMyx-GLuc (panels A and B) or VACV-FLuc (panels C and D), at 4°C, room temperature, or 37°C for 1 hour, and pH 7.4. After this, cells were washed and incubated with neutral pre-warmed media. Cells then were incubated at 37°C and the infection was allowed to proceed for 2 hours prior measuring luciferase activity.
{kind=link}
Fig. S2. Brief exposure at low-pH followed by virus adsorption specifically enhances entry by VACV but not MYXV. Before virus adsorption to the cells, vMyx-GLuc and VACV-FLuc MVs were exposed to pH 5.0 for 3 min at 37°C and subsequently neutralized with excess of media. Subsequently each virus was adsorbed to the cells for 1 hour. Cells were then washed and treated with either pH 7.4, or pH 5.0 for 3 min at 37°C. Cells were infected with either, (A) vMyx-GLuc, or with (B) VACV-FLuc and incubated at 37°C for 2 hours, then lysed, and assayed for luciferase activity.
{kind=link}
Fig. S3. Genistein blocks the synthesis of early and late VACV mRNAs but only reduces the expression of late MYXV genes. HeLa cells were mock treated (no inhibitor) or treated with genistein for 1 hr, then infected with MYXV-GFP (panel A) or VACV-GFP (panel B) At the given time points, RNA was extracted from each sample and used to synthesize cDNA. The expression of each viral early gene (M-T7 for MYXV or F11L for VACV) or late gene (SERP-1 for MYXV or F17R for VACV) at each time point was assayed with gene-specific primers using Sybr green-based real-time PCR as detailed in Materials and Methods. The graphs in panels A and B show the average of experiments in triplicate.
{kind=link}
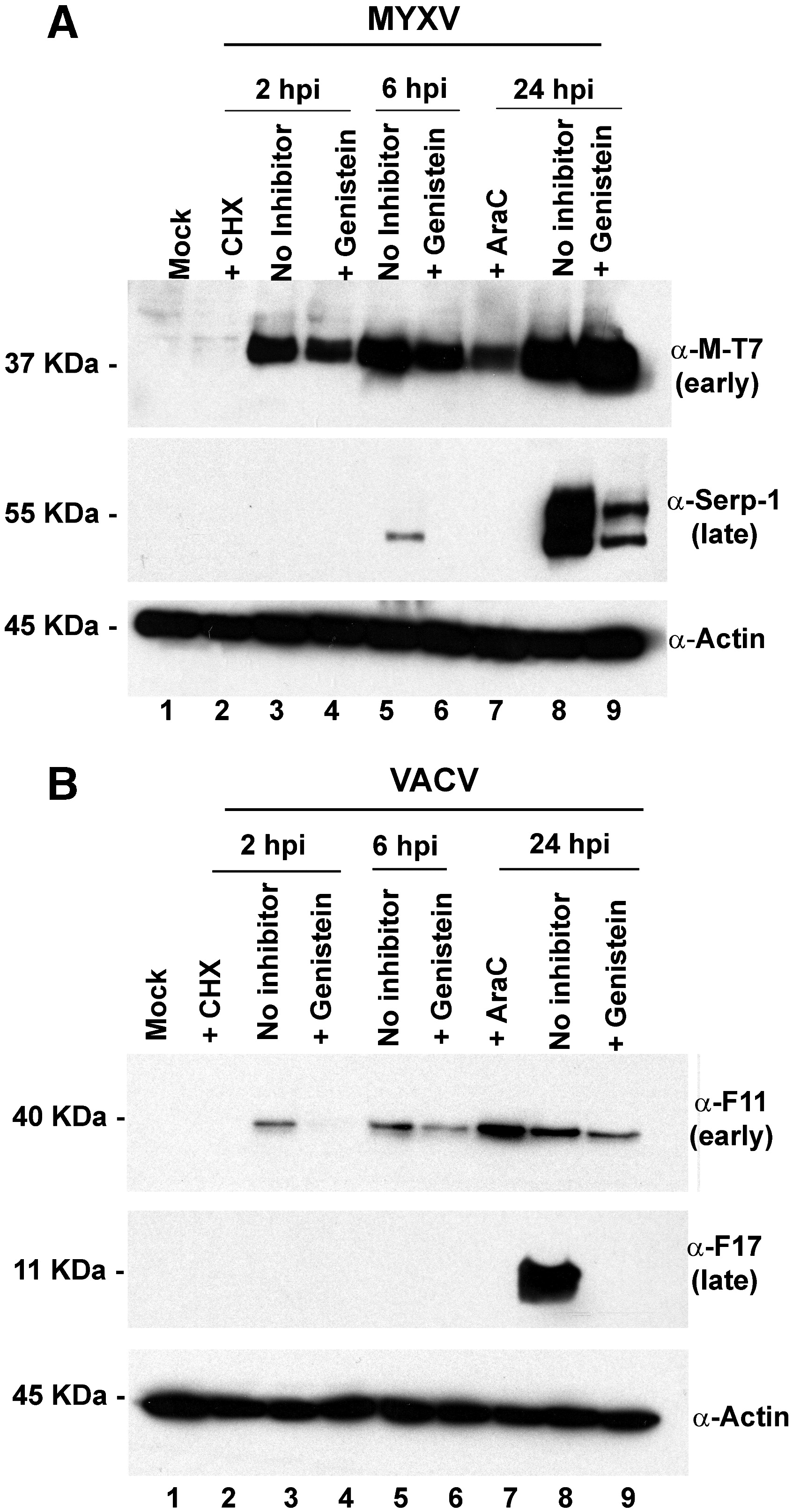
Fig. S4. Genistein blocks early and late VACV protein synthesis but only reduces late MYXV protein expression. HeLa cells were mock treated (no inhibitor) or treated with 100 μM genistein, and then infected with (A) vMyx-GFP-TrFP, or (B) VACV-GFP-TrFP at a MOI of 5.0. At the given time points post-infection, cells were harvested and lysed with RIPA buffer. Representative early and late MYXV and VACV protein synthesis was analyzed by Western blots. When used, 100 μg/mL cycloheximide (CHX) (lane 2), and 50 μg/mL cytosine arabinoside (AraC) (lane 7) were added to the cells to prevent early and late protein synthesis, respectively. Untreated cells (no inhibitor), are shown in lanes 3, 5, and 8; and lanes 4, 6, and 9 show the cells treated with 100 μg/mL genistein. Equal sample loading was confirmed by detection of actin.
{kind=link}
Acknowledgements
We are very grateful with the following researchers for providing reagents, Dr. Peter Turner and Dr. Dick Moyer for the VACV firefly-luciferase construct, Dr. Richard Condit for BSC-40 cells and the antibodies against vaccinia F11 and F17, Dr. Stojdl for the plasmid pT-REx RFP2AGLuc, Dr. Sankar Swaminathan for BJAB cell line. We are grateful to D. Smith for technical support. This work was supported by the NIH and by start-up funds (to GM) from the College of Medicine of the University of Florida.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
MYXV and VACV entry into cancer cells
References
