

Abstract
Porphyria cutanea tarda (PCT) is caused by inhibition of uroporphyrinogen decarboxylase (URO-D) activity in hepatocytes. Subnormal URO-D activity results in accumulation and urinary excretion of uroporphyrin and heptacarboxyl porphyrin. Heterozygosity for mutations in the URO-D gene is found in the familial form of PCT (F-PCT). Over 70 mutations of URO-D have been described but very few have been characterized structurally. Here we characterize 3 mutations in the URO-D gene found in patients with F-PCT, G318R, K297N, and D306Y. Expression of the D306Y mutation results in an insoluble recombinant protein. G318R and K297N have little effect on the structure or activity of recombinant URO-D, but the proteins display reduced stability in vitro.
Keywords: Porphyria cutanea tarda, uroporphyrinogen decarboxylase
INTRODUCTION
Porphyria cutanea tarda (PCT), the most common porphyric disorder in humans, is characterized clinically by skin fragility and bullae on light exposed areas (1). The biochemical characteristics include the accumulation of uroporphyrin and heptacarboxyl porphyrin in the liver and excretion of these compounds in the urine. The biochemical findings are due to markedly diminished activity of uroporphyrinogen decarboxylase (URO-D) in the liver (10).
Mutations in the gene encoding URO-D are found in patients with the familial form of PCT (F-PCT) (1). F-PCT, representing 20–30 percent of PCT cases, is transmitted as an autosomal dominant trait, although mutant alleles of URO-D are not sufficient to produce the disease phenotype (4). Heterozygotes for URO-D mutations have approximately half-normal URO-D activity in all tissues but phenotypic expression of PCT requires a reduction of URO-D activity in hepatocytes to approximately 20 percent of normal (9). Reduced activity of URO-D in hepatocytes is mediated by generation of a competitive inhibitor of the enzyme, namely uroporphomethene (18). Uroporphomethene differs from the fully reduced uroporphyrinogen substrate of URO-D only by the presence of one oxidized bridge carbon between adjacent pyrrole rings (18). Generation of the inhibitor is dependent on iron and one or more genetic or environmental risk factors including HFE (hereditary hemochromatosis gene) mutations, alcohol abuse, hepatitis C, and use of oral medicinal estrogens (3, 7, 8, 10, 26). The same risk factors and the same uroporphomethene inhibitor cause the more common sporadic PCT (S-PCT), a disorder not associated with URO-D mutations (1).
URO-D functions as a homodimer (21). Each monomer of approximately 40 kDa adopts a TIM-barrel fold with one active site (25). The enzyme catalyzes the sequential decarboxylation of the four acetate side chains of uroporphyrinogen to form coproporphyrinogen (13). Both uroporphyrinogen I and uroporphyrinogen III serve as substrates for URO-D but the stereospecificity of the subsequent enzyme in the biosynthetic pathway, coproporphyrinogen oxidase, permits only coproporphyrinogen III to be converted to protoporphyrin IX and heme.
Seventy-one URO-D mutations in patients with F-PCT have been reported in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk). Forty-five are missense or nonsense mutations, 10 are splicing mutations, 6 are small deletions, 4 are gross deletions, 4 are small insertions and one represents a micro indel (insertion and deletion). Catalytic effects of many of the mutations have been determined in red cell lysates from patients with F-PCT or with recombinant proteins (4). Effects of mutations on enzyme stability have been measured in 12 cases although structural effects of naturally occurring mutations have been determined in only 4 cases. Here we report the catalytic and structural consequences of 3 URO-D mutations. Two, K279N, G318R, and D306Y are novel mutations. One, G318R, was previously reported by McMannus et al (15) but was not structurally characterized.
MATERIALS AND METHODS
Human subjects
Three patients with F-PCT were evaluated at the General Clinical Research Center of the University of Utah Medical Center under a protocol approved by the Institutional Review Board. All patients gave written informed consent. All had skin lesions characteristic of PCT and markedly increased secretion of porphyrins in the urine (range 1072 to 4426 μg/24 hr). Uroporphyrin and heptacarboxyl porphyrin were identified as the dominant urinary porphyrins by high-performance liquid chromatography. The diagnosis of F-PCT was based on the demonstration of approximately half-normal URO-D activity in erythrocyte lysates (2) (range 45% to 69%).
