

Alzheimer’s is a progressive and fatal brain disease that is the most common form of dementia. Its characteristic pathology includes extracellular amyloid plaques that form as a result of abnormal clearance and/or increased production of amyloid-β peptides (Aβ) that are released from the amyloid precursor protein (APP).[1, 2] Metal ions, particularly Cu+/2+ and Zn2+ but also Fe2+/3+, have been implicated in two processes related to Aβ pathology: peptide aggregation and formation of reactive oxygen species (ROS).[3]
It is speculated that both APP and Aβ may have normal roles in copper homeostasis.[4, 5] It has also been shown in vitro that Aβ can act as an antioxidant by quenching free radicals and/or by chelating copper.[6, 7] Other evidence, however, suggests that Aβ-Cu complexes are pro-oxidant and directly culpable of neurotoxicity.[8] In vitro, Aβ in the presence of copper or iron and reducing agents like ascorbate produces H2O2,[9–11] which can subsequently react with the reduced metal ions to produce OH• via the Fenton reaction (Eq. 1).[12, 13] Metal-mediated H2O2 generation appears at an early stage during in vitro Aβ aggregation,[10, 11] which supports the notion that soluble Aβ-Cu species are responsible for the oxidative damage that is one of the earliest pathological events in Alzheimer’s disease.[14] Furthermore, copper has been shown to intensify Aβ toxicity in primary cortical neurons.[9, 10, 15] Like Cu2+, Zn2+ also promotes Aβ aggregation in vitro, but the Zn-induced aggregates may be neuroprotective.[16–18]
| 1 |
One hypothesis to reconcile the seemingly contradictory evidence related to metals, Aβ, and oxidative stress is that metal binding and Aβ aggregation may represent an initial, protective response to dampen production of ROS. Excessive H2O2 and an overburden of copper could eventually push the system into a vicious cycle that switches Aβ-Cu activity from antioxidant to pro-oxidant.[19] During this stage, metal exchange with Zn2+ could promote further Aβ aggregation as a defense against copper-induced damage. While chelating agents are known to reverse metal-induced aggregates, this model suggests that disaggregating plaques alone could have the unintended consequence of exacerbating oxidative damage. [19]
Metal chelating agents have appeared as a compelling strategy for Alzheimer’s therapies.[20] In particular, 8-hydroxyquinoline (8HQ) derivatives clioquinol and PBT2 have shown promising results in mouse models and phase IIa clinical trials of Alzheimer’s patients.[21, 22] These compounds inhibit metal-induced Aβ aggregation and ROS generation. While these reports encourage further development of metal-targeted compounds for neurodegenerative disease, there remain significant concerns about manipulating metal distribution in the brain.[23, 24] Given the complexity of the metallobiology in Alzheimer’s, it is particularly challenging to design metal-binding agents that can mitigate the damaging effects of metals while preserving their beneficial properties. In our lab, we are developing prochelators that are designed to bind metals only under conditions of oxidative stress.[25–27] The indications that elevated production of H2O2 by deviant Cu-Aβ interactions may trigger neurodegeneration suggested to us that prochelators activatable by H2O2 may be beneficial for managing a metal burden at locations of disease progression without stimulating widespread metal redistribution. Here, we present a boronic ester-masked 8-hydroxyquinoline derivative called QBP that converts to 8HQ in the presence of H2O2. Once converted to 8HQ, it is available for coordinating metal ions, as shown in Scheme 1 for Cu2+. The protecting group is ultimately released as pinanediol and non-toxic boric acid.[28, 29]
Scheme 1.

Oxidation of QBP by H2O2 and subsequent binding of copper by 8-hydroxyquinoline (8HQ).
QBP was synthesized by reacting commercially available quinoline boronic acid (QBA) with pinanediol in a Dean Stark apparatus. The X-ray crystal structure is shown in Figure 1. QBP is stable in aqueous solution between pH 5–8 over the course of 10 h, although some hydrolysis to QBA occurs at lower and higher pH values, as monitored by UV-Vis and mass spectrometry (data not shown).
Figure 1.