- Andres A, Donovan SM, Kuhlenschmidt MS. Soy isoflavones and virus infections. J. Nutr, Biochem. 2009;20:563–569. doi: 10.1016/j.jnutbio.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres A, Donovan SM, Kuhlenschmidt TB, Kuhlenschmidt MS. Isoflavones at concentrations present in soy infant formula inhibit rotavirus infection in vitro. J Nutr. 2007;137:2068–73. doi: 10.1093/jn/137.9.2068. [DOI] [PubMed] [Google Scholar]
- Barrett JW, Alston LR, Wang F, Stanford MM, Gilbert PA, Gao X, Jimenez J, Villeneuve D, Forsyth P, McFadden G. Identification of host range mutants of myxoma virus with altered oncolytic potential in human glioma cells. J. Neurovirol. 2007a;13:549–560. doi: 10.1080/13550280701591526. [DOI] [PubMed] [Google Scholar]
- Barrett JW, Sypula J, Wang F, Alston LR, Shao Z, Gao X, Irvine TS, McFadden G. Myxoma virus M063R is a host range gene essential for virus replication in rabbit cells. Virology. 2007b;361:123–132. doi: 10.1016/j.virol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Bartee E, Mohamed RM, Lopez MC, Baker HV, McFadden G. The Addition of Tumor Necrosis Factor plus Beta Interferon Induces a Novel Synergistic Antiviral State against Poxviruses in Primary Human Fibroblasts. J. Virol. 2009;83:498–511. doi: 10.1128/JVI.01376-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengali Z, Townsley AC, Moss B. Vaccinia virus strain differences in cell attachment and entry. Virology. 2009;389:132–140. doi: 10.1016/j.virol.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binkert C, Frigerio M, Jones A, Meyer S, Pesenti C, Prade L, Viani F, Zanda M. Replacement of isobutyl by trifluoromethyl in pepstatin A selectively affects inhibition of aspartic proteinases. Chembiochem. 2006;7:181–186. doi: 10.1002/cbic.200500180. [DOI] [PubMed] [Google Scholar]
- Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci U S A. 1988;85:7972–7976. doi: 10.1073/pnas.85.21.7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron C, Hota-Mitchell S, Chen L, Barrett J, Cao JX, Macaulay C, Willer D, Evans D, McFadden G. The complete DNA sequence of myxoma virus. Virology. 1999;264:298–318. doi: 10.1006/viro.1999.0001. [DOI] [PubMed] [Google Scholar]
- Carter GC, Law M, Hollinshead M, Smith GL. Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J. Gen. Virol. 2005;86(Pt 5):1279–90. doi: 10.1099/vir.0.80831-0. [DOI] [PubMed] [Google Scholar]
- Chen X, Wang Z. Regulation of intracellular trafficking of the EGF receptor by Rab5 in the absence of phosphatidylinositol 3-kinase activity. EMBO. 2001;15:68–74. doi: 10.1093/embo-reports/kve005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WL, Lin CL, Yang MH, Tzou DL, Chang W. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J. Virol. 2007;81:2149–2157. doi: 10.1128/JVI.02302-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CS, Hsiao JC, Chang YS, Chang W. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 1998;72:577–585. doi: 10.1128/jvi.72.2.1577-1585.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CS, Huang CY, Chang W. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 2005;79:1623–1634. doi: 10.1128/JVI.79.3.1623-1634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinescua SN, Cernescub CD, Popescu LM. Effects of protein kinase C inhibitors on viral entry and infectivity. FEBS Letters. 1991;292:31–33. doi: 10.1016/0014-5793(91)80826-o. [DOI] [PubMed] [Google Scholar]
