

Abstract
Bone marrow-derived cells (BMDC) have been shown to graft injured tissues, differentiate in specialized cells, and participate in repair. The importance of these processes in acute lung bacterial inflammation and development of fibrosis is unknown. The goal of this study was to investigate the temporal sequence and lineage commitment of BMDC in mouse lungs injured by bacterial pneumonia. We transplanted GFP-tagged BMDC into 5-Gy-irradiated C57BL/6 mice. After 3 months of recovery, mice were subjected to LD50 intratracheal instillation of live E. coli (controls received saline) which produced pneumonia and subsequent areas of fibrosis. Lungs were investigated by immunohistology for up to 6 months. At the peak of lung inflammation, the predominant influx of BMDC were GFP+ leukocytes. Postinflammatory foci of lung fibrosis were evident after 1–2 months. The fibrotic foci in lung stroma contained clusters of GFP+ CD45+ cells, GFP+ vimentin-positive cells, and GFP+ collagen I-positive fibroblasts. GFP+ endothelial or epithelial cells were not identified. These data suggest that following 5-Gy irradiation and acute bacterial pneumonia, BMDC may temporarily participate in lung postinflammatory repair and stromal remodeling without long-term engraftment as specialized endothelial or epithelial cells.
Keywords: Lung, Stem cells, Bone marrow, Pneumonia, Fibrosis
Introduction
Acute inflammation in the lung is a complex multistage process in which the initial inflammatory reaction involving necrosis and apoptosis of the lung cells is followed by a recovery phase. After the initial inflammatory phase, remodeling can result in the formation of pulmonary fibrosis [1–5]. Both inflammatory cells and different populations of fibroblasts participate in these phases of the process [6].
Transformation of bone marrow-derived stem cells (BMDC) into different types of cells in the lung has been noted in several in vivo observations [7–10]. Krause et al. [10] showed that marrow engraftment in the lung resulted in an epithelial-like transformation. These results were not confirmed by the Weissman group [11], who observed the engraftment of only leukocyte lineages in their experiments. Further studies suggested marrow engraftment in the lungs as epithelial cells, which was facilitated by inflammatory reactions [12–15].
To achieve significant chimerism in the lung, high doses of radiation were required. Such high doses, however, produced substantial injury to the lung epithelium [7]. Yamada et al. [15] found stem cells differentiated into endothelial and epithelial cells in the lung after lethal irradiation and bone marrow reconstitution with liver-derived stem cells, which was followed by LPS instillation. Additional evidence for lung engraftment following bone marrow or lung transplantation comes from biopsies of human lung [16–19]. Other aspects of adult stem cell biology in lung disease have been thoroughly reviewed recently [20, 21].
We recently reported that intratracheal instillation of bone marrow-derived mesenchymal stem cells provides survival advantage in endotoxin-induced lung injury [22], although the benefit was not related to engraftment of these cells to the lung. In 5-Gy-irradiated chimeric mouse subjected to naphthalene-induced lung injury, we found very limited engraftment of bone marrow-derived mesenchymal stem cells in the bronchial epithelium [23], although in vitro these cells were capable of endodermal differentiation [24]. Homing of BMDC to the lung and their possible differentiation into specialized cells of endothelial or epithelial lining, as well as participation in postinflammatory lung fibrosis following acute bacterial pneumonia, have not yet been studied.
The goal of this study was to investigate the temporal sequence of appearance of transplanted BMDC in the lungs of 5.05-Gy-irradiated animals. Our hypothesis was that BMDC may participate in lung inflammation and postinflammatory repair and remodeling following bacterial pneumonia. The first aim was to determine the extent of infiltration by BMDC in noninjured lungs subjected to low-level irradiation. The second aim was to determine the temporal pattern of participation of BMDC in lung repair following acute bacterial pneumonia. The third aim was to characterize the lineage commitment of BMDC, incorporated in the lung during the development of fibrosis.
Methods
Animal Experiments and Preliminary Studies
In preliminary studies whole bone marrow was first harvested from 6–12-month-old C57BL/6 male GFP+ Tg mice (C57BL/6 TgN[ACT6EGFP], The Jackson Laboratory, Bar Harbor, ME) and 106 cells per animal were transplanted into nonirradiated newborn (age = 24–48 h, n = 8, injection performed into temporal vein) mice and irradiated (5.05-Gy dose of total body irradiation given at one time 24 h before transplantation) 3–5-week-old (n = 4) C57BL/6 mice. As no incorporation of BMDC into the lung structures up to 6 months of observation was observed in these experiments (data not shown), further experiments were performed using bone marrow from donors aged 2–4 weeks. Similarly, additional preliminary experiments (n = 6) revealed that incorporation of BMDC into the lungs 2 months after whole bone marrow transplantation occurred in 5.05-Gy-irradiated recipients 3–5 weeks old, but not 1 year old (data not shown). Therefore, further experiments were performed with donors 2–4 weeks old and recipients 3–5 weeks old.
