

Abstract
Previously, we demonstrated (17) that 11,12- and 14,15-epoxyeicosatrienoic acids (EETs) produce marked reductions in myocardial infarct size. Although it is assumed that this cardioprotective effect of the EETs is due to a specific interaction with a membrane-bound receptor, no evidence has indicated that novel EET antagonists selectively block the EET actions in dogs. Our goals were to investigate the effects of 11,12- and 14,15-EET, the soluble epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA), and the putative selective EET antagonist, 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE), on infarct size of barbital anesthetized dogs subjected to 60 min of coronary artery occlusion and3hof reperfusion. Furthermore, the effect of 14,15-EEZE on the cardioprotective actions of the selective mitochondrial ATP-sensitive potassium channel opener diazoxide was investigated. Both 11,12- and 14,15-EET markedly reduced infarct size [expressed as a percentage of the area at risk (IS/AAR)] from 21.8 ± 1.6% (vehicle) to 8.7 ± 2.2 and 9.4 ± 1.3%, respectively. Similarly, AUDA significantly reduced IS/AAR from 21.8 ± 1.6 to 14.4 ± 1.2% (low dose) and 9.4 ± 1.8% (high dose), respectively. Interestingly, the combination of the low dose of AUDA with 14,15-EET reduced IS/AAR to 5.8 ± 1.6% (P < 0.05), further than either drug alone. Diazoxide also reduced IS/AAR significantly (10.2 ± 1.9%). In contrast, 14,15-EEZE had no effect on IS/AAR by itself (21.0 ± 3.6%), but completely abolished the effect of 11,12-EET (17.8 ± 1.4%) and 14,15-EET (19.2 ± 2.4%) and AUDA (19.3 ± 1.6%), but not that of diazoxide (10.4 ± 1.4%). These results suggest that activation of the EET pathway, acting on a putative receptor, by exogenous EETs or indirectly by blocking EET metabolism, produced marked cardioprotection, and the combination of these two approaches resulted in a synergistic effect. These data also suggest that 14,15-EEZE is not blocking the mitochondrial ATP-sensitive potassium channel as a mechanism for antagonizing the cardioprotective effects of the EETs.
Keywords: cytochrome P-450 epoxygenase, epoxyeicosatrienoic acids, soluble epoxide hydrolase, ATP-sensitive potassium channel opener
arachidonic acid is metabolized to various bioactive eicosanoids by cyclooxygenase, lipoxygenase, and the cytochrome P-450 (CYP) monooxygenase pathways (21). We and others have recently found that inhibition of the CYP ω-hydroxylase pathway (6, 7, 15) and activation of the CYP epoxygenase pathway by overexpressing CYP2J2 (26, 27) or by administering exogenous epoxyeicosatrienoic acids (EETs) result in an improvement in the recovery of contractile function in isolated, perfused mouse hearts (19, 27), subjected to global ischemia and reperfusion, and a reduction in infarct size (IS) in intact canine or rat hearts (7, 17), subjected to regional ischemia and reperfusion. These protective effects were mediated by activation of the sarcolemmal (sarc) or the mitochondrial (mito) ATP-sensitive potassium (KATP) channel (7, 12, 17, 19). Similarly, exogenous EETs protect endothelial cells from apoptosis produced by TNF-α via a MAPK and phosphatidylinositol 3-kinase/Akt signaling pathway (25, 28), and similar protective effects have been reported by Dhanasekaran et al. (2) in human endothelial cells obtained from the pulmonary and coronary vasculature and cardiac myocytes exposed to anoxia (3). Although it is assumed that the beneficial effects of CYP ω-hydroxylase inhibition and CYP2J2 overexpression are the result of an increase in endogenous EETs, there is no direct evidence that the protective actions of these two interventions are actually the result of the actions of EETs on a putative membrane receptor.
In this regard, Gauthier et al. (4, 5) recently identified and characterized a unique EET antagonist, 14,15-epoxyeicosa-5(Z)-enoic acid (EEZE). 14,15-EEZE behaves as a direct EET antagonist at a receptor binding site on the cell membrane or intracellularly (30) and antagonizes selectively many of the vascular actions of the EETs (4, 5, 29), although one study in mesenteric artery demonstrated that 14,15-EEZE had intrinsic vasodilator activity (8). However, the effects of 14,15-EEZE on the cardioprotective actions of the EETs have not been thoroughly addressed and are a major focus of the present study.
Another method that has been shown to be cardioprotective in mice involves inhibition of the major enzymatic hydrolysis pathway of the EETs, soluble epoxide hydrolase (EPHX2) (9, 10, 18, 20). This tactic has been used effectively to increase endogenous EET concentrations under basal conditions and particularly during ischemia-reperfusion. Recently, Seubert et al. (20) studied the functional role of deleting the EPHX2 gene, Ephx2, on cardioprotection in isolated wild-type (C57BL6) and Ephx2 null mouse hearts subjected to 20 min of ischemia and 40 min of reperfusion. The Ephx2 null hearts had a better recovery of contractile function and a modest reduction in IS compared with the wild-type hearts. These results suggest that enhancing cellular concentrations of EETs by blocking their hydrolysis may be a potential new therapeutic way to produce cardioprotection. We have further addressed the beneficial effects of inhibiting this pathway with a pharmacological blocker of EPHX2, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid (AUDA) (10), to determine the cardioprotective potential of inhibiting EPHX2 (18), in a large animal model of ischemia-reperfusion injury, and also to determine the possibility that EPHX2 inhibition, in combination with exogenous EETs, might have additive or synergistic effects on irreversible injury.