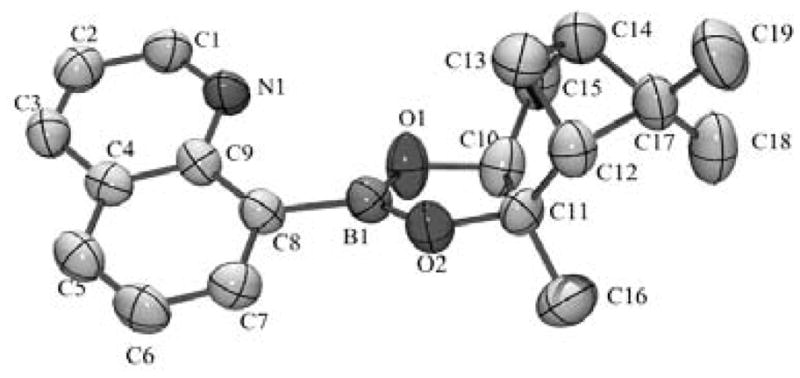
Xray structure of QBP. Thermal ellipsoids are shown at 50% probability. Hydrogen atoms have been omitted for clarity.
With the phenol of 8HQ masked by the pinanediol boronic ester, the QBP prochelator should have little to no affinity for metal ions. A comparison of the UV-vis spectra in Figure 2 of QBP alone or in the presence of Cu2+ for an hour reveals no change in spectral features and validates the assumption that QBP does not interact with Cu2+ in its prochelator form. Addition of H2O2, however, causes a new spectrum to appear that matches that of [Cu(8HQ)2], consistent with the reaction in Scheme 1.
Figure 2.

UV-vis spectra of 100 μM QBP in PBS pH 7.4 in the absence ( ) and presence (
) and presence ( ) of 50 μM Cu(Gly)2, and 60 min after treatment of the QBP/Cu sample with 4 mM H2O2 (
) of 50 μM Cu(Gly)2, and 60 min after treatment of the QBP/Cu sample with 4 mM H2O2 ( ) The resulting spectrum is nearly identical to that of independently prepared [Cu(8HQ)2] (
) The resulting spectrum is nearly identical to that of independently prepared [Cu(8HQ)2] ( ).
).
In order to determine the rate of conversion of QBP to 8HQ by H2O2, reactions were monitored spectrophotometrically under pseudo first-order conditions of excess H2O2. The observed rate constants (kobs) were plotted against peroxide concentration to give a rate constant k of 0.25 M−1s−1 (see the Supporting Information).
Chelators, including those based on the 8HQ motif, have been shown to have a dramatic effect on metal-induced Aβ aggregation.[30] Here, we monitored the metal-induced aggregation of Aβ by two complementary methods: light scattering, which reports the change in solution turbidity as a result of precipitate formation, and soluble protein concentration, which ascertains the fraction of protein that did not precipitate from solution. These methods provide a preliminary assessment of aggregation propensity, but do not report on detailed morphological changes of aggregates or fibrils.[31] In the light scattering assay, turbidity is assessed as the difference in absorbance at 405 nm between the sample and its matched control that contains the same components but without Aβ. The black, left-hand bars in Figure 3 show that, as previously observed by others,[32, 33] both Cu2+ and Zn2+ increase the turbidity of Aβ samples, with Zn2+ causing a more profound effect. The presence of 2 equiv of 8HQ inhibits the Cu2+-induced aggregation and significantly reduces Zn2+-induced aggregation. After an hour of incubation, the aggregated peptide was removed by centrifugation and the amount of soluble peptide remaining was determined by a BCA assay. The gray, right-hand bars in Figure 3 show that samples treated with Cu2+ alone have only 60% soluble peptide, while those containing 8HQ have greater than 90% soluble Aβ. Similar results are seen for Zn2+, with greater than 80% of the peptide remaining soluble when 8HQ is present. These data corroborate the turbidity assay and show that 8HQ present at the outset of metal–Aβ incubations prevents aggregation. If 8HQ is added to pre-formed metal–Aβ aggregates, the turbidity of the solutions decreases (see Supp. Info.), demonstrating that 8HQ can both prevent and reverse Aβ aggregation under these conditions.