Donor bone marrow was harvested from 2–4-week-old C57BL/6 male GFP+ Tg mice (C57BL/6 TgN[AC-T6EGFP], The Jackson Laboratory). Recipients were strain-matched 3–5-week-old C57BL/6 mice.
Bone Marrow (BM) Preparation
Animals were euthanized by CO2 inhalation and BM was harvested by flushing tibias and femurs with Ca++Mg++-free phosphate-buffered saline (PBS) with penicillin-streptomycin. Cells were washed, dispersed through a 27-G needle, filtered through 100-μm-pore filter, and washed twice in PBS. Cell were checked for viability with Trypan blue stain and were more than 90% viable.
Recipients
Thirty-five recipients received whole-body irradiation (5.05 Gy) 24 h before transplantation. Under general anesthesia (pentobarbital, 10 mg/kg), an incision was made on the neck and animals were infused with 106 BM cells in 0.2 ml of PBS into the jugular vein. In three additional controls, whole BM from non-GFP mice was transplanted into 5.05-Gy-irradiated recipients.
In the experimental group, mice were subjected to development of E. coli pneumonia by intratracheal instillation of washed E. coli JM109 (107 cfu/animal). This dose produced approximately 50% lethality. The LD50 dose of E. coli was determined in separate experiments (n = 20). Under general anesthesia, 50 μl of E. coli suspension was instilled into the trachea via a 25-G needle. The mice were allowed to recover in 100% O2. Death occurred within 1 week in 15 of 25 instilled animals; 10 animals survived up to 6 months and were used for morphologic analyses. In the control mice (n = 10), PBS was infused intratracheally.
Immunohistologic Analysis of the Lung
Animals were euthanized in 100% CO2. The chest was opened and an incision was made in the left atrium. The pulmonary artery was flushed with ice-cold PBS with 10% sucrose (5 ml) under pressure of 20 cmH2O. A catheter was inserted into the trachea and the lungs were fixed through the airways with 200-proof ethanol at 25 cmH2O. The lungs were then removed from the chest and the lung lobes were maintained in pure ethanol at 4°C for further analyses. Before cryosectioning, lungs were washed in a large volume of PBS for 24 h, placed in 30% sucrose (in PBS) for 12 h, frozen in liquid nitrogen, embedded in OCT compound, and cut in a Tissue-Tek cryostat at −25°C. Tissue slices (10 μm) were mounted on polylysine-coated glass and kept at −20°C for immunostaining.
Paraffin Sections and Immunostaining
Paraffin sectionin and hematoxylin and eosin staining of the lung were performed at the Pathology Department, University of California, San Francisco. Paraffin sections were deparaffinized in xylene and stained for GFP as described below, with costaining by propidium iodine (PI, 0.5 μg/ml, 3 min). Trichrome staining was performed by Excalibur Pathology Inc. (Moore, OK).
Immunofluorescence
Slides were washed with PBS, postfixed with ethanol, washed twice, exposed to 0.2% Triton X-100 in PBS for 5 min, and then washed with PBS twice again. Tissues were further blocked with 3% goat serum, 2% horse serum, and 3% BSA/0.1% Tween 20 in 4× SSC for 20 min. Appropriate primary antibody (in 2× SSC/1% BSA/0.1% Tween 20) was added for 1 h at 37°C, and slices were washed three times with PBS and blocked with 3% goat serum, 2% horse serum, 3% BSA/0.1% Tween 20 in 4× SSC for 20 min. Red fluorescent dye (Rhodamine, Texas Red, Alexa Fluor® 633, or Cy5)-conjugated secondary antibody (in 2× SSC/1% BSA/0.1% Tween 20) was added for 1 h at 37°C. Control tissues were stained with isotype mouse or rabbit primary antibody. Slides were washed three times with PBS, mounted on glass in Fluoro-Guard antifade reagent (BioRad, Hercules, CA), and coverslips were applied. Laser confocal fluorescence microscopy (Axiovert 100, LSM 510, Zeiss, Germany) was performed at the UC Davis Department of Medicine Confocal Imaging Core Facility or at Confocal Imaging Facility (LSM Paskal, Zeiss, Germany) Institute of Cytology, Russian Academy of Science.
Antibodies
The following antibodies were used: anti-green fluorescent protein rabbit IgG, 1:50 (Sigma, St. Lous, MI); for staining epithelial cells, mouse monoclonal anti-pan-cytokeratin, 1:50 (Calbiochem, San Diego, CA, clone B311.1); mouse monoclonal anti-cytokeratin 14, 1:50 (Harlan Sera-Labs, clone LL002); for staining endothelial cells, monoclonal anti-mouse CD31 (PECAM 1), 1:100 (BD Biosciences, clone 390); rabbit polyclonal factor VIII–related antigen/von Willebrand factor Ab-1, 1:50 (NeoMarkers, Fremont, CA); fibroblasts and myofibroblasts, mouse monoclonal anti-actin smooth muscle Ab-1, 1:200 (NeoMarkers, clone 1A4); mouse monoclonal vimentin Ab-2, 1:400 (NeoMarkers, clone V9); rabbit polyclonal anti-collagen I, 1:10 (Chemicon Millipore, Billerica, MA); staining for leukocytes, rat anti-mouse CD45, 1:20 (BD Biosciences, clone 30-F11); mouse monoclonal anti-CD3, CD11b, CD34 were all used at 1:50 dilution (Serotec, England). Secondary rhodamine, Cy-5, and Texas Red or Alexa-Fluor® 633-labeled anti-mouse, anti-rat, and anti-rabbit antibodies were from Molecular Probes (Eugene, OR).