MATERIALS AND METHODS
This study was approved by performed in accordance with the guidelines of the Animal Care Committee of the Medical College of Wisconsin, which is accredited by the American Association of Laboratory Animal Care.
Materials
11,12-EET, 14,15-EET, and 14,15-EEZE were synthesized in the laboratory of Dr. J. R. Falck. AUDA was synthesized as described (13). All other chemicals were of the highest analytic or purity grades. Distilled, deionized water was used in all experiments.
General preparation of dogs
The protocol used has been thoroughly described in detail in previous publications from our laboratory (15, 17). Briefly, adult mongrel dogs of either sex, weighing 15–25 kg, were fasted overnight, anesthetized with the combination of barbital sodium (200 mg/kg) and pentobarbital sodium (15 mg/kg), and ventilated with room air supplemented with 100% oxygen. Body temperature was carefully controlled at 38 ± 1°C with a heating pad. Atelectasis was prevented by maintaining an end-expiratory pressure of 5–7 cm with a trap. Arterial blood pH, Pco2, and Po2 were monitored at selected intervals by an AVL blood-gas analyzer and maintained within normal physiological limits (pH = 7.35–7.45, Pco2 30–40 Torr, and Po2 85–130 Torr) by adjusting the respiratory rate and the oxygen flow rate or by adding 1.5% sodium bicarbonate intravenously if necessary. An electromagnetic flowmeter (Statham 2202) was used to measure left anterior descending (LAD) blood flow. A mechanical occluder was placed distal to the flow probe and the occluder, such that there were no branches between the flow probe and occluder. A double-tipped Millar (model 770) catheter was placed into the carotid artery and left ventricle (LV) to measure aortic and LV pressures and to determine LV change in pressure over time (positive and negative). The left atrium was cannulated via the appendage for radioactive microsphere injections.
Experimental protocol
Dogs were sequentially assigned to 12 groups for different treatments (Fig. 1). In all groups, including the AUDA-treated dogs, eight dogs were included for statistical analysis. At 15 min before the 60-min LAD occlusion period, 11,12-EET (0.128 mg/kg), 14,15-EET (0.128 mg/kg), 14,15-EEZE (0.128 mg/kg), or vehicle was administered by a 2- to 3-min intracoronary infusion, as shown in protocol 1 (Fig. 1, top). In three subsequent series of dogs, 14,15-EEZE was administered intracoronary 5 min before 11,12- or 14,15-EET administration to determine whether the EET antagonist could block the cardioprotective effect of the EETs to reduce IS. In protocol 2, the EPHX2 inhibitor AUDA (0.157 or 0.314 mg/kg) was administered intracoronary 15 min before the 60-min coronary artery occlusion. In subsequent experiments, the low dose of AUDA (0.157 mg/kg) was administered 5 min before 14,15-EET to determine whether AUDA would enhance the effect of 14,15-EET by inhibiting its breakdown to 14,15-dihydroxyeicosatrienoic acid (DHET). To further determine the selectivity of 14,15-EEZE to block endogenous EETs, in a final series (protocol 3, Fig. 1, bottom), 14,15-EEZE was administered 15 min before high-dose AUDA and the mito KATP opener diazoxide (3 mg/kg). Diazoxide was given by itself in another group. Since the KATP channel is thought to be an important downstream component of EET-induced cardioprotection (7, 12, 19), we wanted to determine whether 14,15-EEZE could block the cardioprotective effect of the EETs at this level in the signaling pathway. In all groups, hemodynamic measurements, blood-gas analyses, and regional myocardial blood flow (radioactive microspheres) measurements were performed at baseline and at 30 min into the 60-min occlusion period. Following reperfusion, hemodynamics and blood gases were measured every hour, and regional myocardial blood flow was determined at the end of3hof reperfusion. At the end of the experiment, the hearts were electrically fibrillated, removed, and prepared for IS determination and regional myocardial blood flow calculations the following day.
Fig. 1.

Protocols used in the 3 series of canine experiments (N = 8/group). EET, epoxyeicosatrienoic acids; EEZE, epoxyeicosa-5(Z)-enoic acid; OCC, occlusion; REP, reperfusion; AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; LD, low dose; HD, high dose.
IS determination
IS was determined as previously described by our laboratory (17). Briefly, at the end of the 3-h reperfusion period, the LAD was reoccluded and cannulated distal to the occlusion site. To determine the anatomic area at risk (AAR) and the nonischemic area, 5 ml of Patent Blue dye and 5 ml of saline were injected at equal pressure into the LAD and left atrium, respectively. The heart was then immediately fibrillated and removed. The LV was dissected and sliced into serial transverse sections 6–7 mm in width. The nonstained ischemic area and the blue-stained normal area were separated and incubated in a solution containing 1% 2,3,5-triphenyltetrazolium chloride (Sigma) in 0.1 mol/l phosphate buffer, pH 7.4, at 37°C for 15 min. After incubation overnight in 10% formaldehyde, the noninfarcted and infarcted tissues within the AAR were separated and determined gravimetrically. IS was expressed as a percentage of the AAR (IS/AAR).