Case 1
A 65 year old Caucasian woman. Erythrocyte URO-D activity 45% of normal. Serum ferritin 397 ng/mL. Risk factors: oral post-menopausal estrogen replacement therapy; HFE genotype H63D/H63D.
Case 2
A 54 year old Caucasian man. Erythrocyte URO-D activity 69% of normal. Serum ferritin 564 ng/mL. Risk factors: HFE genotype H63D/S65C. A younger brother, age 48, also developed PCT. Serum ferritin 464 ng/mL. Risk factors: HFE genotype H63D/S65C; alcohol abuse.
Case 3
A 44 year old Caucasian man. Erythrocyte URO-D activity 67% of normal. Serum ferritin 390 ng/mL. Risk factors: hepatitis C; alcohol abuse; HFE genotype H63D/wild type.
URO-D loci sequencing
Genomic DNA was extracted from peripheral blood leukocytes and sequenced after PCR amplification using conditions previously reported (20).
Plasmids and protein expression
The pHT77 plasmid (21) was mutagenized using a QuikChange II Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). Mutations were confirmed by sequencing. Mutant recombinant proteins were expressed in Rosetta2 (DE3) pLysS (Novagen, Madison, WI, USA) in 2 L cultures by autoinduction (24), purified as previously described (20) and dialyzed into 50 mM Tris pH 7.4, 100 mM NaCl, 5% glycerol and 1 mM β-mercaptoethanol.
URO-D activity assays and kinetic measurements
Recombinant wild type and mutant URO-D proteins were assayed for activity as previously described (19). The URO-D assay for kinetic studies was modified as described below. The substrates, uroporphyrinogen I or III were synthesized enzymatically and then adjusted to pH 6.8 (22). The resulting stock solutions were diluted to contain 40, 20, 10, 5.0, 2.5, 2.0 and 0.0 μM uroporphyrinogen in the URO-D assay mixture. A 120 μL aliquot of substrate solution was added to a 1.5 mL microfuge tube containing 70 μL 50 mM KH2PO4 pH 6.8. The mixture was incubated at 37 °C for 3 min and then mixed with 0.4 μg recombinant URO-D in 10 μL of 50 mM K2HPO4 pH 6.8 and 1 mg/mL bovine serum albumin. The decarboxylation reaction was incubated for 10 min at 37 °C and then stopped by the addition of 200 μL 3 M HCl. The resulting acidified mixture was exposed to UV light for 30 min and then centrifuged at 13,000 rpm for 15 minutes. Porphyrins in the supernatant were analyzed by HPLC (19). All steps until the addition of HCl were performed under minimal illumination.
The kinetic parameters Km, Vmax and kcat for the various URO-D samples were determined using both Michaelis-Menten and Eadie-Hofstee plots. The lines obtained used points that included at least the top three highest concentrations of uroporphyrinogen I or III (40, 20 and 10 μM) to ensure that accurate initial rates were used in the calculations of the kinetic parameters.
Protein crystallization
Recombinant URO-D proteins were crystallized as previously described (21, 22, 25). X-ray diffraction data were collected from crystals maintained at 100K using a Rigaku MicroMax-007HF rotating anode generator with copper anode and a Rigaku Raxis-4 image plate detector. Data were integrated and scaled using HKL2000 (17). Crystal structures were determined as previously described (22). A starting model of apo-URO-D (pdb coordinates 1uro.pdb) was refined against the data using the maximum likelihood target function in REFMAC5 (16). Electron density maps were used to evaluate and rebuild the structures in the program O (14). Refinement and model building provided a model for each mutant protein that was in good agreement with the diffraction data and stereochemical expectations (Table 1). Pymol was used to prepare figures (6). The PBD codes for the refined structures are 3gw3: K297R and 3gw0: G318R.
Table 1.