Statistical Analyses
At least six different sections from each lung were used for analyses. Cell counting was performed on 20 different randomly selected visual fields; numbers of GFP+ cells were determined as percentage of the total number of cells (counted by numbers of PI-stained nuclei). In antibody-specific staining, the numbers of ligand-positive cells were expressed as percentage of total numbers of GFP+ cells in 20 different visual fields. All data are presented as the mean ± SE.
Results
Incorporation of BMDC in the Lung Following 5-Gy Radiation Doses
Total body irradiation by 5.05 Gy, not followed by BM transplantation, resulted in 40% mortality within 1 month. A dose of 5.05-Gy total body irradiation followed by transplantation of 106 whole BM cells resulted in 10% mortality within 1 month. Our experience demonstrated that mortality following irradiation depended upon the age of recipients, housing conditions, and treatment. The animals we used were young, kept in a nonsterile environment, and were not treated with antibiotics; therefore, in our opinion the 10% that died did so from infections before engraftment occurred. We did not study animals that died for chimerism. Following BM cell transplantation, some mice were killed at 1, 2, or 3 months before the instillation of bacteria. Three months post-transplantation, the experimental group received an intratracheal instillation of E. coli and the control group received an intratracheal instillation of PBS. The mice were then killed at 1 week, 1.5–2 months, 4 months, and 6 months following instillation.
Specific GFP fluorescence was confirmed by staining paraffin-sectioned and cryosectioned tissues with antibodies against GFP. Processing for paraffin sectioning significantly quenched native GFP fluorescence, but in paraffin sections fluorescence of GFP+ cells stained with anti-GFP antibody was clearly identified and observed only in chimeric animals transplanted with GFP+ bone marrow cells (Fig. 1), but not in mice transplanted with GFP− bone marrow cells. We found cells expressing GFP in all recipients and structures containing GFP protein in multiple organs, including the liver, spleen, bone marrow, heart, and brain. The percentage of GFP+ cells in bone marrow was 25–50% in all animals at the time of sacrifice, which occurred up to 9 months after BM transplantation. There was evidence of GFP+ cells in the noninjured lungs of all the examined mice, but only a small percentage of the cells were GFP+ (Fig. 2A, B).
Fig. 1.

Staining for GFP in paraffin and cryosections in the mouse lung at 6–8 weeks following GFP+ BMDC transplantation and E. coli pneumonia. A, B Confocal microscopy images of mouse lungs, paraffin-sectioned and stained for GFP with FITC-labeled antibody (green fluorescence) and costained for DNA with propidium iodine (PI). Original magnification ×40. A Control staining with isotype rabbit primary antibody, no GFP signal is present. B Staining with anti-GFP antibody. Upper-left panel: red fluorescence image (PI); upper-right panel: green fluorescence image (FITC); lower-left panel: image of tissue in transmitted laser light; lower-right panel: combined image. C, D Preservation of specific GFP native fluorescence in cryosectioned tissues and conformation of GFP fluorescence. Confocal microscopy images of mouse lungs transplanted with GFP+ bone marrow. Staining with anti-GFP antibody, labeled with Alexa Fluor® 633 (Red). C Control staining with rabbit isotype antibody. D Staining with anti-GFP antibody only. In C and D, signals from GFP (green) and anti-GFP antibody (red) colocalize. Upper-left panel: red fluorescence image (Alexa Fluor® 633); upper-right panel: green fluorescence image (GFP); lower-left panel: image of tissue in transmitted laser light; lower-right panel: combined image
Fig. 2.

Chimerism in noninjured and injured lung. Confocal images of cryosectioned lung, nuclear stained with propidium iodine (red fluorescence). GFP+ cells are clearly visible as indicated by bright green fluorescence. A, B Lungs from mice after 1 month of 5.05-Gy irradiation and GFP+ BM cell transplantation (control group). Original magnification ×20. C–E Progeny of transplanted BMDC participate in acute lung inflammation. Confocal images of the lung, costaining PI (red), GFP+ cells are bright green (arrows). C After intratracheal administration of E. coli, lungs are infiltrated with GFP+ BMDC (bacterial pneumonia, 1 week). Abscess (bright red E. coli) is surrounded by GFP+ BMDC (arrow, original magnification ×40), costaining PI (red). D Multiple GFP+ BMDC are present in infected alveoli (original magnification ×40), costaining PI (red). E GFP+ cells in the peribronchial interstitium (arrows), original magnification ×20, costaining PI (red)
Incorporation of BMDC in the Lung Following Acute Pneumonia
Intratracheal instillation of E. coli resulted in the development of severe pneumonia with approximately 50% mortality within 1 week. Examples of lung inflammation in these animals are shown in Fig. 2C–E. There was massive infiltration by GFP+ cells, most of which were leukocytes. We also observed formation of abscesses in the lung tissue (Fig. 2C). There was also participation of GFP+ cells in the inflammatory responses in the alveoli (Fig. 2D). In the following 2-month period, the degree of inflammation decreased in survivors as determined by postmortem analyses. Later, GFP+ cells were predominantly located in the perivascular-peribronchial interstitial spaces (Fig. 2E).