Regional myocardial blood flow
Regional myocardial blood flow, expressed as milliliters per minute per gram of tissue, was measured by the radioactive microsphere technique, as previously reported (17). Microspheres were administered 30-min into the 60-min occlusion period and at the end of 3 h of reperfusion. Transmural blood flow was calculated as the weighted average of the subepicardium, midmyocardium, and subendocardium of each region.
Statistical analysis
All values are expressed as means ± SE (N = 8/group). Differences between groups in hemodynamics and blood gases were compared by using a two-way ANOVA. Differences between groups in tissue blood flows, AAR, and IS/AAR were compared by a one-way ANOVA followed by Tukey’s post hoc test. Differences between groups were considered significant if P < 0.05. Linear regression analysis was performed to determine the correlation between transmural collateral blood flow in the ischemic area and myocardial IS (IS/AAR). Analysis of covariance, with collateral blood flow as the covariate, was used to determine whether differences in this relationship were observed among the treatment groups analyzed.
RESULTS
Hemodynamics
Heart rate and mean arterial blood pressure at baseline and at 30 min of ischemia or at3hof reperfusion were not different between all of the experimental groups (Table 1). These data suggest that changes in hemodynamics were not responsible for the differences in IS/AAR observed between control dogs and those treated with the various EET mimetics, diazoxide, or the EET antagonist EEZE. Blood gases and pH were also monitored throughout the experiments and were not different between groups at any time points measured (data not shown).
Table 1. Hemodynamic values.
| Control | Occlusion (30 min) | Reperfusion (3 h) | |
|---|---|---|---|
| Heart rate, beats/min | |||
| Control | 156±4 | 155±4 | 155±6 |
| 11,12-EET | 154±4 | 156±4 | 154±2 |
| 14,15-EET | 153±3 | 158±4 | 148±3 |
| AUDA (LD) | 154±8 | 153±3 | 153±7 |
| AUDA (HD) | 156±2 | 151±6 | 153±6 |
| AUDA (LD) + 14,15-EET | 146±2 | 146±3 | 149±2 |
| 14,15-EEZE | 152±2 | 152±2 | 153±4 |
| 14,15-EEZE + 14,15-EET | 160±6 | 160±8 | 154±4 |
| 14,15-EEZE + 11,12-EET | 152±5 | 151±3 | 154±4 |
| Diazoxide | 151±5 | 154±7 | 151±4 |
| 14,15-EEZE + Diazoxide | 152±3 | 158±7 | 153±4 |
| 14,15-EEZE + AUDA (HD) | 155±4 | 152±1 | 144±7 |
| Mean arterial pressure, mmHg | |||
| Control | 100±4 | 104±6 | 110±4 |
| 11,12-EET | 97±3 | 100±5 | 104±5 |
| 14,15-EET | 92±6 | 90±7 | 99±5 |
| AUDA (LD) | 97±7 | 95±8 | 101±7 |
| AUDA (HD) | 94±8 | 88±8 | 99±6 |
| AUDA (LD) + 14,15-EET | 108±5 | 104±4 | 110±4 |
| 14,15-EEZE | 115±8 | 110±7 | 117±4 |
| 14,15-EEZE + 14,15-EET | 130±5 | 120±8 | 123±4 |
| 14,15-EEZE + 11,12-EET | 115±2 | 108±3 | 114±4 |
| Diazoxide | 99±3 | 96±4 | 107±5 |
| 14,15-EEZE + Diazoxide | 105±5 | 96±7 | 100±4 |
| 14,15-EEZE + AUDA (HD) | 102±6 | 98±5 | 101±8 |
All values are means ± SE; N = 8 dogs. There were no significant differences between groups by ANOVA followed by Newman-Keuls post hoc test. EET, epoxyeicosatrienoic acids; AUDA, 12-(3-adamantan-1-yl-ureido)-dodecanoic acid; LD, low dose; HD, high dose; EEZE, epoxyeicosa-5(Z)-enoic acid.
Regional myocardial blood flow
Transmural blood flow in the nonischemic (left circumflex coronary bed) and the ischemic (LAD bed) were measured at 30 min of occlusion and at 3 h of reperfusion with radioactive microspheres. There were no significant differences in nonischemic transmural blood flows between groups or transmural collateral blood flow (ischemic bed) at the measured points. Most importantly, there were no differences in flow in the ischemic reperfused area during occlusion, which suggested that all groups were subjected to similar degrees of ischemia (Table 2). There were also no differences in AAR between groups (data not shown). Since the two major determinants of the ultimate IS/AAR in dogs are the AAR and collateral blood flow, these data suggest that changes in these two parameters were not responsible for any differences in IS/AAR observed between control and drug-treated dogs.