Data collection and refinement statistics for K297N and G318R URO-D. mutants.
| Data set | K297N | G318R |
|---|---|---|
| Wavelength (Å) | 1.5418 | 1.5418 |
| Resolution (Å)a | 30.0–1.70 (1.76–1.70) | 30.0–2.00 (2.07–2.00) |
| # Reflections Measured | 258,037 | 182,847 |
| # Unique Reflections | 48,712 | 31,077 |
| Completeness (%) | 99.3 (96.9) | 100.0 (100.0) |
| <I/σI> | 8.5 (2.1) | 9.0 (1.9) |
| Mosaicity (°) | 0.79 | 0.81 |
| Rsymb (%) | 0.076 (0.351) | 0.084 (0.451) |
| # Protein residues modeled | 11–366 | 11–366 |
| # Water molecules | 364 | 180 |
| <B>protein (Å2) | 30.5 | 40.8 |
| <B>main-chain (Å2) | 28.8 | 38.9 |
| <B>water (Å2) | 44.8 | 50.7 |
| Rcrystc (%) | 0.184 (0.359) | 0.194 (0.244) |
| Rfree (%)d | 0.238 (0.351) | 0.239 (0.256) |
| RMSD bond lengths (Å)/angles (°) | 0.016/1.533 | 0.016/1.977 |
Values in parentheses refer to the high-resolution shell.
Rsym = Σ|I−<I>|/ΣI where I is the intensity of an individual measurement and <I> is the corresponding mean value.
Crystallographic R value (Rcryst) = Σ||Fo|−|Fc||/Σ|Fo|, where |Fo| is the observed and |Fc| the calculated structure factor amplitude.
Rfree is the same as Rcryst calculated with a randomly selected test set of reflections that were never used in refinement calculations. For each refinement the following number of reflections were chosen for the test set; K297N, 1507; G318R, 1257.
RESULTS
Recombinant protein expression and characterization
Three URO-D mutations were identified in PCT patients: G318R (case 1), D306Y (case 2) and K297N (case 3). Each of these mutations was introduced into a URO-D expression plasmid and the recombinant proteins were expressed and purified. G318R and K297N were soluble but D306Y proved to be insoluble (Fig. 1). Attempts at refolding the purified insoluble D306Y protein were unsuccessful. Activities of the soluble purified recombinant mutant proteins G318R and K279N were assayed using uroporphyrinogen I and III as substrates and compared to the activity of wild-type URO-D. Relative enzyme activities are shown in Table 2 and kinetic parameters based on Michaelis-Menten plots are shown in Table 3. Decreased activity was observed at saturating substrate concentrations, however, the substrate binding varied little. Values for the biologically relevant isomer III substrate differed compared to the isomer I substrate as previously reported (5, 12, 27). Nearly identical results were obtained using Eadie-Hofstee plots (data not shown). The Vmax, Km and kcat varied slightly but the Vmax/Km ratio remained constant (Table 3).
Figure 1. SDS-PAGE analysis of soluble and insoluble fractions from mutant URO-D expression.

Mutant recombinant URO-Ds were expressed in Rosetta2(λDE3) pLysS cells (Novagen). Cells were pelleted and resuspended in 50 mM Tris pH 8.0 and 100 mM NaCl. Cells were lysed by sonication and centrifuged 45 min at 11,000 × g. The supernatants are presented as “S” (soluble fraction) and the pellets as “I” (insoluble fraction). A sample of each preparation containing 10 μg of protein was separated by 10% SDS-PAGE. Every pellet contained small amounts of recombinant URO-D. In all mutants except D306Y URO-D was present in the soluble fraction. Purified wild type URO-D (rhUroD) is shown in the far right lane as a standard. Molecular weight markers are shown in the far left lane.
Table 2.
Activity of recombinant wild type and mutant URO-D proteins.
| Activity as percent of wild type | ||
|---|---|---|
| Substrate | Uroporphyrinogen I | Uroporphyrinogen III |
| Wild Type | 100 | 100 |
| G318R | 60.9 ± 26.3 | 76.2 ± 34.5 |
| D306Ya | ND | ND |
| K297N | 63.1 ± 13.5 | 73.7 ± 15.9 |
No recombinant protein recovered.
Table 3.