We further analyzed the differentiation fate of GFP+ cells. Because cell fusion may be responsible for the display of tissue-specific cell proteins by BMDC, attention was given to determine ploidy of GFP+ cells by examining most GFP+ cells in the lungs of chimeric animals costained for DNA. We did not observe polyploid GFP+ cells in the lung tissues. Tissues were costained with the following: antibodies to leukocytes, CD45, CD3, CD11B, and CD34; antibodies to epithelial cells, cytokeratin 14, pan-cytokeratin; antibodies to endothelial cells, factor VIII (VonWillebrand factor) and CD31 (PE-CAM-1); and antibodies to fibroblasts and myofibroblasts, vimentin, collagen I, and smooth-muscle actin. Positive staining for leukocytes, epithelial cells, and fibroblast GFP+ cells was found in different regions of the lung (alveolar spaces or peribronchial interstitium). Typical images are shown in Fig. 3. Clones of CD45+ and vimentin-positive GFP+ cells only were found, but these findings were rare.
Fig. 3.

Examples of incorporation of different cell types of GFP+ cells in the lung 6 weeks after induction of pneumonia. A GFP+ cells in the bronchial wall, costaining PI (original magnification ×25). B CD45+ GFP+ cells in the bronchial wall. Lung was costained for CD45 common leukocyte antigen with red-fluorescent secondary antibody. Split image: upper-left: red fluorescence only; upper-right: green fluorescence only; lower-left: combined image, original magnification ×65. Some GFP+ cells displayed pan-cytokeratin in bronchial and alveolar epithelium following acute inflammation. C–F Confocal images of the lung tissue, stained for pan-cytokeratin (red fluorescence: Texas Red). GFP+ cells have bright green fluorescence. C Orthogonal view of bronchial wall. Z stack of images of the bronchial wall. GFP+ cells are clearly present inside epithelium (magnification ×40). D Image of bronchial wall, examples of GFP+ cells expressing pan-cytokeratin (blue arrows) and nonexpressing cytokeratin (green arrow, “positive control” cell, magnification ×65). E GFP+ pan-cytokeratin-positive cells in the alveolar walls, magnification ×25. F Image of alveolar wall, examples of GFP+ cells expressing pan-cytokeratin (blue arrows) and GFP− epithelial cells expressing pan-cytokeratin (“negative control” cell, pink arrow), magnification ×65. D, F Split images: upper-left: red fluorescence only; upper-right: green fluorescence only; lower-left: combined image
Lungs from mice exposed only to radiation but not bacterial inflammation were subjected to histologic analyses and signs of lung damage were not observed. Postinflammatory lung fibrosis was evident in pneumonia survivors after 1–2 months. There were multiple foci of fibrosis that tested positive for collagen by trichrome staining and were located predominantly in the peribronchial interstitial spaces (Fig. 4). Multiple GFP+ cells, which were present in these regions, were primarily positive for vimentin and CD45 (Fig. 5). Also, 20% of the GFP+ cells were also positive for collagen I, indicating that a fraction of transplanted BM cells engrafted as fibroblasts at foci of fibrosis (Fig. 5).
Fig. 4.

GFP+ cells present in areas of focal pulmonary fibrosis. Representative histologic samples with Trichrome Masson staining for collagen (blue, A–D) in paraffin-sectioned lungs and confocal microscopy images (E, F) of lung cryosections 2 months after instillation of bacteria into the lungs. A Lungs of control mice instilled with PBS. No signs of fibrosis and collagen deposition. B–D Lungs from mice instilled with bacteria, deposition of collagen and dense cell infiltration are present in peribronchial and perivascular interstitium, magnification ×25. E–G Confocal microscopy images of the same lungs as presented in B–D. GFP+ cells are bright green. Foci of fibrosis with multiple GFP+ cells in perivascular and peribronchial interstituim in E and F (costaining PI, red, magnification ×25). G GFP+ cells form clones of vimentin-positive cells. Tissues were stained for vimentin with Texas Red secondary antibody (red). Split image: upper-left: red fluorescence (vimentin) only; upper-right: green fluorescence (GFP) only; lower-left: combined image, magnification ×65
Fig. 5.