Table 2. Transmural blood flow values.
| Group | Nonischemic Region |
Ischemic Region |
||
|---|---|---|---|---|
| 30-min Occlusion | 3-h Reperfusion | 30-min Occlusion | 3-h Reperfusion | |
| Control | 0.85±0.16 | 0.98±0.16 | 0.07±0.01 | 0.67±0.09 |
| 11,12-EET | 0.65±0.11 | 0.85±0.10 | 0.12±0.01 | 0.86±0.07 |
| 14,15-EET | 0.78±0.06 | 1.09±0.08 | 0.12±0.01 | 0.81±0.10 |
| AUDA (LD) | 1.17±0.11 | 1.29±0.19 | 0.11±0.01 | 1.53±0.18 |
| AUDA (HD) | 1.03±0.08 | 1.31±0.20 | 0.10±0.02 | 1.06±0.14 |
| AUDA(LD) + 14,15-EET | 1.10±0.08 | 1.46±0.11 | 0.14±0.02 | 1.05±0.10 |
| 14,15-EEZE | 1.16±0.08 | 1.04±0.08 | 0.11±0.02 | 1.15±0.11 |
| 14,15-EEZE + 14,15-EET | 1.19±0.17 | 1.21±0.11 | 0.11±0.02 | 0.82±0.15 |
| 14,15-EEZE + 11,12-EET | 1.14±0.16 | 1.20±0.08 | 0.13±0.03 | 0.81±0.06 |
| Diazoxide | 1.62±0.18 | 1.67±0.19 | 0.13±0.03 | 1.55±0.18 |
| 14,15-EEZE + Diazoxide | 1.60±0.17 | 1.42±0.12 | 0.07±0.03 | 1.00±0.13 |
| 14,14-EEZE + AUDA (HD) | 1.14±0.11 | 0.92±0.08 | 0.07±0.02 | 0.60±0.04 |
Values are means ± SE in ml·min−1·g−1; N = 8 dogs.
Effects of 11,12-EET and 14,15-EET on IS/AAR in the absence and presence of the EET antagonist 14,15-EEZE
Both 11,12-EET and 14,15-EET produced a marked reduction in IS/AAR compared with the vehicle-treated dogs (Fig. 2). Control IS/AAR was 21.8 ± 1.6%, and 11,12- and 14,15-EET reduced IS/AAR to 8.7 ± 2.2 and 9.4 ± 1.3%, respectively (P < 0.001). These results are in agreement with previously published results (17) with this same dose of EETs (0.128 mg/kg) in dogs. However, in the presence of the selective EET antagonist (4, 5) 14,15-EEZE, the cardioprotective effects of 11,12- and 14,15-EET were abolished (IS/AAR = 19.2 ± 2.4 and 17.8 ± 1.4%, respectively, Fig. 2). 14,15-EEZE had no effect on IS/AAR when administered alone (21.0 ± 3.6%). To further confirm that the reductions in IS were not the result of an EET-induced increase in collateral blood flow, we performed a further statistical test on four groups of dogs shown in Fig. 2. Figure 3 shows the relationship between myocardial IS and transmural collateral blood flow measured at 30 min into the ischemic period. In the four groups analyzed, there is an inverse relationship between these two parameters, as shown by linear regression analysis. In the 14,15-EET group, this relationship was shifted downward compared with the control, 14,15-EEZE group, and the 14,15-EEZE + 14,15-EET group (P < 0.05 by analysis of covariance). These data suggest that the effect of 14,15-EET to reduce IS is independent of its effect on coronary collateral blood flow (Fig. 3).
Fig. 2.
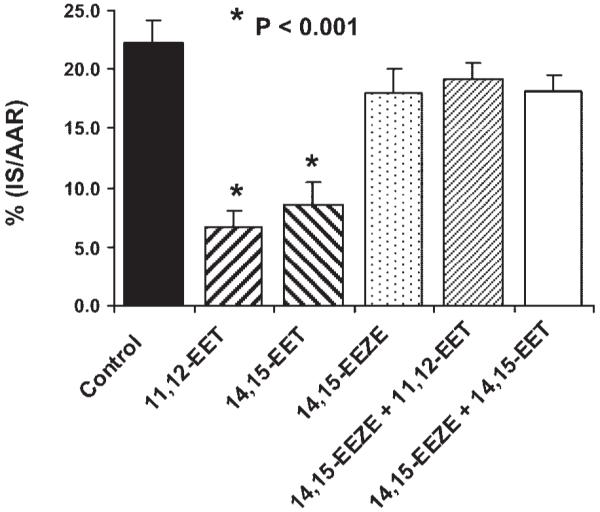
Effect of 11,12-EET and 14,15-EET (0.128 mg/kg) alone or in the presence of the putative EET receptor antagonist 14,15-EEZE on myocardial infarct size [expressed as a percentage of the area at risk (IS/AAR)] in dogs subjected to 60 min of coronary artery occlusion and 3 h of reperfusion. 14,15-EEZE had no effect on IS/AAR by itself, but completely abolished the cardioprotective effect of the 2 EETs. *P < 0.001 vs. control.
Fig. 3.
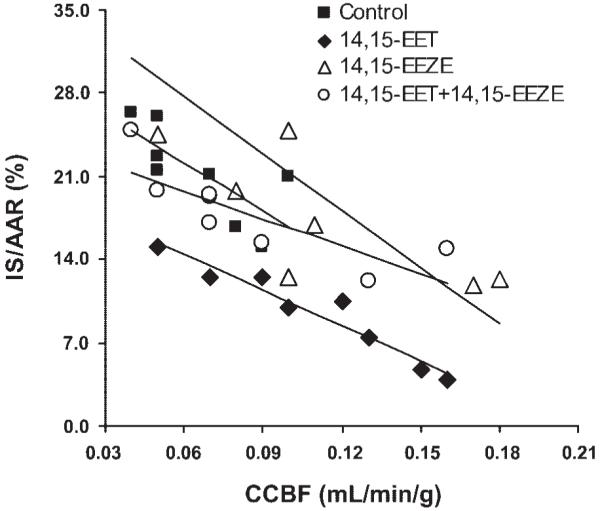
The relationship between transmural collateral blood flow in the ischemic area at 30 min of occlusion and myocardial infarct size (IS/AAR). In all 4 groups, there is an inverse relationship between collateral blood flow and infarct size (control: y = 29–124x; 14,15-EET: y = 20–93x; 14,15-EEZE: y = 39–163x; 14,15-EEZE + 14,15-EET: y = 24–78x). CCBF, coronary collateral blood flow.
