

Here we have demonstrated an apparently general method for the electrochemical detection of macromolecules that interact strongly with specific small molecule targets. Using the small molecule recognition elements biotin and digoxigenin, we demonstrate detection of subnanomolar to low nanomolar concentrations of streptavidin and anti-digoxigenin antibodies directly in blood serum and other complex sample matrices.
Our approach is based on a relatively new class of reagentless, electrochemical sensors that utilize electrode-bound, redox-tagged oligonucleotide probes as their sensing elements.1 Prior to this work, these electrochemical DNA (E-DNA) and electrochemical, aptamer-based (E-AB) sensors have been reported against specific DNA and RNA sequences,2 proteins,3,4 small molecules,5–7 and inorganic ions.8,9 Because all of the sensing components in the E-DNA/E-AB platform are covalently attached to the interrogating electrode, the approach requires neither exogenous reagents nor labeling of the target. Likewise, because their signaling is linked to specific, binding-induced changes in the dynamics of the probe DNA (rather than changes in adsorbed mass, charge, etc.), these sensors function well when challenged with complex, contaminant-ridden samples such as blood serum, soil extracts, and foodstuffs.5,7,9,10 These attributes render the E-DNA/E-AB platform an appealing approach for the specific detection of oligonucleotides and other targets that bind DNA or RNA.11–13
Despite their promise, however, sensors in this class have only been reported for the detection of targets that bind to unmodified DNA or RNA. And while the use of aptamer receptors has broadened the range of targets that can be detected using unmodified oligonucleotides,14 further expansion of the platform to targets that, for example, bind specific small molecules should increase the approach's utility. Fortunately, the observation that E-DNA/E-AB signaling requires only that target binding alters the dynamics of the DNA-bound redox tag15,16 suggests a means of detecting proteins that bind small molecules by simply appending the small molecule to the DNA probe. In support of this hypothesis we demonstrate here two new sensors that combine the selectivity and convenience of E-DNA sensors with the expanded recognition properties of small molecule receptors.
The new sensor platform comprises a small-molecule recognition element appended onto a relatively rigid, partially double-stranded DNA scaffold that, in turn, is chemi-adsorbed to an interrogating gold electrode (Figure 1, left). One of the two scaffold strands, the anchoring strand, is linked to the electrode using self-assembled monolayer chemistry via a 5′ thiol group and is modified with a redox tag (here methylene blue) at its 3′ terminus. The second strand, the recognition strand, is complementary to a region of the anchoring strand and is covalently modified with the relevant small molecule recognition element at one of its termini. In the absence of target, the modified, double-stranded scaffold is free to collide with the electrode surface (Figure 1, left) and, thus, produces a large faradaic current at the redox potential expected for methylene blue (Figure 1, right). Upon target binding, this current is reduced (Figure 1, right), presumably because the bulky target reduces the efficiency with which the redox tag collides with the electrode (Figure 1, center). Alternatively the reduced electron transfer may be due to target-induced cross-linking between adjacent scaffolds (antibodies contain two binding sites).
Figure 1.