Kinetic parameters of recombinant wild type and mutant URO-D proteins.
| Uroporphyrinogen I | ||||
|---|---|---|---|---|
| Vmaxa | Kmb | kcatc | Vmax/Km | |
| WT | 4390 ± 99 | 38.1 ± 2.1 | 0.124 ± 0.003 | 115 ± 4 |
| K297N | 3679 ± 323 | 33.0 ± 1.3 | 0.104 ± 0.009 | 112 ± 14 |
| G318R | 3558 ± 28 | 27.7 ± 3.5 | 0.102 ± 0.001 | 131 ± 17 |
| Uroporphyrinogen III | ||||
| WT | 11175 ± 1194 | 10.8 ± 1.1 | 0.317 ± 0.034 | 1039 ± 3 |
| K297N | 12754 ± 3103 | 14.4 ± 5.9 | 0.361 ± 0.088 | 919 ± 161 |
| G318R | 14981 ± 808 | 14.8 ± 1.4 | 0.425 ± 0.023 | 1013 ± 41 |
pmol/hour
μM
sec−1
Recombinant D306Y URO-D proved to be insoluble. Aspartic acid 306 is located next to a highly conserved proline at position 307 on βα loop 7 at the dimer interface (Fig. 2) (25). The carboxylate side chain forms a hydrogen bond with tyrosine 182 of the adjacent monomer, thereby stabilizing the dimeric structure. The D306Y mutation therefore disrupts the intersubunit hydrogen bond and introduces a larger side chain. We also substituted a large positively charged residue at position 306 (D306K) and found that this mutant protein was also insoluble (data not shown). A neutral residue (alanine) was also substituted at position 306. This D306A mutant protein appeared to be soluble but quickly formed catalytically inactive aggregates when eluted from the Ni-NTA purification column (data not shown). These observations presumably reflect the importance of the appropriate subunit-subunit interactions involving Asp306 for maintenance of the correctly folded and assembled URO-D protein.
Figure 2. Overlay of wild type URO-D and mutants G318R and K297N.
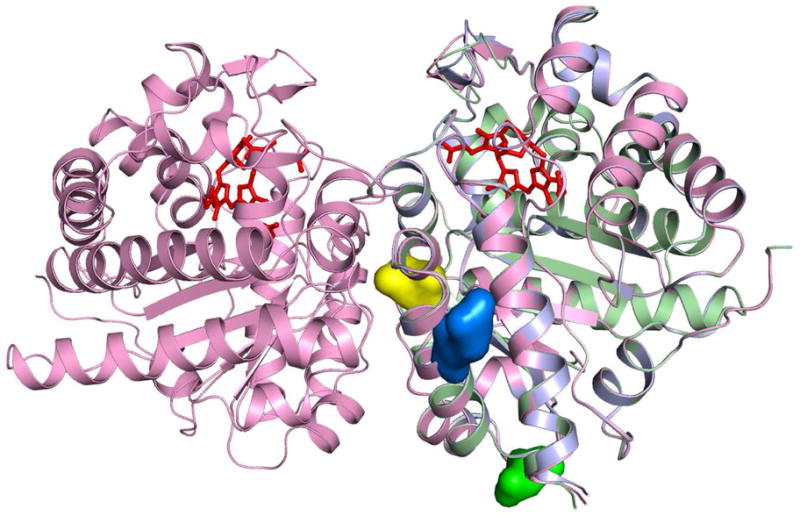
A ribbon diagram of the wild type URO-D homodimer is shown in magenta. Monomers of G318R (slate) and K297N (lime) were aligned to wild type URO-D by least-squares overlap of all C-alpha atoms. The product of the URO-D reaction, coproporphyrinogen III (red) is shown in each of the active sites (22). The side chains R318 (blue) and K297 (green) are highlighted as space filling models. The side chain D306 (yellow) lies at the dimer interface.
Crystal structures
Both the G318R and the K297N recombinant mutant proteins crystallized under conditions previously optimized for wild type URO-D. A well-refined model for each mutant protein was obtained (G318R dmin = 2.0 Å, Rfree = 0.238; K297N dmin = 1.7 Å, Rfree = 0.234). Both mutant models superimposed closely with the wild type structure (G318R root mean square deviation (RMSD) = 0.349, K297N RMSD = 0.201). Both mutations map to residues on the protein surface, greater than 18 Å distant from the active site. In each case, examination of the structures revealed no apparent global or local structural changes resulting from the mutation (Fig. 2).
DISCUSSION
Heterozygosity for loss-of-function alleles of URO-D can be detected by finding sub-normal URO-D activity in erythrocyte lysates (7, 11). This reduced URO-D activity is widely utilized as the criterion for distinguishing F-PCT from the more common S-PCT. The means by which identified missense mutations affect URO-D activity have been defined in very few cases, with only four of these mutations characterized structurally (4, 23). One such mutant is G156D which showed a small distortion of the structure and altered the position of residue D86. D86 is a key residue that has been seen to interact with the four pyrrole NH groups of the bound tetrapyrrole product and presumably makes equivalent interactions with the uroporphyrinogen substrate to promote the electron withdrawal needed for catalytic activity (22). All of the structurally characterized mutants, G168R, I260T and F232L, resulted in decreased enzymatic activity and decreased protein stability even though structural changes were minor (4).