Effect of the EPHX2 inhibitor AUDA on IS/AAR alone and in combination with 14,15-EET
AUDA produced a dose-dependent reduction in IS/AAR (control: 21.8 ± 1.6%; AUDA: 0.157 mg/kg, 14.4 ± 1.2%; AUDA: 0.314 mg/kg, 9.4 ± 1.8%; Fig. 4). When the low dose of AUDA was combined with 0.128 mg/kg of 14,15-EET, IS/AAR was further reduced to 5.8 ± 1.3%, which was significantly less than the small dose of AUDA given alone (P < 0.05). This combination was also smaller than the high dose of AUDA or 14,15-EET alone, although this effect did not reach statistical significance. Analysis of 14,15-EET in plasma samples at 5- and 30-min reperfusion by liquid chromatography-electrospray ionization-mass spectrometry (16) indicates that 14,15-EET concentrations increased with AUDA administrations, as shown in Fig. 5. At 5-min reperfusion, AUDA at 0.157 and 0.314 mg/kg increased 14,15-EET concentrations from 9.2 ± 7.7 pg/ml (control) to 147.2 ± 39.0 and 211.0 ± 12.3 pg/ml, respectively. At 30-min reperfusion, AUDA increased 14,15-EET concentrations from 9.6 ± 6.4 pg/ml (control) to 153.3 ± 41.0 and 226.0 ± 54.7 pg/ml, respectively.
Fig. 4.
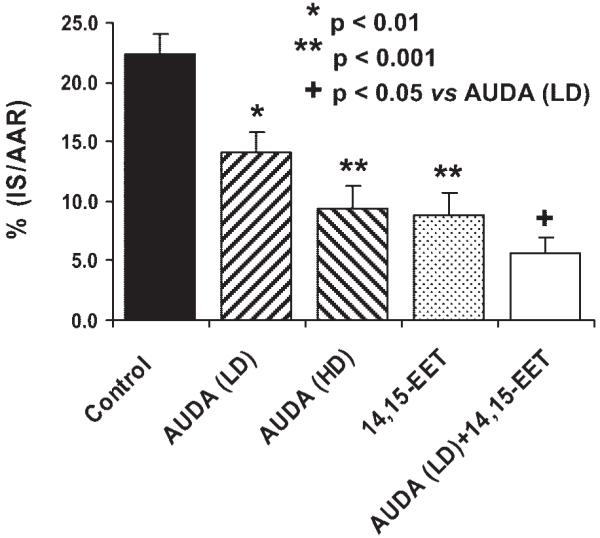
Effects of LD (0.157 mg/kg) and HD (0.314 mg/kg) AUDA and 14,15-EET (0.128 mg/kg) alone or in combination with LD AUDA on myocardial IS/AAR in dogs subjected to 60 min of left anterior descending coronary artery occlusion and3hof reperfusion. *P < 0.01 vs. control. **P < 0.001 vs. control. +P < 0.05 vs. AUDA (LD).
Fig. 5.
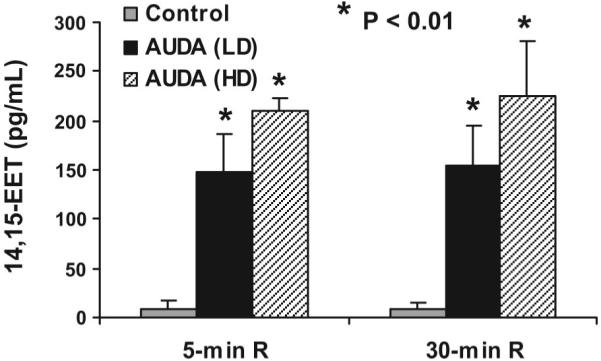
14,15-EET concentrations (pg/ml) in coronary venous plasma at 5 and 30 min of reperfusion (R) following treatment with either the LD of AUDA (0.157 mg/kg) or the HD of AUDA (0.314 mg/kg) compared with predrug control values. *P < 0.01 vs. control.
Effect of 14,15-EEZE on AUDA and diazoxide-induced cardioprotection
Diazoxide and high-dose AUDA produced similar reductions in IS; however, 14,15-EEZE pretreatment only abolished AUDA-induced cardioprotection and was without effect on diazoxide-induced cardioprotection (Fig. 6). These results support the selectivity of 14,15-EEZE for a putative EET receptor, independent of the mito KATP channel.
Fig. 6.