Figure 3.
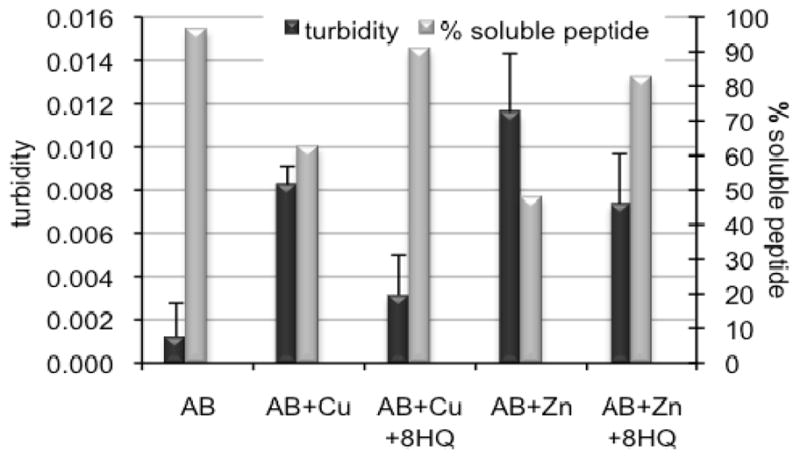
Turbidity assay, as monitored by the difference in absorbance at 405 nm between the sample and its matched control that does not contain Aβ. Samples containing 10 μM Aβ were incubated with 10 μM Cu(Gly)2 or ZnCl2 in the presence or absence of 20 μM 8HQ for 1 h at 37 °C. A405 readings were taken 1 h after mixing (black, left-hand bars). The % soluble peptide remaining after 1 h was determined by a BCA protein concentration assay (gray, right-hand bars) on centrifuged samples.
QBP, on the other hand, does not interfere with metal-induced Aβ aggregation. The black, left-hand bars of Figure 4 show that the presence of QBP does not change the turbidity of solutions containing Aβ alone or Aβ plus Cu2+. These results show that QBP itself neither induces nor prevents metal-induced Aβ aggregation, as predicted based on its lack of metal-binding ability.
Figure 4.

Turbidity assay, as monitored by the difference in absorbance at 405 nm between the sample and its matched control that does not contain Aβ. Samples contain combinations of 10 μM Aβ, 10 μM Cu2+ (provided as Cu(Gly)2), 20–100 μM QBP, or 10–20 μM 8HQ, as indicated. A405 readings were taken 1 h after mixing (black bars), then again 30 min after addition of 1 mM H2O2 (gray bars).
In order to show that the 8HQ that is generated in situ from the reaction of QPB and H2O2 is capable of reversing Aβ aggregation, 1 mM H2O2 was added to each of the samples in Figure 4 that already contained Cu2+-aggregated Aβ. The gray bars in Figure 4 show that H2O2 alone does not reduce the turbidity of samples containing Aβ and Cu2+, nor does it increase the turbidity of samples containing Aβ alone or Aβ/Cu2+/8HQ. These results show that H2O2 itself does not influence the aggregation state of Aβ. In contrast, samples that contain Aβ/Cu2+/QBP show a significant decrease in turbidity 30 min following H2O2 addition. This result is consistent with conversion of QBP to 8HQ, which can subsequently bind Cu2+ and reverse Aβ aggregation. Given the concentrations of QBP and H2O2 present in the samples and the rate constant for prochelator-to-chelator conversion, this reaction is predicted to generate 9–45 μM 8HQ, depending on the initial QBP concentration. The highest concentration is certainly sufficient for complete binding of Cu2+ in a 1:2 complex, although as shown in the Figure, even the lower concentration is effective.
Confirmation that [Cu(8HQ)2] is generated in the reaction of Aβ/Cu/QBP/H2O2 described in Figure 4 comes from mass spectral detection of 352 m/z, which is consistent with [Cu(8HQ)2]. Further evidence comes from the UV-vis spectrum of the reaction mixture, shown in Figure 5. The complex [Cu(8HQ)2] has a characteristic absorbance band at 375 nm, which is clearly visible in samples that contain Aβ, Cu2+ and 8HQ. Samples that contain Aβ/Cu/QBP/H2O2 also show this characteristic peak, verifying that [Cu(8HQ)2] has indeed been generated. This absorbance band tails into the 405 nm region used for monitoring turbidity, but its contribution was appropriately accounted for by subtracting the matched control from the sample value, ensuring that residual A405 reported in Figures 3 and 4 can be attributed to turbidity.