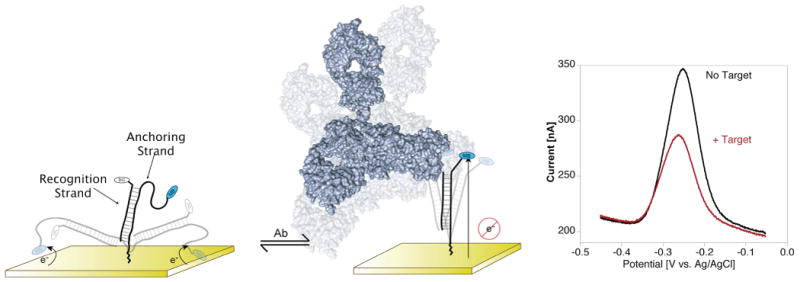
Here we demonstrate a novel electrochemical sensing architecture that retains the selectivity and convenience of E-DNA sensors1–10 while expanding its range to the detection of macromolecules that bind to specific small molecules. The new architecture utilizes a largely double-stranded DNA as a rigid-but-dynamic scaffold to support a small-molecule recognition element. One strand of the scaffold, the “anchoring strand”, is attached to the electrode surface at its thiol-modified 5′ terminus and labeled with a redox tag (here methylene blue) at its 3′ terminus. The second strand, the “recognition strand”, is modified either at its 3′ terminus or, as shown, its 5′ terminus with a small-molecule recognition element. (Left) In the unbound state, the scaffold supports efficient electron transfer between the redox label and the electrode. (Center) The binding of the macromolecular target to this recognition element reduces the transfer efficiency, thus significantly reducing the observed faradaic current. (Right) Shown here are representative square wave voltammograms of the free and target-bound sensor (for the detection of 30 nM anti-digoxigenin antibody in 50% blood serum).
We fabricated our first sensor by appending a biotin to the 5′ terminus of the recognition strand, which places it distal to the electrode (Figure 1, left). In the presence of the protein target streptavidin, this leads to a significant decrease in faradaic current (Figure 2, top). However, the magnitude of this signal suppression (under, for example, saturating target) depends on the precise configuration of the DNA scaffold. A series of sensors containing recognition strands ranging from 13 to 27 bases in length (all centered on the middle of the same, 27-base anchoring strand, Figure 2, top) produce signal suppression of 10% to 45% (50 nM target). The best signaling is observed using a 19-base recognition strand (sequence 19B5) that leaves a 4-base single-stranded element on each end of the scaffold. By comparison, a shortened, 10-base recognition strand that is offset to place the biotin in the same position (producing a longer single-stranded element proximal to the electrode) leads to poorer signal change (data not shown), suggesting that optimal signaling reflects tuning of the flexibility of the scaffold (the length of the single-stranded region) rather than the precise placement of the recognition element. Finally, no signal suppression is observed with a 17-base recognition strand lacking biotin (ctrl in Figure 2).
Figure 2.
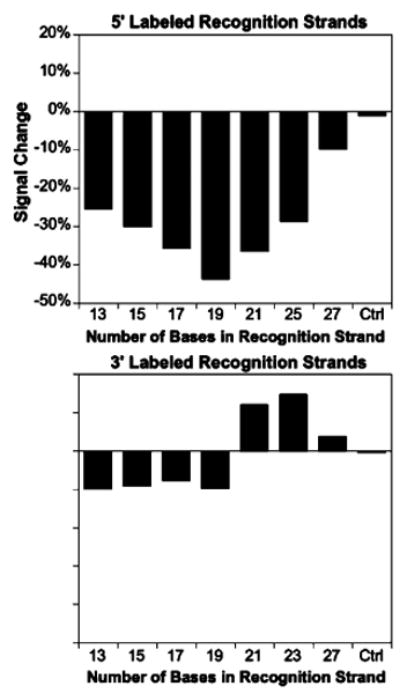
Sensor response depends on the flexibility of the DNA scaffold. Using a 27 base anchoring strand and 5′ placement of the biotin recognition element (distal to the electrode), we achieve optimal signaling with a recognition strand of 19 bases (centered on the middle of the anchoring strand). In contrast, 3′ placement of the recognition element produces a sharp transition from signal-off behavior at short lengths to signal-on behavior for recognition strands of 21 or more bases. Double-stranded scaffolds 17 bases in length lacking the small molecule recognition element (Ctrl) do not respond to target. All data represent addition of saturating (50 nM) streptavidin target.