The K297N and G318R described here do not alter the catalytic activity of URO-D (Table 3), yet do cause reduced enzyme activity in vivo. As expected neither mutation significantly distorts the structure (Fig. 2). Both K297N and G318R map to the surface of the protein where side chain substitutions are tolerated. The mutations do, however, result in a change in charge at the altered residue. The K297N mutation substitutes a small neutral residue for the larger positively charged lysine. The G318R mutation introduces a large positively charged side chain in place of the minimal side chain of the glycine residue. We suggest that altered surface charges resulting from both mutations affect stability of the enzyme in vivo. This suggestion is supported by a previous description of a patient with F-PCT associated with the G318R mutation, where immunoquantification of URO-D protein in erythrocytes from that patient showed a 60% reduction compared to normal (15). Similar results would be expected in reference to the K297N mutant.
Our data demonstrate that missense mutations may have minor effects on the structure of URO-D, however, there may be no effect on the catalytic activity of the enzyme when assayed in vitro. In clinical practice distinguishing between a true mutant, that affects substrate binding, turnover rate changes or protein stability and a missense mutation that only alters the primary sequence can often only be done using in vitro assays. Alterations in enzyme stability are most easily shown by demonstrating that enzyme activity and URO-D protein concentrations are sub-normal in red cell lysates. Without these measurements it would not be possible to distinguish between a simple polymorphism and a disease-causing mutation.
Acknowledgments
These studies were supported by NIH grants RO1 DK02503, P30 DK072437, RO1 GM56775 and the UL1 RR025764.
Abbreviations
- Fig
figure
- HFE
hemochromatosis gene
- PCT
porphyria cutanea tarda
- S-PCT
sporadic porphyria cutanea tarda
- F-PCT
familial porphyria cutanea tarda
- URO-D
uroporphyrinogen decarboxylase
References
- 1.Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of Heme Biosynthesis: X-linked Sideroblastic Anemia and the Porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 2991–3062. [Google Scholar]
- 2.Badenas C, To-Figueras J, Phillips JD, Warby CA, Munoz C, Herrero C. Identification and characterization of novel uroporphyrinogen decarboxylase gene mutations in a large series of porphyria cutanea tarda patients and relatives. Clin Genet. 2009;75:346–353. doi: 10.1111/j.1399-0004.2009.01153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulaj ZJ, Franklin MR, Phillips JD, Miller KL, Bergonia HA, et al. Transdermal estrogen replacement therapy in postmenopausal women previously treated for porphyria cutanea tarda. J Lab Clin Med. 2000;136:482–488. doi: 10.1067/mlc.2000.111024. [DOI] [PubMed] [Google Scholar]
- 4.Bulaj ZJ, Phillips JD, Ajioka RS, Franklin MR, Griffen LM, et al. Hemochromatosis genes and other factors contributing to the pathogenesis of porphyria cutanea tarda. Blood. 2000;95:1565–1571. [PubMed] [Google Scholar]
- 5.de Verneuil H, Grandchamp B, Nordmann Y. Some kinetic properties of human red cell uroporphyrinogen decarboxylase. Biochim Biophys Acta. 1980;611:174–186. doi: 10.1016/0005-2744(80)90053-4. [DOI] [PubMed] [Google Scholar]
- 6.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
- 7.Elder GH. Porphyria cutanea tarda. Semin Liver Dis. 1998;18:67–75. doi: 10.1055/s-2007-1007142. [DOI] [PubMed] [Google Scholar]
- 8.Elder GH. Alcohol intake and porphyria cutanea tarda. Clin Dermatol. 1999;17:431–436. doi: 10.1016/s0738-081x(99)00029-2. [DOI] [PubMed] [Google Scholar]
- 9.Elder GH. Porphyria cutanea tarda and related disorders. In: Kadish KM, Smith KM, Guilard R, editors. Porphyrin Handbook, Part II. Academic Press; San Diago: 2003. pp. 67–92. [Google Scholar]