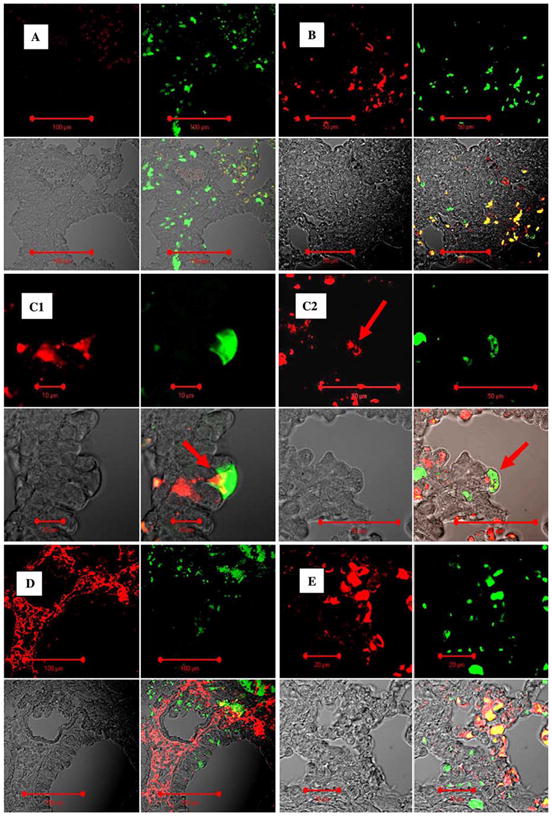
GFP+ cells present in lung interstitial spaces were predominantly leukocytes. Representative confocal microscopy images of lung sections (A–C, paraffin sections) from mice transplanted with BM 2 months after instillation of bacteria into the lungs. A Negative control isotype staining for common leukocyte antigen CD45 stained with red fluorescent antibody. Split image: upper-left: CD45; upper-right: GFP (green); lower-left: DIC image; lower-right: combined image. B GFP+ cells present in interstitium are predominantly positive for CD45. Split image: upper-left: CD45; upper-right: GFP (green); lower-left: DIC image; lower-right: combined image. C1, C2 GFP+ cell located in bronchial epithelial layer is positive for CD45 (arrows). Split image: upper-left: CD45; upper-right: GFP (green); lower-left: DIC image; lower-right: combined image. In D and E the same regions of these lungs were stained for collagen I (red fluorescence). Lungs were stained with anticollagen I antibody (Alexa Fluor® 633, red) and anti-GFP FITC-labeled antibody (green). Twenty percent of GFP+ cells in the lung interstitium were positive for collagen I. Upper-left panel: red fluorescence image (collagen I); upper-right panel: green fluorescence image (GFP); lower-left panel: image of tissue in transmitted laser light; lower-right panel: combined image
Temporal Patterns of Engraftment
The temporal sequence of lung engraftment of GFP+ cells in control animals and in the animals that developed pneumonia is shown in Fig. 6. In control animals, the percentage of engraftment did not exceed 0.1% during the periods of observation that continued up to 6 months. In animals with pneumonia, the development of lung inflammation was associated with a large influx of bone marrow-derived cells, which disappeared with the resolution of the inflammation. Only a few GFP+ cells were observed in the lung at 6 months.
Fig. 6.
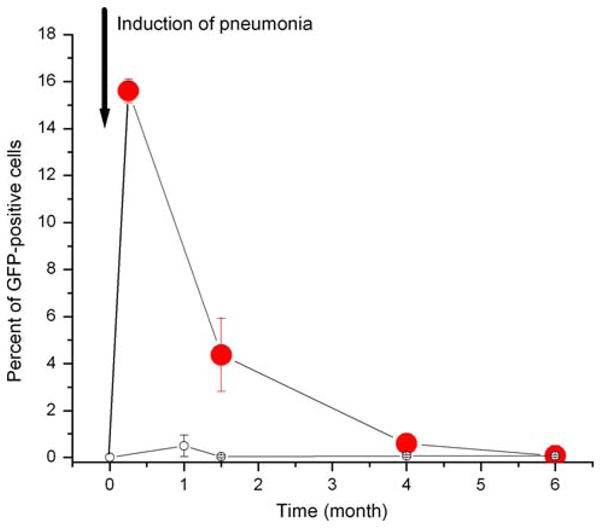
Dynamics of lung engraftment by marrow cells following pneumonia. The time courses of lung engraftment in control animals (empty circles) and in animals following acute bacterial pneumonia (filled circles) are shown
The percentage of GFP+ cells displaying ligands, which are characteristic of leukocytes, epithelial and endothelial cells, and fibroblasts (in the group of animals transplanted with granulocyte fraction of BM), is given in Table 1. No trends of time-dependent engraftment were noted for CD34-, CD3-, CD11b-, or CK14-positive GFP+ cells.
Table 1.