Figure 5.

UV-vis analysis of H2O2-treated Aβ samples from Figure 4.  : Aβ+H2O2;
: Aβ+H2O2; : Aβ+Cu+8HQ+H2O2;
: Aβ+Cu+8HQ+H2O2; : Aβ+Cu+QBP+H2O2.
: Aβ+Cu+QBP+H2O2.
The previous experiment contained a relatively high concentration (1 mM) of H2O2 that was added in a single bolus. Several groups have shown that combinations of Aβ, Cu2+ and reductants produce significant amounts of H2O2 from O2.[10, 34] Therefore, to investigate whether these conditions of more biologically relevant H2O2 production are sufficient for activating QBP, samples of Aβ, Cu2+, ascorbic acid, and either 8HQ or QBP were monitored for H2O2 production with the Amplex Red/horseradish peroxidase (HRP) assay. Figure 6 shows the concentration of H2O2 detected by Amplex Red after a 1-h incubation of 200 nM Cu(Gly)2, 200 nM Aβ, 10 μM ascorbic acid and either 8HQ or QBP over a range of concentrations in sodium phosphate buffer at pH 7.4. Samples that contain Cu2+ and ascorbate (with or without Aβ) provide just over 300 nM detectable H2O2 when the Amplex Red reagents are added at the end of the 1-h incubation period. This result shows that Aβ neither prevents nor promotes H2O2 production by copper under these conditions when compared to copper in the presence of glycine as a carrier ligand, a result also seen by others.[12] In contrast, 8HQ inhibits H2O2 production, even when present at only a 0.25 equiv of Cu2+. When 8HQ is present at a 2:1 ratio to Cu2+, the amount of detectable H2O2 diminishes to one third that detected in the absence of 8HQ. This result confirms that 8HQ coordinates Cu2+ in a manner that diminishes its ability to catalyze the formation of H2O2 from O2 in the presence of reductant.
Figure 6.

Hydrogen peroxide detection by the Amplex Red/HRP assay for samples containing various combinations of 200 nm Aβ, 200 nm Cu(Gly)2, 10 μM ascorbic acid, 2–200 μM QBP, 200–400 nM 8HQ, in a total volume of 50 μL buffer (50 mM hepes, 150 mM NaCl). Samples were incubated at 37 °C for 1 h before 50 μL of Amplex Red/HRP reagent was added. Fluorescence readings were taken with λex = 485 and λem = 590 nm.
When QBP is added to the reaction mixture in place of 8HQ, similar results are obtained. As shown in Figure 6, samples that contain 200 μM QBP along with Cu2+, ascorbate, and Aβ result in detection of ~100 nM H2O2, which is a third of the concentration obtained in the absence of chelator.
If the Amplex Red/HRP reagents are included at the beginning of the incubations, then up to 500 nM H2O2 is detected (not shown). These data suggest that H2O2 degrades during the incubation period, possibly via Fenton reaction or hydrolysis. This experiment also provides the maximum amount of H2O2 produced by the Cu/Aβ/ascorbic acid system under these conditions. Given this number and the rate constant for the QBP-to-8HQ conversion, the 200 μM QBP sample in Figure 6 gives a calculated yield of 90 nM 8HQ. The results in Figure 6 are consistent with this prediction, as they indeed show that 200 μM QBP provides protection against H2O2 formation that is similar to that of 50–200 nM 8HQ.