- Damm E, Pelkmans L, Kartenbeck J, Mezzacasa A, Kurzchalia T, Helenius A. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J. Cell. Biol. 2005;168:477–488. doi: 10.1083/jcb.200407113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröse S, Altendorf K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 1997;200(Pt 1):1–8. doi: 10.1242/jeb.200.1.1. [DOI] [PubMed] [Google Scholar]
- Earl PL, Cooper N, Wyatt S, Moss B, Carroll MW. Preparation of cell cultures and vaccinia virus stocks. In: R. B., Ausubel FM, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current protocols in molecular biology. Vol. 2. John Wiley and Sons; New York, N.Y.: 1998. pp. 16.16.1–16.16.3. [DOI] [PubMed] [Google Scholar]
- Evers DL, Chao CF, Wang X, Zhang Z, Huong SM, Huang ES. Human cytomegalovirus-inhibitory flavonoids: studies on antiviral activity and mechanism of action. Antiviral Res. 2005;68:124–134. doi: 10.1016/j.antiviral.2005.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner F, Ratcliffe FN. Myxomatosis. Cambridge University Press; Cambridge, UK: 1965. [Google Scholar]
- Foo CH, Lou H, Whitbeck JC, Ponce-de-León M, Atanasiu D, Eisenberg RJ, Cohen GH. Vaccinia virus L1 binds to cell surfaces and blocks virus entry independently of glycosaminoglycans. Virology. 2009;385:368–382. doi: 10.1016/j.virol.2008.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fretz M, Jin J, Conibere R, Penning NA, Al-Taei S, Storm G, Futaki S, Takeuchi T, Nakase I, Jones AT. Effects of Na+/H+ exchanger inhibitors on subcellular localisation of endocytic organelles and intracellular dynamics of protein transduction domains HIV-TAT peptide and octaarginine. J Control Release. 2006;116:247–254. doi: 10.1016/j.jconrel.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Hayashi T, Otsuka H, Takeda Y. Antiviral activity of 5,6,7-trimethoxyflavone and its potentiation of the antiherpes activity of acyclovir. J Antimicrob Chemother. 1997;39:821–824. doi: 10.1093/jac/39.6.821. [DOI] [PubMed] [Google Scholar]
- Hsiao JC, Chung CS, Chang W. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 1999;73:8750–8761. doi: 10.1128/jvi.73.10.8750-8761.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB, Barrett JW, Chang W, Chung CS, Zeng W, Masters J, Mann M, Wang F, Cao J, McFadden G. Role of the Serine-Threonine Kinase PAK-1 in Myxoma Virus Replication. J. Virol. 2003;77:5877–5888. doi: 10.1128/JVI.77.10.5877-5888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee R, Heinemann U, Eder C. Voltage-gated proton currents in microglia of distinct morphology and functional state. Neuroscience. 1999;4:1415–1424. doi: 10.1016/s0306-4522(98)00710-6. [DOI] [PubMed] [Google Scholar]
- Kim SY, Kim DH, Hyun JW, Henson JW, Kim HS. Irisolidone, an isoflavone metabolite, represses JC virus gene expression via inhibition of Sp1 binding in human glial cells. Biochem Biophys Res Commun. 2006;344:3–8. doi: 10.1016/j.bbrc.2006.03.165. [DOI] [PubMed] [Google Scholar]
- Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Ishimoto A, Amanuma H. Genistein, a protein tyrosine kinase inhibitor, suppresses the fusogenicity of Moloney murine leukemia virus envelope protein in XC cells. Arch Virol. 2003;148:1899–914. doi: 10.1007/s00705-003-0164-z. [DOI] [PubMed] [Google Scholar]