Percentage of different lineages of GFP+ cells in the lung following transplantation of BM and acute bacterial pneumonia
| Time | CD45+ | Vimentin | SMA | Col I | PCK | CD31 |
|---|---|---|---|---|---|---|
| 1 week (n = 4) | 61 ± 5 | 0 | 0.1 ± 0.1 | 0 | 1 ± 1 | 0 |
| 6 weeks (n = 6) | 74 ± 9 | 33 ± 7 | 5 ± 3 | 20 ± 6 | 8 ± 1 | 8 ± 3 |
| 16 weeks (n = 6) | 61 ± 10 | 50 ± 5 | 26 ± 20 | 35 ± 5 | 0 | 0 |
CD45 = common leukocyte antigen; Vimentin = marker of fibroblasts and mesenchymal cells, SMA = smooth muscle actin; PCK = pan-cytokeratin; CD31 = PECAM-1; Col I = collagen I Data are mean ± SE
Discussion
Our preliminary experiments demonstrated that incorporation of BMDC into the lung occurred when the age of the donor and the recipient was less than 5 weeks. We did not observe incorporation of BMDC from 6–12-month-old donors even in newborn mice. Though mice of this age are more sensitive to radiation than older animals, we used 3–5-week-old recipients to achieve meaningful quantities of BMDC incorporation into the lung. These data are consistent with previously described age-related differences in homing and engraftment of mouse hematopoietic cells [25–27]. Liang et al. [25] reported a dramatic (80%) decline in homing and engraftment with increasing donor and recipient ages. Mechanisms of these phenomena are complex, poorly understood, and include numbers of hematopoietic progenitors in donor marrow, expression of CXCR4, clonogenic potential, as well as capacity of recipient stroma to capture and support stem cells, all decreasing with age.
After transplanting whole BM into irradiated recipients, there was very limited incorporation of cells in the non-injured lung. The method of whole-BM transplantation that we used resulted in both myeloid and mesenchymal engraftment of GFP+ cells to the bone marrow.
Significant numbers of BMDC progeny in the lung were observed only at the peak of acute inflammation from bacterial pneumonia, which was predominantly associated with an influx of GFP+ leukocytes that persisted up to 6 months at some sites. During the subacute phase of inflammation (1–2 weeks), there was an increase in the incorporation of epithelial-like cells and endothelial-like cells. This increase was followed by the appearance of vimentin-positive cells at the sites of fibrosis, some of which were collagen I-positive. During the following 6 months, there was a decrease in the number of cells bearing proteins specific for epithelial and endothelial cells. These data, combined with other observations that lethal irradiation results in a large percentage of epithelial-like features [10], suggest that acute injury of the lung initially promotes the appearance of BMDC in the lung as epithelial-like or endothelial-like cells, which are then replaced by undifferentiated or fibroblast-like mesenchymal cells and finally disappear when the lung architecture is restored to normal in surviving animals.
We used GFP as a cell tag, and immunologic conflict in our transplantation experiments was minimal. We confirmed the specificity of GFP fluorescence in paraffin sections by staining with antibody for GFP and in cryosections by staining with a secondary antibody bearing a red fluorescent signal, which demonstrated colocalization of these two fluorescent probes. No GFP signal was observed in the lungs from control animals that had been transplanted with GFP− cells. Previously, staining for Y chromosome [12] or for both Y and X chromosomes [28] was effectively used to determine the possibility of cell fusion. In this study we did not perform this analysis because we previously had shown that GFP is a reliable tag of engrafted cells by Y chromosome staining in sex-mismatched transplantations [23]; therefore, we checked only GFP-positive cells for polyploidy, which was not observed. Because we were not able to demonstrate physiologically important or long-term engraftment of bone marrow cells as specialized epithelial or endothelial cells, analysis of the possibility of cell fusion was not warranted. At 6 months post-transplantation, we observed 20–50% bone marrow engraftment and the presence of GFP+ cells in the liver, spleen, and other organs. There is a possibility that GFP may not be highly expressed in differentiated epithelial and endothelial cells, although GFP is expressed in GFP+ Tg mouse epithelium and endothelium.
We used lungs of mice that exhibited minimal radiation damage and observed only very limited incorporation in the absence of additional injury from bacteria. This finding is consistent with previous reports. Kotton et al. [14] reported that plastic-adherent marrow-derived cells engrafted only as type I pneumocytes in nonradiated mice. Engraftment was found in only two of nine animals with noninjured lungs, although bleomycin-induced injury resulted in engraftment in all animals while no grafting to other organs was seen. Similarly, our results are in agreement with studies of nonirradiated animals by Ortiz et al. [9]. In that study, mesenchymal stem cells from bone marrow localized to areas of bleomycin-induced injury. Interstitial monocytes/macrophages, subepithelial fibroblast-like interstitial cells, and type I alveolar epithelial cells were observed among the engrafted cells. High levels of engraftment in the lung were observed only following damaging levels of radiation [7, 8], which not only produced significant injury to alveolar cells, but were also likely to eliminate the lung’s own progenitor cells, thus allowing repopulation of epithelium by circulating stem cells.