A current hypothesis about Alzheimer’s is that oxidative stress is an early event in disease progression and that copper may be a culprit in promoting further oxidative damage. The results presented here indicate that prochelator QBP can be activated under conditions that mimic early Alzheimer’s pathology where copper, Aβ, and biological reductants exacerbate ROS formation. Importantly, the prochelator itself does not prevent or disaggregate metal-promoted Aβ aggregates if they are not accompanied by elevated H2O2. This feature may be beneficial as it may not be desirable to disaggregate stable plaques in the absence of ROS. Once activated to its unmasked form, however, the released 8HQ diminishes copper’s ROS-forming reactivity, resolubilizes existing metal-associated aggregates, and inhibits further Aβ aggregation. The H2O2 trigger incorporated into QBP provides just one level of specificity for targeting a chelating agent to a local environment with an elevated H2O2 concentration; it does not target the agent to amyloid-dense regions. Future work, therefore, will focus on incorporating amyloid-binding units[35, 36] into the prochelator structure to create multifunctional agents directed to both the structure and reactivity of amyloid-beta.
Experimental Section
Experimental procedures, including synthesis, turbidity and hydrogen peroxide assays, peptide preparation and oxidation kinetic studies, are available in the Supporting Information. CCDC 743258 (QBP) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Supplementary Material
Acknowledgments
KJF thanks the Sloan Foundation, the Camille and Henry Dreyfus Foundation, and the National Institutes of Health (grant GM084176) for supporting this work; MGD acknowledges a C.R. Hauser Fellowship from Duke University.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org.
References
- 1.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. J Biol Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bush AI. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 4.Gaggelli E, Kozlowski H, Valensin D, Valensin G. Chem Rev. 2006;106:1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 5.Treiber C, Simons A, Strauss M, Hafner M, Cappai R, Bayer TA, Multhaup G. J Biol Chem. 2004;279:51958–51964. doi: 10.1074/jbc.M407410200. [DOI] [PubMed] [Google Scholar]
- 6.Zou K, Gong JS, Yanagisawa K, Michikawa M. J Neurosci. 2002;22:4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baruch-Suchodolsky R, Fischer B. Biochemistry. 2009;48:4354–4370. doi: 10.1021/bi802361k. [DOI] [PubMed] [Google Scholar]
- 8.Rauk A. Dalton Trans. 2008:1273–1282. doi: 10.1039/b718601k. [DOI] [PubMed] [Google Scholar]
- 9.Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JDA, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, Scarpa RC, Saunders AJ, Lim J, Moir RD, Glabe C, Bowden EF, Masters CL, Fairlie DP, Tanzi RE, Bush AI. J Biol Chem. 1999;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 10.Opazo C, Huang X, Cherny RA, Moir RD, Roher AE, White AR, Cappai R, Masters CL, Tanzi RE, Inestrosa NC, Bush AI. J Biol Chem. 2002;277:40302–40308. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 11.Tabner BJ, El-Agnaf OMA, Turnbull S, German MJ, Paleologou KE, Hayashi Y, Cooper LJ, Fullwood NJ, Allsop D. J Biol Chem. 2005;280:35789–35792. doi: 10.1074/jbc.C500238200. [DOI] [PubMed] [Google Scholar]
- 12.Nadal RC, Rigby SEJ, Viles JH. Biochemistry. 2008;47:11653–11664. doi: 10.1021/bi8011093. [DOI] [PubMed] [Google Scholar]
- 13.Guilloreau L, Combalbert S, Sournia-Saquet A, Mazarguil H, Faller P. Chem BioChem. 2007;8:1317–1325. doi: 10.1002/cbic.200700111. [DOI] [PubMed] [Google Scholar]