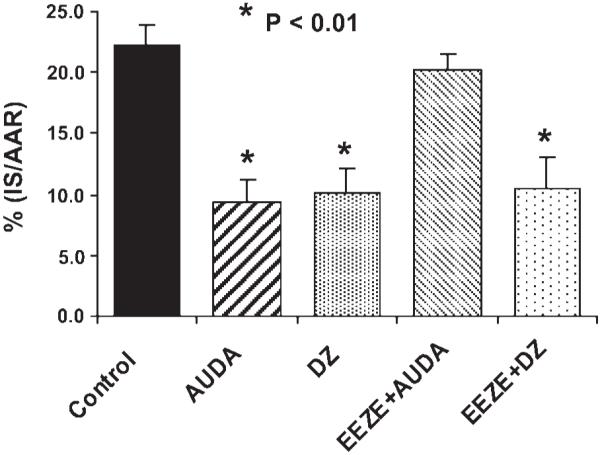
Effects of AUDA (HD) and the mitochondrial ATP-sensitive potassium channel opener diazoxide (DZ) on myocardial IS/AAR alone or in the presence of 14,15-EEZE in dogs subjected to 60 min of coronary artery occlusion and3hof reperfusion. 14,15-EEZE completely abolished the effect of AUDA, but had no effect on the cardioprotective effect of DZ. *P < 0.01 vs. control.
DISCUSSION
The present study confirms the potent cardioprotective efficacy of administering exogenous EETs to the ischemic heart (17). This study demonstrates that an analog of 14,15-EET, 14,15-EEZE (4, 5), is a potent antagonist of 11,12- and 14,15-EET to reduce IS in the canine heart at a dose that had no effect on IS alone. These data and those obtained in previous studies from our laboratory and those of others suggest that 14,15-EEZE selectively blocks the effects of EETs in various species and organs, most importantly in the vasculature and heart (4, 5, 20, 21). Furthermore, our results indicate that inhibition of the EPHX2 by AUDA produced a reduction in IS when administered alone and was able to potentiate the effect of 14,15-EET when these two agents were given together. These results also suggest that one may be able to combine lower doses of EETs and EPHX2 inhibitors to reduce ischemia-reperfusion injury, which would lessen the worry about adverse side effects that might possibly occur when higher doses of either drug are given alone. Finally, the effect of 14,15-EEZE appeared to be selective for a direct EET-mediated action at a receptor, since it did not block the downstream cardioprotection produced via activation of the mito KATP channel.
The 14,15-EET analog 14,15-EEZE was synthesized in 2002 (4) and was tested in a number of in vitro systems to determine its activity as an EET antagonist. This compound blocks EET-induced vascular relaxations produced by all four EET regioisomers in bovine coronary artery rings (4). Furthermore, it also inhibited indomethacin-resistant relaxations to methacholine and arachidonic acid and indomethacin-resistant and l-nitroarginine-resistant relaxations to bradykinin. However, this compound did not block relaxations to sodium nitroprusside, iloprost, or the potassium channel openers, bimakalim or NS1619. The results obtained with diazoxide support the previous ones obtained in the presence of the above-mentioned K+ channel openers, bimakalim and NS1619. In rat renal microsomes, 14,15-EEZE did not affect EET synthesis or 20-HETE synthesis (4, 5, 9). A more recent study, in which the characterization of several 14,15-epoxyeicosatrienyl-sulfonamides as full 14,15-EET agonists, suggested that the EETs may actually bind to a specific membrane binding site or receptor (29, 30). A membrane receptor for the EETs or 14,15-EEZE has not been definitively identified; however, the ability of 14,15-EEZE to block a number of biological responses to the EETs, including IS reduction in mice (21) and dogs (this study) and myocardial contractile dysfunction in mice (26, 27), strongly suggests this possibility.
Another possibility that we considered is that 14,15-EEZE is aKATP channel antagonist, since it has been well established that the EETs are mito and sarc KATP channel openers (11, 12, 17, 19). In this regard, the cardioprotective effects of the EETs were blocked by glibenclamide, a nonselective KATP antagonist (17, 19), to a similar degree to that observed with 14,15-EEZE. However, the results obtained with diazoxide in the present study and bimakalim in previous studies (4, 5) suggest that 14,15-EEZE is not blocking mito KATP.
Although there are several pathways involved in the metabolism of the EETs, the conversion to vicinal-diols by EPHX2 appears to be the major pathway for inactivation (22). This enzyme is found in the cytosol, and the human epoxide hydrolase is the product of a single copy gene, Ephx2, which is found on chromosome 8 (9). This enzyme is found in almost all mammalian tissues, including heart, and metabolizes epoxide-containing compounds to diols or glycols by adding water to the epoxide moiety (9, 22). The diols and glycols are relatively inactive. Several epoxide hydrolase inhibitors have been developed and tested for their vasodilator and anti-inflammatory activities. They have been shown to lower blood pressure and reduce renal damage in models of hypertension (9) and to reduce the inflammatory response to inhaled tobacco smoke and to injection of LPS. Recently, Seubert et al. (20) studied the cardiovascular effects of mice with a targeted deletion of EPHX2. The Ephx2 null mice appeared normal anatomically and functionally under basal conditions, but they exhibited a much better recovery of myocardial contractile function following 20 min of ischemia and reperfusion compared with the wild-type mice. It is interesting that, while the Ephx2 null mice have markedly increased plasma ratios of EETs to DHETs than wild-type mice, they possessed only slightly smaller infarcts (20). The present results with the EPHX2 inhibitor AUDA in dog hearts show that administration of AUDA produced a dose-dependent reduction in IS. Although EETs were not measured for the whole duration of occlusion and reperfusion, the concentrations of 14,15-EET in plasma at 5 and 30 min of reperfusion support the suggestion that AUDA is producing its cardioprotective effect by enhancing, at least in part, the concentrations of endogenous EETs during ischemia and/or early reperfusion. Despite species differences, the selective EPHX2 inhibitor may be more specific for reducing EET hydrolysis than the Ephx2 knockout and may provide more definitive conclusions. EPHX2 is a bifunctional enzyme with both epoxide hydrolase activity and lipid phosphatase activity (1, 14). Targeted deletion of the enzyme eliminates both activities and cannot be ascribed to inhibition of epoxide hydrolysis only. The lipid phosphatase activity may have an unidentified role(s) in cardioprotection. This possibility needs further investigation.