In contrast to the above-described sensors, sensors fabricated with biotin on the 3′ terminus of the recognition strand, which places the recognition element proximal to the electrode, display rather more complex behavior (Figure 2, bottom). As was true of all of the 5′ recognition strands we investigated, short 3′ recognition strands generate “signal-off” sensors in which target binding suppresses the signaling current. 3′ recognition strands of 21 or more bases, however, produce “signal-on” sensors in which target binding increases the faradaic current, culminating in the 15% increase observed for a 23-base, 3′ recognition strand (centered on the 27-base anchoring strand). We presume this signal-on behavior arises when target binding to the proximal side of the double-stranded scaffold forces its distal end (and thus the methylene blue) toward the electrode surface, increasing electron transfer. A shorter, 17-base recognition strand that retains the same recognition element location (sequence 23S17B3) yields a still greater signal increase (∼45%, see Figure 3), supporting the argument that scaffold flexibility is a key design parameter.
Figure 3.
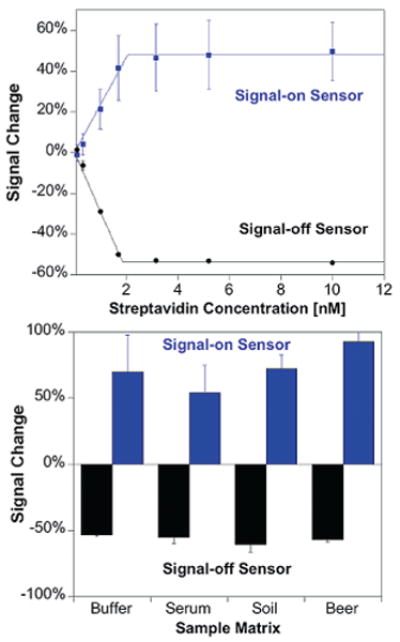
Both the signal-on and signal-off streptavidin sensors achieve subnanomolar detection limits and are able to function in complex samples. Shown on the top are titrations of signal-on (23S17B3) and signal-off (19B5) streptavidin sensors in buffer (the biphasic nature of the curves arises due to the subnanomolar dissociation constant of the streptavidin–biotin interaction). The sensors function comparably in complex samples such as blood serum, soil suspensions, and beer (bottom), yielding similar signals upon addition of saturating (30 nM) streptavidin target. The error bars in this and the following figure represent the standard deviations of measurements conducted using three separately fabricated electrodes. The signal-on construct has large electrode-to-electrode variability in gain, although detection limits are similar for each individual electrode. (Figure S7 presents titration data for individual electrodes.)
The new sensor platform is sensitive, selective, reusable, and rapid. Both the signal-on and signal-off streptavidin sensors respond sensitively to their target, exhibiting detection limits below 1 nM (Figure 3). Neither architecture responds significantly to nontargeted proteins, such as a mixture of IgG antibodies (Figure S2), and both architectures support the detection of their targets directly in complex sample matrices, such as 50% blood serum, 5% (w/v) soil in buffer, and foodstuffs (Figure 3, bottom and Figure S3). The sensors are also readily regenerable: a short rinse with deionized water to disrupt hybridization and remove the recognition strand before the addition of fresh recognition strand allows reuse for more than five cycles (Figure S4). Finally, both sensors equilibrate rapidly, achieving saturation with exponential time constants of less than 4 min (10 nM target; Figure S5).
To test the generality of our approach, we have also fabricated sensors employing the steroid digoxigenin as the recognition element. Using a 19-base recognition strand with a 5′ digoxigenin label (sequence 19D5), we obtain a limit of detection of ∼5 nM for monoclonal anti-digoxigenin antibodies in both buffer (Figure 4) and 50% blood serum (Figure S3). Unlike the streptavidin sensor, however, recognition strands employing a 3′ (i.e., electrode proximal) digoxigenin do not produce “signal-on” sensors (Figure S6), suggesting that the signal-on mechanism is sensitive to the precise geometry of the target–receptor complex and may not be as general as the “signal-off” architecture. Like its predecessor, the digoxigenin-based sensor is specific, selective, and rapid: it does not respond to other, nontargeted proteins (Figure S2); performs well when deployed directly in buffer, blood serum, soil suspensions, and foodstuffs (Figure 4); and exhibits exponential equilibration with a time constant of 10 min (30 nM target; Figure S5).