Acute and chronic inflammatory lung diseases often result in the formation of pulmonary fibrosis [1–3]. In fibrosis, accumulation of fibroblasts and myofibroblasts, which synthesize and organize extracellular matrix, leads to the formation of “scars.” Fibrous tissue transforms normal lung architecture and impairs some lung functions. Both inflammatory cells and fibroblasts with altered functional characteristics participate in the formation of fibroblastic foci [3, 5]. It is hypothesized that in chronic inflammation, the population of the lung fibroblasts is distinct from normal lung fibroblasts [6]. Bone marrow-derived circulating fibroblasts may participate in chronic lung inflammation [30]. In mice, reovirus 1/L-induced pneumonia resulted in histopathologic changes similar to human acute respiratory distress syndrome (ARDS), with healing via resolution and/or repair with fibrosis [31]. T cells were shown to play a distinct role in the development of intraluminal fibrosis associated with either bronchiolitis obliterans-organizing pneumonia or ARDS [32]. The role of BMDC in the development of lung fibrosis following bacterial pneumonia has not been addressed before. Our study demonstrates that BMDC may differentiate into lung mesenchymal fibroblasts during development of postinflammatory areas of focal fibrosis in the lung. This observation is consistent with a report [33] describing large numbers of GFP+ fibroblasts, the progeny of transplanted bone marrow, in active lung fibrotic lesions induced by endotracheal bleomycin.
We found cells displaying proteins specific for epithelial and endothelial cells in the lung, although we did not find evidence that these cells form clones or distinct groups in tissues. It should be noted that in previous studies of lung grafting, there also were no reports of extensive clonal formation at the sites of engraftment. Kotton et al. [14] specifically state that multicellular engraftment in the lung was not due to clonal formation. In this study, the BMDC that participated in lung repair as epithelial-like cells began to appear 2–4 days following acute injury; their numbers reached a maximum at 2–4 weeks, but they eventually disappeared or were replaced by fibroblasts. The patterns of BMDC appearance in the lung were associated with the different stages of injury; the commitment of these BMDC changed during the transition to subsequent phases. However, it remains unclear whether this transition was the result of the appearance of newly infiltrated cells or represented the transition of previously homed cells. We cannot rule out the possibility that in this model of recovery from acute bacterial pneumonia, BMDC, initially incorporated as epithelial-like cells, transformed into mesenchymal cells. However, in any case, BMDC did not show long-term engraftment as specialized differentiated endothelial or epithelial cells and, therefore, would be unlikely to have physiologic significance as substitute cells.
Our results suggest that multiple BMDC can participate in lung inflammatory repair and remodeling, and they may participate in different ways during different phases. We propose that BMDC home to the site of acute lung injury temporarily, probably to perform a substitute or regulatory role. After a specific period of time, this population of BMDC is replaced by a new population or else disappears. Mesenchymal stem cells of BM are known to be precursors of different cell types [34], and their ability to replace or restore airway epithelium in vivo and in vitro has been reviewed [35, 36]. The stability of this replacement has not been previously addressed.
Limitations of this study include the use of an irradiated chimeric model and transplantation of whole bone marrow rather than a specific subset of BM cells. Radiation is likely to damage airway epithelial and lung progenitor cells. However, we used minimal doses of radiation required for achievement of chimerism. Because we observed the presence of minimal amounts of GFP+ cells in the lungs of animals without pneumonia, this result indicates that radiation damage to the lungs was negligible. Whole bone marrow contains multiple stem cell subsets, including hematopoietic progenitors and mesenchymal stem cells. Our findings indicate that only hematopoietic lineages participated in the long-term population of the lung. Therefore, BM is an unlikely source of physiologically significant epithelial or endothelial regeneration after acute bacterial pneumonia. Thus, attempts to isolate subsets of BMDC that may become progenitors of epithelium or endothelium may not be of major value.
In summary, acute inflammation from bacterial pneumonia in the lung triggers incorporation of BMDC at the sites of lung injury. During the acute phase of inflammation, there is evidence that a small number of BMDC display proteins specific for endothelial and epithelial cells. Later, these BMDC are replaced by undifferentiated BMDC, leukocytes, and fibroblasts during the formation of some areas of focal fibrosis. BMDC may play a transient and possibly regulatory role in stromal remodeling after bacterial pneumonia, but probably not as a pathway to permanently replace injured endothelial or epithelial barriers of the lung.
Acknowledgments
Antibodies were a gift from Dr. J. H. Widdicombe (UC Davis). V. Mikhaylov supported by RFBR 06-0406388 OFI.