- Laliberte JP, Moss B. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. U S A. 2009;106:17517–17521. doi: 10.1073/pnas.0909376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Stupack DG, Brown SL, Klemke R, Schlaepfer DD, Nemerow GR. Association of p130CAS with phosphatidylinositol-3-OH kinase mediates adenovirus cell entry. J Biol Chem. 2000;275:14729–35. doi: 10.1074/jbc.275.19.14729. [DOI] [PubMed] [Google Scholar]
- Lun X, Yang W, Alain T, Shi ZQ, Muzik H, Barrett JW, McFadden G, Bell J, Hamilton MG, Senger DL, Forsyth PA. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65:9982–9990. doi: 10.1158/0008-5472.CAN-05-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun XQ, Zhou H, Alain T, Sun B, Wang L, Barrett JW, Stanford MM, McFadden G, Bell J, Senger DL, Forsyth PA. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67(18):8818–8827. doi: 10.1158/0008-5472.CAN-07-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden G. Poxvirus tropism. Nat Rev Microbiol. 2005;3:201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- Mercer J, Helenius A. Virus entry by macropinocytosis. Nat Cell Biol. 2009;11:510–520. doi: 10.1038/ncb0509-510. [DOI] [PubMed] [Google Scholar]
- Morotti A, Mila S, Accornero P, Tagliabue E, Ponzetto C. K252a inhibits the oncogenic properties of Met, the HGF receptor. Oncogene. 2002;21:4885–4893. doi: 10.1038/sj.onc.1205622. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxvirus entry and membrane fusion. Virology. 2006;344:48–54. doi: 10.1016/j.virol.2005.09.037. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5th ed. Vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 2905–2946. [Google Scholar]
- Nakase I, Niwa M, Takeuchi T, Sonomura K, Kawabata N, Koike Y, Takehashi M, Tanaka S, Ueda K, Simpson JC, Jones AT, Sugiura Y, Futaki S. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004;10:1011–1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Okada M, Irie S, Sawada M, Urae R, Urae A, Iwata N, Ozaki N, Akazawa K, Nakanishi H. Pepstatin A Induces Extracellular Acidification Distinct From Aspartic Protease Inhibition in Microglial Cell Lines. Glia. 2003;43:167–174. doi: 10.1002/glia.10237. [DOI] [PubMed] [Google Scholar]
- Opgenorth A, Graham K, Nation N, Strayer D, McFadden G. Deletion analysis of two tandemly arranged virulence genes in myxoma virus, M11L and myxoma growth factor. J. Virol. 1992;66:4720–4731. doi: 10.1128/jvi.66.8.4720-4731.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo GA, Yarom N, Gazit A, Sandalon Z, Baniyash M, Kleinberger-Doron N, Levitzki A, Ben-Yehuda D. The tryphostin AG17 induces apoptosis and inhibition of cdk2 activity in a lymphoma cell line that overexpresses bcl-2. Cancer Res. 1997;57:2434–2439. [PubMed] [Google Scholar]
- Robin V, Irurzun A, Amoros M, Boustie J, Carrasco L. Antipoliovirus flavonoids from Psiadia dentata. Antiviral Chem Chemother. 2001;12:283–291. doi: 10.1177/095632020101200503. [DOI] [PubMed] [Google Scholar]