Figure 4.
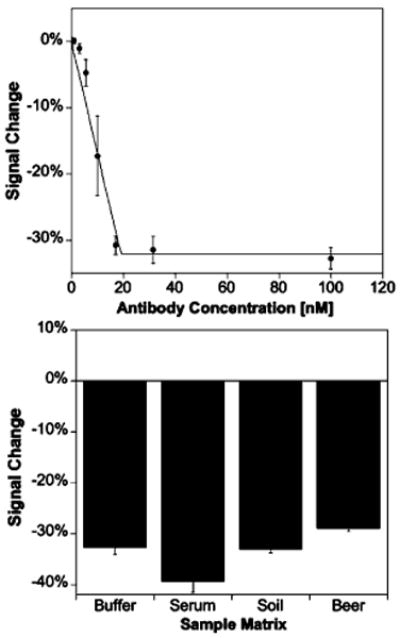
The new sensing architecture appears to be general. For example, employing digoxigenin as the recognition element we readily detect anti-digoxigenin antibodies at low nanomolar concentrations in buffer (top; the biphasic curve arises due to the subnanomolar dissociation constant of the antibody–digoxigenin interaction). Antibody detection also functioned in complex sample matrices (bottom), with comparable signals obtained upon addition of saturating (30 nM) antibody target. Unlike the streptavidin sensor, no “signal-on” sensors are observed, suggesting that the “signal-off” approach may be more general.
This new sensing approach offers several significant advantages over other methods for monitoring protein–small molecule interactions or detecting proteins that bind to specific small molecule targets. For example, while standard methods for protein detection, such as ELISAs and western blots, can be several orders of magnitude more sensitive,17 they are time and capital intensive and typically involve reagent-intensive amplification steps and multiple washings.18,19 Less cumbersome methods for monitoring protein–small molecule interactions, such as fluorescence polarization and surface plasmon resonance (SPR) and quartz crystal microbalance (QCM) approaches, often fail in complex samples because of background fluorescence20 or nonspecific adsorption.21,22 Finally, while our approach is intellectually related to the promising new method of Rant and co-workers23,24 in which binding-induced changes in the electrostatically driven dynamics of DNA are monitored optically, our approach appears less technically complex and, because of the relative paucity of electroactive interferants, may be better suited for deployment in complex samples.
In short, the approach we have demonstrated here is rapid and convenient and functions even when challenged directly in clinically and environmentally relevant samples. Moreover, it appears that this new approach may also be rather versatile; in principle, given a target macromolecule large enough to alter the collision dynamics of the scaffold, the only limiting factor is the ability to effectively conjugate the relevant small recognition molecule to a rigid-yet-dynamic scaffold.
Supplementary Material
Acknowledgments
This work was supported by the NIH through Grant GM062958-01 and by the Institute for Collaborative Biotechnologies through Grant DAAD19-03-D-0004 from the U.S. Army Research Office. K.J.C. is supported by funds from the California HIV/AIDS Research Program of the University of California, Grant No. D07-SB-417.