- 14.Castellani RJ, Honda K, Zhu XW, Cash AD, Nunomura A, Perry G, Smith MA. Age Res Rev. 2004;3:319–326. doi: 10.1016/j.arr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Smith DP, Smith DG, Curtain CC, Boas JF, Pilbrow JR, Ciccotosto GD, Lau TL, Tew DJ, Perez K, Wade JD, Bush AI, Drew SC, Separovic F, Masters CL, Cappai R, Barnham KJ. J Biol Chem. 2006;281:15145–15154. doi: 10.1074/jbc.M600417200. [DOI] [PubMed] [Google Scholar]
- 16.Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang X, Farrag YW, Perry G, Bush AI. J Biol Chem. 2000;275:19439–19442. doi: 10.1074/jbc.C000165200. [DOI] [PubMed] [Google Scholar]
- 17.Cardoso SM, Rego AC, Pereira C, Oliveira CR. Neurotox Res. 2005;7:273–281. doi: 10.1007/BF03033885. [DOI] [PubMed] [Google Scholar]
- 18.Meloni G, Sonois V, Delaine T, Guilloreau L, Gillet A, Teissie J, Faller P, Vasak M. Nat Chem Biol. 2008;4:366–372. doi: 10.1038/nchembio.89. [DOI] [PubMed] [Google Scholar]
- 19.Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, Smith MA, Martins RN. Brain Res Rev. 2003;43:1–16. doi: 10.1016/s0165-0173(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 20.Bush AI. J Alzheimer’s Dis. 2008;15:223–240. doi: 10.3233/jad-2008-15208. [DOI] [PubMed] [Google Scholar]
- 21.Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 22.Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- 23.Madsen E, Gitlin JD. Annu Rev Neurosci. 2007;30:317–337. doi: 10.1146/annurev.neuro.30.051606.094232. [DOI] [PubMed] [Google Scholar]
- 24.Benvenisti-Zarom L, Chen J, Regan RF. Neuropharmacol. 2005;49:687–694. doi: 10.1016/j.neuropharm.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 25.Charkoudian LK, Dentchev T, Lukinova N, Wolkow N, Dunaief JL, Franz KJ. J Inorg Biochem. 2008;102:2130–2135. doi: 10.1016/j.jinorgbio.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charkoudian LK, Pham DM, Franz KJ. J Am Chem Soc. 2006;128:12424–12425. doi: 10.1021/ja064806w. [DOI] [PubMed] [Google Scholar]
- 27.Charkoudian LK, Pham DM, Kwan A, Vangeloff A, Franz KJ. Dalton Trans. 2007:5031–5042. doi: 10.1039/b705199a. [DOI] [PubMed] [Google Scholar]
- 28.Weir RJ, Fisher RS. Toxicol Appl Pharmacol. 1972;23:351–364. doi: 10.1016/0041-008x(72)90037-3. [DOI] [PubMed] [Google Scholar]
- 29.Rezanka T, Sigler K. Phytochem. 2008;69:585–606. doi: 10.1016/j.phytochem.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 30.Scott LE, Orvig C. Chem Rev. 2009;109:4885–4910. doi: 10.1021/cr9000176. [DOI] [PubMed] [Google Scholar]
- 31.Mancino AM, Hindo SS, Kochi A, Lim MH. Inorg Chem. 2009;48:9596–9598. doi: 10.1021/ic9014256. [DOI] [PubMed] [Google Scholar]
- 32.Huang X, Atwood CS, Moir RD, Hartshorn MA, Vonsattel JP, Tanzi RE, Bush AI. J Biol Chem. 1997;272:26464–26470. doi: 10.1074/jbc.272.42.26464. [DOI] [PubMed] [Google Scholar]
- 33.Storr T, Scott LE, Bowen ML, Green DE, Thompson KH, Schugar HJ, Orvig C. Dalton Trans. 2009:3034–3043. doi: 10.1039/b902545f. [DOI] [PubMed] [Google Scholar]
- 34.Deraeve C, Boldron C, Maraval A, Mazarguil H, Gornitzka H, Vendier L, Pitié M, Meunier B. Chem Eur J. 2008;14:682–696. doi: 10.1002/chem.200701024. [DOI] [PubMed] [Google Scholar]
- 35.Dedeoglu A, Cormier K, Payton S, Tseitlin KA, Kremsky JN, Lai L, Li XH, Moir RD, Tanzi RE, Bush AI, Kowall NW, Rogers JT, Huang XD. Exp Gerontol. 2004;39:1641–1649. doi: 10.1016/j.exger.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Rodriguez C, Sanchez de Groot N, Rimola A, Alvarez-Larena A, Lloveras V, Vidal-Gancedo J, Ventura S, Vendrell J, Sodupe M, Gonzalez-Duarte P. J Am Chem Soc. 2009;131:1436–1451. doi: 10.1021/ja806062g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.