- 10.Elder GH, Lee GB, Tovey JA. Decreased activity of hepatic uroporphyrinogen decarboxylase in sporadic porphyria cutanea tarda. N Engl J Med. 1978;299:274–278. doi: 10.1056/NEJM197808102990603. [DOI] [PubMed] [Google Scholar]
- 11.Elder GH, Roberts AG, de Salamanca RE. Genetics and pathogenesis of human uroporphyrinogen decarboxylase defects. Clin Biochem. 1989;22:163–168. doi: 10.1016/s0009-9120(89)80072-4. [DOI] [PubMed] [Google Scholar]
- 12.Elder GH, Tovey JA, Sheppard DM. Purification of uroporphyrinogen decarboxylase from human erythrocytes. Immunochemical evidence for a single protein with decarboxylase activity in human erythrocytes and liver. Biochem J. 1983;215:45–55. doi: 10.1042/bj2150045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson AH, Sancovich HA, Ferrmola AM, Evans N, Games DE, et al. Macrocyclic intermediates in the biosynthesis of porphyrins. Philos Trans R Soc Lond. 1976;273:191–206. doi: 10.1098/rstb.1976.0009. [DOI] [PubMed] [Google Scholar]
- 14.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47 (Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 15.McManus JF, Begley CG, Sassa S, Ratnaike S. Five new mutations in the uroporphyrinogen decarboxylase gene identified in families with cutaneous porphyria. Blood. 1996;88:3589–3600. [PubMed] [Google Scholar]
- 16.Murshudov GN. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 17.Otwinowski Z, Minor W. Processing X-ray diffraction data in oscillation mode. In: Carter C, Sweet R, editors. Methods in Enzymology. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 18.Phillips JD, Bergonia HA, Reilly CA, Franklin MR, Kushner JP. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc Natl Acad Sci U S A. 2007;104:5079–5084. doi: 10.1073/pnas.0700547104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips JD, Kushner JP. Measurement of Uroporphyrinogen Decarboxylase Activity. In: Maines MD, Costa LG, Reed DJ, Sassa S, Sipes IG, editors. Current Protocols in Toxicology. John Wiley & Sons, Inc; New York: 1999. pp. 8.4.1–8.4.13. [DOI] [PubMed] [Google Scholar]
- 20.Phillips JD, Parker TL, Schubert HL, Whitby FG, Hill CP, Kushner JP. Functional consequences of naturally occurring mutations in human uroporphyrinogen decarboxylase. Blood. 2001;98:3179–3185. doi: 10.1182/blood.v98.12.3179. [DOI] [PubMed] [Google Scholar]
- 21.Phillips JD, Whitby FG, Kushner JP, Hill CP. Characterization and crystallization of human uroporphyrinogen decarboxylase. Protein Sci. 1997;6:1343–1346. doi: 10.1002/pro.5560060624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips JD, Whitby FG, Kushner JP, Hill CP. Structural basis for tetrapyrrole coordination by uroporphyrinogen decarboxylase. EMBO J. 2003;22:6225–6233. doi: 10.1093/emboj/cdg606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips JD, Whitby FG, Stadtmueller BM, Edwards CQ, Hill CP, Kushner JP. Two novel uroporphyrinogen decarboxylase (URO-D) mutations causing hepatoerythropoietic porphyria (HEP) Transl Res. 2007;149:85–91. doi: 10.1016/j.trsl.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Whitby FG, Phillips JD, Kushner JP, Hill CP. Crystal structure of human uroporphyrinogen decarboxylase. EMBO J. 1998;17:2463–2471. doi: 10.1093/emboj/17.9.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wyckoff EE, Kushner JP. Heme Biosynthesis, the Porphyrias, and the Liver. In: Arias IM, Boyer JL, Fausto N, Jakoby WB, Schachter DA, Shafritz DA, editors. The Liver: Biology and Pathobiology. Raven Press, Ltd; New York: 1994. pp. 505–527. [Google Scholar]
- 27.Wyckoff EE, Phillips JD, Sowa AM, Franklin MR, Kushner JP. Mutational analysis of human uroporphyrinogen decarboxylase. Biochimica et Biophysica Acta. 1996;1298:294–304. doi: 10.1016/s0167-4838(96)00148-3. [DOI] [PubMed] [Google Scholar]