Interestingly, when combining the EPHX2 inhibitor with 14,15-EET, a greater protective effect was observed than that observed with the EET administered alone. This is not surprising, since preventing the metabolism and inactivation of 14,15-EET would effectively increase the concentration of EETs in the heart and produce a greater effect on reducing IS/AAR. Since EPHX2 inhibitors are antihypertensive in models of hypertension and protect the kidney from end-organ damage, the present results and those of Seubert et al. (20) demonstrate an efficacious effect to reduce reversible and irreversible cardiac damage, resulting from ischemia and/or reperfusion. It also suggests that EPHX2 inhibitors or the administration of more long-acting epoxygenase products (EET derivatives) or the combination of the two may have potential benefit in patients with coronary artery disease. In this regard, several studies have shown that the risk of coronary atherosclerosis is increased in patients with polymorphisms of the CYP2J2, which further supports the importance and protective potential of this system at several points in the heart and coronary vasculature (23, 24).
Acknowledgments
GRANTS
This work was supported by National Institutes of Health (NIH) Grants HL-74314-04 and HL-51055. Partial support was provided by NIH Grant GM-31278 and the Robert A. Welch Foundation (J. R. Falck), National Institute of Environmental Health Sciences (NIEHS) Grant R37 ES02710, and NIEHS Superfund Basic Research Program Grant P42 ES004699 (B. D. Hammock).
REFERENCES
- 1.Cronin A, Mowbray S, Durk H, Homburg S, Fleming I, Fisslthaler B, Oesch F, Arand M. The N-terminal domain of mammalian soluble epoxide hydrolase is a phosphatase. Proc Natl Acad Sci USA. 2003;100:1552–1557. doi: 10.1073/pnas.0437829100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dhanasekaran A, Al-Saghir R, Lopez B, Zhu D, Gutterman DD, Jacobs ER, Medhora M. Protective effects of epoxyeicosatrienoic acids on human endothelial cells from the pulmonary and coronary vasculature. Am J Physiol Heart Circ Physiol. 2006;291:H517–H531. doi: 10.1152/ajpheart.00953.2005. [DOI] [PubMed] [Google Scholar]
- 3.Dhanasekaran A, Gruenloh SK, Buonaccorsi JN, Zhang R, Gross GJ, Falck JR, Patel PK, Jacobs ER, Medhora M. Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am J Physiol Heart Circ Physiol. 2008;294:H724–H735. doi: 10.1152/ajpheart.00979.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gauthier KM, Deeter C, Krishna UM, Reddy YK, Bondlela M, Falck JR, Campbell WB. 14,15-Epoxyeicosa-5(Z)-enoic acid: a selective epoxyeicosatrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries. Circ Res. 2002;90:1028–1036. doi: 10.1161/01.res.0000018162.87285.f8. [DOI] [PubMed] [Google Scholar]
- 5.Gauthier KM, Jagadeesh SG, Falck JR, Campbell WB. 14,15-Epoxyeicosa-5(Z)-enoic-mSI: a 14,15- and 5,6-EET antagonist in bovine coronary arteries. Hypertension. 2003;42:555–561. doi: 10.1161/01.HYP.0000091265.94045.C7. [DOI] [PubMed] [Google Scholar]
- 6.Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth P, Jr, Yeager M, Gottlieb RA. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome P450 inhibitors. Proc Natl Acad Sci USA. 2004;101:1321–1326. doi: 10.1073/pnas.0308185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gross GJ, Hsu A, Falck JR, Nithipatikom K. Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol. 2007;42:687–691. doi: 10.1016/j.yjmcc.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrington LS, Falck JR, Mitchell JA. Not so EEZE: the “EDHF” antagonist 14,15 epoxyeicosa-5(Z)-enoic acid has vasodilator properties in mesenteric arteries. Eur J Pharmacol. 2004;506:165–168. doi: 10.1016/j.ejphar.2004.09.062. [DOI] [PubMed] [Google Scholar]
- 9.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:F496–F503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- 10.Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs) Prostaglandins Other Lipid Mediat. 2007;82:42–49. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ke Q, Xiao YF, Bradbury JA, Graves JP, Degraff LM, Seubert JM, Zeldin DC. Electrophysiological properties of cardiomyocytes isolated from CYP2J2 transgenic mice. Mol Pharmacol. 2007;72:1063–1073. doi: 10.1124/mol.107.035881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu T, Ye D, Wang X, Seubert JM, Graves JP, Bradbury JA, Zeldin DC, Lee HC. Cardiac and vascular KATP channels in rats are activated by endogenous epoxyeicosatrienoic acids through different mechanisms. J Physiol. 2006;575:627–644. doi: 10.1113/jphysiol.2006.113985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morisseau C, Goodrow MH, Newman JW, Wheelock CE, Dowdy DL, Hammock BD. Structural refinement of inhibitors of urea-based soluble epoxide hydrolases. Biochem Pharmacol. 2002;63:1599–1608. doi: 10.1016/s0006-2952(02)00952-8. [DOI] [PubMed] [Google Scholar]