- Satheshkumar PS, Moss B. Characterization of a newly identified 35-amino-acid component of the vaccinia virus entry/fusion complex conserved in all chordopoxviruses. J. Virol. 2009;83:12822–12832. doi: 10.1128/JVI.01744-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senkevich TG, Koonin EV, Bugert JJ, Darai G, Moss B. The genome of molluscum contagiosum virus: analysis and comparison with other poxviruses. Virology. 1997;233:19–42. doi: 10.1006/viro.1997.8607. [DOI] [PubMed] [Google Scholar]
- Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L, Chandran B. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 2004;78:4207–23. doi: 10.1128/JVI.78.8.4207-4223.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford MM, McFadden G. Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer. Expert. Opin. Biol. Ther. 2007;7:1415–1425. doi: 10.1517/14712598.7.9.1415. [DOI] [PubMed] [Google Scholar]
- Stanford MM, Werden SJ, McFadden G. Myxoma virus in the European rabbit: interactions between the virus and its susceptible host. Vet. Res. 2007;38:299–318. doi: 10.1051/vetres:2006054. [DOI] [PubMed] [Google Scholar]
- Stevenson BR, Begg DA. Concentration-dependent effects of cytochalasin actin filaments in MDCK epithelial cells. J. Cell. Sci. 1994;107:367–375. doi: 10.1242/jcs.107.3.367. [DOI] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting Temporal and Spatial Control of Cytokinesis with a Myosin II Inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Thorne S. Oncolytic vaccinia virus: from bedside to benchtop and back. Curr Opin Mol Ther. 2008;10:387–392. [PubMed] [Google Scholar]
- Townsley AC, Moss B. Two Distinct Low-pH Steps Promote Entry of Vaccinia Virus. J. Virol. 2007;81:8613–8620. doi: 10.1128/JVI.00606-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley AC, Weisberg AS, Wagenaar TR, Moss B. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J. Virol. 2006;80:8899–8908. doi: 10.1128/JVI.01053-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner PC, Moyer RW. The vaccinia virus fusion inhibitor proteins SPI-3 (K2) and HA (A56) expressed by infected cells reduce the entry of superinfecting virus. Virology. 2008;380:226–233. doi: 10.1016/j.virol.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderplasschen A, Hollinshead M, Smith GL. Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J. Gen. Virol. 1998;79:877–887. doi: 10.1099/0022-1317-79-4-877. [DOI] [PubMed] [Google Scholar]
- Zaidi N, Burster T, Sommandas, Herrmann V, Boehm T, B.O., Driessen C, Voelter W, Kalbacher H. A novel cell penetrating aspartic protease inhibitor blocks processing and presentation of tetanus toxoid more efficiently than pepstatin A. Biochem. Biophys. Res. Commun. 2007;364:243–249. doi: 10.1016/j.bbrc.2007.09.114. [DOI] [PubMed] [Google Scholar]
- Wang G, Barrett JW, Stanford M, Werden SJ, Johnston JB, Gao X, Sun M, Cheng JQ, McFadden G. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. U S A. 2006;103:4640–4645. doi: 10.1073/pnas.0509341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilenko ST, Meyers AF, Vander Helm K, Barry M. Vaccinia virus infection disarms the mitochondrion-mediated pathway of the apoptotic cascade by modulating the permeability transition pore. J. Virol. 2001;75:11437–11448. doi: 10.1128/JVI.75.23.11437-11448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo Y, Kelly KJ, Stanford MM, Galanis C, Chun YS, Fong Y, McFadden G. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann. Surg. Oncol. 2008;15:2329–2335. doi: 10.1245/s10434-008-9924-z. [DOI] [PubMed] [Google Scholar]