Footnotes
Supporting Information Available: Materials and methods, including fabrication of sensors and all DNA sequences used; further data on sensor optimization (probe packing density as well as recognition strands for digoxigenin sensors), sensor equilibration kinetics, sensor regeneration, and information on sensor specificity (testing sensor constructs against single incorrect targets instead of mixtures of targets such as blood serum). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Fan C, Plaxco KW, Heeger AJ. Proc Natl Acad Sci U S A. 2003;100:9134–9137. doi: 10.1073/pnas.1633515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ricci F, Plaxco KW. Microchim Acta. 2008;163:149–155. [Google Scholar]
- 3.Radi AE, Sánchez JLA, Baldrich E, O'Sullivan CK. J Am Chem Soc. 2006;128:117–124. doi: 10.1021/ja053121d. [DOI] [PubMed] [Google Scholar]
- 4.Lai RY, Plaxco KW, Heeger AJ. Anal Chem. 2007;79:229–233. doi: 10.1021/ac061592s. [DOI] [PubMed] [Google Scholar]
- 5.Baker BR, Lai RY, Wood MS, Doctor EH, Heeger AJ, Plaxco KW. J Am Chem Soc. 2006;128:3138–3139. doi: 10.1021/ja056957p. [DOI] [PubMed] [Google Scholar]
- 6.Zuo X, Song S, Zhang J, Pan D, Wang L, Fan C. J Am Chem Soc. 2007;129:1042–1043. doi: 10.1021/ja067024b. [DOI] [PubMed] [Google Scholar]
- 7.Ferapontova EE, Olsen EM, Gothelf KV. J Am Chem Soc. 2008;130:4256–4258. doi: 10.1021/ja711326b. [DOI] [PubMed] [Google Scholar]
- 8.Radi AE, O'Sullivan CK. Chem Commun. 2006:3432–3434. doi: 10.1039/b606804a. [DOI] [PubMed] [Google Scholar]
- 9.Xiao Y, Rowe AA, Plaxco KW. J Am Chem Soc. 2007;129:262–263. doi: 10.1021/ja067278x. [DOI] [PubMed] [Google Scholar]
- 10.Lubin AA, Lai RY, Heeger AJ, Plaxco KW. Anal Chem. 2006;78:5671–5677. doi: 10.1021/ac0601819. [DOI] [PubMed] [Google Scholar]
- 11.Thorpe HH. Trends Biotechnol. 2003;21:522–524. doi: 10.1016/j.tibtech.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Palecek E. Trends Biotechnol. 2004;22:55–58. doi: 10.1016/j.tibtech.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 13.Eisenstein M. Nat Methods. 2006;3:244. doi: 10.1038/nmeth0406-244b. [DOI] [PubMed] [Google Scholar]
- 14.Xiao Y, Plaxco KW. In: Functional Nucleic Acids for Sensing and Other Applications. Lu Y, Li Y, editors. Kluwer/Springer; New York: 2009. pp. 179–198. [Google Scholar]
- 15.Ricci F, Lai RY, Heeger AJ, Plaxco KW, Sumner JJ. Langmuir. 2007;23:6827–6834. doi: 10.1021/la700328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ricci F, Lai RY, Plaxco KW. Chem Commun. 2007:3768–3770. doi: 10.1039/b708882e. [DOI] [PubMed] [Google Scholar]
- 17.Adler M, Wacker R, Niemeyer CM. Analyst. 2008;133:702–718. doi: 10.1039/b718587c. [DOI] [PubMed] [Google Scholar]
- 18.Crowther JR. In: The ELISA Guidebook. Walker JM, editor. Vol. 149 Humana; Totawa, NJ: 2001. [Google Scholar]
- 19.Wild D, editor. The Immunoassay Handbook. 2nd. Nature; London: 2001. [Google Scholar]
- 20.Owicki JC. J Biomol Screen. 2000;5:297–306. doi: 10.1177/108705710000500501. [DOI] [PubMed] [Google Scholar]
- 21.Homola J. Anal Bioanal Chem. 2003;377:528–539. doi: 10.1007/s00216-003-2101-0. [DOI] [PubMed] [Google Scholar]
- 22.Cooper MA. Anal Bioanal Chem. 2003;377:834–842. doi: 10.1007/s00216-003-2111-y. [DOI] [PubMed] [Google Scholar]
- 23.Rant U, Arinaga K, Scherer S, Pringsheim E, Fujita S, Yokoyama N, Tornow M, Abstreiter G. Proc Natl Acad Sci U S A. 2007;104:17364–17369. doi: 10.1073/pnas.0703974104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rant U, Pringsheim E, Kaiser W, Arinaga K, Knezevic J, Tornow M, Fujita S, Yokoyama N, Abstreiter G. Nano Lett. 2009;9:1290–1295. doi: 10.1021/nl8026789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.