- 14.Newman JW, Morisseau C, Harris TR, Hammock BD. The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc Natl Acad Sci USA. 2003;100:1558–1563. doi: 10.1073/pnas.0437724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nithipatikom K, DiCamelli RF, Kohler S, Gumina RJ, Falck JR, Campbell WB, Gross GJ. Determination of cytochrome P450 metabolites of arachidonic acid in coronary venous plasma during ischemia and reperfusion in dogs. Anal Biochem. 2001;292:115–124. doi: 10.1006/abio.2001.5044. [DOI] [PubMed] [Google Scholar]
- 16.Nithipatikom K, Grall AJ, Holmes BB, Harder DR, Falck JR, Campbell WB. Liquid chromatographic-electrospray ionization-mass spectrometric analysis of cytochrome P450 metabolites of arachidonic acid. Anal Biochem. 2001;298:327–336. doi: 10.1006/abio.2001.5395. [DOI] [PubMed] [Google Scholar]
- 17.Nithipatikom K, Moore JM, Isbell MA, Falck JR, Gross GJ. Epoxyeicosatrienoic acids in cardioprotection: ischemic versus reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;291:H537–H542. doi: 10.1152/ajpheart.00071.2006. [DOI] [PubMed] [Google Scholar]
- 18.Olearczyk JJ, Field MB, Kim IH, Morisseau C, Hammock BD, Imig JD. Substituted adamantyl-urea inhibitors of the soluble epoxide hydrolase dilate mesenteric resistance vessels. J Pharmacol Exp Ther. 2006;318:1307–1314. doi: 10.1124/jpet.106.103556. [DOI] [PubMed] [Google Scholar]
- 19.Seubert J, Yang B, Alyce Bradbury J, Graves J, Miller L, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–514. doi: 10.1161/01.RES.0000139436.89654.c8. [DOI] [PubMed] [Google Scholar]
- 20.Seubert JM, Sinal CJ, Graves J, DeGraff LM, Bradbury JA, Lee CR, Goralski K, Carey MA, Luria A, Newman JW, Hammock BD, Falck JR, Roberts H, Rockman HA, Murphy E, Zeldin DC. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99:442–450. doi: 10.1161/01.RES.0000237390.92932.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seubert JM, Zeldin DC, Nithipatikom K, Gross GJ. Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat. 2007;82:50–59. doi: 10.1016/j.prostaglandins.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–C1012. doi: 10.1152/ajpcell.00402.2006. [DOI] [PubMed] [Google Scholar]
- 23.Spiecker M, Darius H, Hankeln T, Soufi M, Sattler AM, Schaefer JR, Node K, Borgel J, Mugge A, Lindpaintner K, Huesing A, Maisch B, Zeldin DC, Liao JK. Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation. 2004;110:2132–2136. doi: 10.1161/01.CIR.0000143832.91812.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spiecker M, Liao J. Cytochrome P450 epoxygenase CYP2J2 and the risk of coronary artery disease. Trends Cardiovasc Med. 2006;16:204–208. doi: 10.1016/j.tcm.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Wei X, Xiao X, Hui R, Card JW, Carey MA, Wang DW, Zeldin DC. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J Pharmacol Exp Ther. 2005;314:522–532. doi: 10.1124/jpet.105.083477. [DOI] [PubMed] [Google Scholar]
- 26.Wu S, Chen W, Murphy E, Gabel S, Tomer KB, Foley J, Steenbergen C, Falck JR, Moomaw CR, Zeldin DC. Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. J Biol Chem. 1997;272:12551–12559. doi: 10.1074/jbc.272.19.12551. [DOI] [PubMed] [Google Scholar]
- 27.Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem. 1996;271:3460–3468. doi: 10.1074/jbc.271.7.3460. [DOI] [PubMed] [Google Scholar]
- 28.Yang S, Lin L, Chen JX, Lee CR, Seubert JM, Wang Y, Wang H, Chao ZR, Tao DD, Gong JP, Lu ZY, Wang DW, Zeldin DC. Cytochrome P-450 epoxygenases protect endothelial cells from apoptosis induced by tumor necrosis factor-alpha via MAPK and PI3K/Akt signaling pathways. Am J Physiol Heart Circ Physiol. 2007;293:H142–H151. doi: 10.1152/ajpheart.00783.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang W, Holmes BB, Gopal VR, Kishore RV, Sangras B, Yi XY, Falck JR, Campbell WB. Characterization of 14,15-epoxyeicosatrienoyl-sulfonamides as 14,15-epoxyeicosatrienoic acid agonists: use for studies of metabolism and ligand binding. J Pharmacol Exp Ther. 2007;321:1023–1031. doi: 10.1124/jpet.107.119651. [DOI] [PubMed] [Google Scholar]
- 30.Yang W, Tuniki VR, Anjaiah S, Falck JR, Hillard CJ, Campbell WB. Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist, 20–125iodo-14,15-epoxyeicosa-8(Z)-enoic acid. J Pharmacol Exp Ther. 2008;324:1019–1027. doi: 10.1124/jpet.107.129577. [DOI] [PubMed] [Google Scholar]