- Wu Y, Lun X, Zhou H, Wang L, Sun B, Bell JC, Barrett JW, McFadden G, Biegel JA, Senger DL, Forsyth PA. Oncolytic efficacy of recombinant vesicular stomatitis virus and myxoma virus in experimental models of rhabdoid tumors. Clin. Cancer, Res. 2008;14:1218–1227. doi: 10.1158/1078-0432.CCR-07-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yura Y, Yoshida H, Sato M. Inhibition of herpes simplex virus replication by genistein, an inhibitor of protein-tyrosine kinase. Arch Virology. 1993;132:451–461. doi: 10.1007/BF01309554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Efficient adsorption MYXV to human and rabbit cell lines requires 37°C. HeLa cells (panels A and C) and rabbit RK13 cells (panels B and D) were incubated with vMyx-GLuc (panels A and B) or VACV-FLuc (panels C and D), at 4°C, room temperature, or 37°C for 1 hour, and pH 7.4. After this, cells were washed and incubated with neutral pre-warmed media. Cells then were incubated at 37°C and the infection was allowed to proceed for 2 hours prior measuring luciferase activity.
Fig. S2. Brief exposure at low-pH followed by virus adsorption specifically enhances entry by VACV but not MYXV. Before virus adsorption to the cells, vMyx-GLuc and VACV-FLuc MVs were exposed to pH 5.0 for 3 min at 37°C and subsequently neutralized with excess of media. Subsequently each virus was adsorbed to the cells for 1 hour. Cells were then washed and treated with either pH 7.4, or pH 5.0 for 3 min at 37°C. Cells were infected with either, (A) vMyx-GLuc, or with (B) VACV-FLuc and incubated at 37°C for 2 hours, then lysed, and assayed for luciferase activity.
Fig. S3. Genistein blocks the synthesis of early and late VACV mRNAs but only reduces the expression of late MYXV genes. HeLa cells were mock treated (no inhibitor) or treated with genistein for 1 hr, then infected with MYXV-GFP (panel A) or VACV-GFP (panel B) At the given time points, RNA was extracted from each sample and used to synthesize cDNA. The expression of each viral early gene (M-T7 for MYXV or F11L for VACV) or late gene (SERP-1 for MYXV or F17R for VACV) at each time point was assayed with gene-specific primers using Sybr green-based real-time PCR as detailed in Materials and Methods. The graphs in panels A and B show the average of experiments in triplicate.
Fig. S4. Genistein blocks early and late VACV protein synthesis but only reduces late MYXV protein expression. HeLa cells were mock treated (no inhibitor) or treated with 100 μM genistein, and then infected with (A) vMyx-GFP-TrFP, or (B) VACV-GFP-TrFP at a MOI of 5.0. At the given time points post-infection, cells were harvested and lysed with RIPA buffer. Representative early and late MYXV and VACV protein synthesis was analyzed by Western blots. When used, 100 μg/mL cycloheximide (CHX) (lane 2), and 50 μg/mL cytosine arabinoside (AraC) (lane 7) were added to the cells to prevent early and late protein synthesis, respectively. Untreated cells (no inhibitor), are shown in lanes 3, 5, and 8; and lanes 4, 6, and 9 show the cells treated with 100 μg/mL genistein. Equal sample loading was confirmed by detection of actin.