References
- 1.Martin C, Papazian L, Payan MJ, Saux P, Gouin F. Pulmonary fibrosis correlates with outcome in adult respiratory distress syndrome. A study in mechanically ventilated patients. Chest. 1995;107:196–200. doi: 10.1378/chest.107.1.196. [DOI] [PubMed] [Google Scholar]
- 2.Molina-Molina M, Badia JR, Marin-Arguedas A, Xaubet A, Santos MJ, Nicolas JM, Ferrer M, Torres A. Outcomes and clinical characteristics of patients with pulmonary fibrosis and respiratory failure admitted to an intensive care unit. A study of 20 cases. Med Clin (Barc) 2003;121:63–67. doi: 10.1016/s0025-7753(03)73856-1. [DOI] [PubMed] [Google Scholar]
- 3.Chapman HA. Disorders of lung matrix remodeling. J Clin Invest. 2004;113:148–157. doi: 10.1172/JCI20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- 5.Antoniou KM, Bouros D, Siafakas NM. Top ten list in idiopathic pulmonary fibrosis. Chest. 2004;125:1885–1887. [Google Scholar]
- 6.Torry DJ, Richards CD, Podor TJ, Gauldie J. Anchorage-independent colony growth of pulmonary fibroblasts derived from fibrotic human lung tissue. J Clin Invest. 1994;93:1525–1532. doi: 10.1172/JCI117131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theise ND, Henegariu O, Grove J, Jagirdar J, Kao PN, Crawford JM, Badve S, Saxena R, Krause DS. Radiation pneumonitis in mice: a severe injury model for pneumocyte engraftment from bone marrow. Exp Hematol. 2002;30:1333–1338. doi: 10.1016/s0301-472x(02)00931-1. [DOI] [PubMed] [Google Scholar]
- 8.Abe S, Boyer C, Liu X, Wen FQ, Kobayashi T, Fang Q, Wang X, Hashimoto M, Sharp JG, Rennard SI. Cells derived from the circulation contribute to the repair of lung injury. Am J Respir Crit Care Med. 2004;170:1158–163. doi: 10.1164/rccm.200307-908OC. [DOI] [PubMed] [Google Scholar]
- 9.Ortiz L, Gambelli F, McBride C, Gaupp D, Baddoo M, Kaminski N, Phinney DG. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci U S A. 2003;100:8407–8411. doi: 10.1073/pnas.1432929100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis S. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–377. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 11.Wagers A, Sherwood R, Christensen J, Weissman IL. Little evidence for developmental plasticity of adult hematopoietic stem cell. Science. 2002;297:2256–2259. doi: 10.1126/science.1074807. [DOI] [PubMed] [Google Scholar]
- 12.Grove J, Lutzko C, Priller J, Henegariu O, Theise ND, Kohn D, Krause DS. Marrow-derived cells as vehicles for delivery of gene therapy to pulmonary epithelium. Am J Respir Cell Mol Biol. 2002;27:645–651. doi: 10.1165/rcmb.2002-0056RC. [DOI] [PubMed] [Google Scholar]
- 13.Abe S, Laube G, Boyer C, Rennard SI, Sharp JG. Transplanted BM and BM side population cells contribute progeny to the lung and liver in irradiated animals. Cytotherapy. 2003;5:523–533. doi: 10.1080/14653240310003576. [DOI] [PubMed] [Google Scholar]
- 14.Kotton DN, Ma BY, Cardoso WV, Sanderson EA, Summer RS, Williams MC, Fine A. Bone-marrow-derived cells as progenitors of lung alveolar epithelium. Development. 2001;128:5181–5188. doi: 10.1242/dev.128.24.5181. [DOI] [PubMed] [Google Scholar]
- 15.Yamada M, Kubo H, Kobayashi S, Ishizawa K, Numasaki M, Ueda S, Suzuki T, Sasaki H. Bone-marrow-derived progenitor cells are important for lung repair after lipopolysaccharide-induced lung injury. J Immunol. 2004;172:1266–1272. doi: 10.4049/jimmunol.172.2.1266. [DOI] [PubMed] [Google Scholar]
- 16.Suratt B, Cool C, Serls A, Chen L, Varella-Garcia M, Shpall E, Brown K, Worthen GS. Human pulmonary chimerisms after hematopoietic stem cell transplantation. Am J Respir Crit Care Med. 2003;168:318–322. doi: 10.1164/rccm.200301-145OC. [DOI] [PubMed] [Google Scholar]
- 17.Kleeberger W, Versmold A, Rothhamel T, Glockner S, Bredt M, Haverich A, Lehmann U, Kreipe H. Increased chimerism of bronchial and alveolar epithelium in human lung allografts undergoing chronic injury. Am J Pathol. 2003;162:1487–1494. doi: 10.1016/S0002-9440(10)64281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zander DS, Baz MA, Cogle CR, Visner GA, Thiese ND, Crawford JM. Bone marrow-derived stem cell repopulation contributes minimally to the type II pneumocyte pool in transplanted human lungs. Transplantation. 2005;80:206–212. doi: 10.1097/01.tp.0000165095.39320.50. [DOI] [PubMed] [Google Scholar]
- 19.Zander DS, Cogle CR, Thiese ND, Crawford JM. Donor-derived type II pneumocytes are rare in the lungs of allogeneic hematopoietic cell transplant recipients. Ann Clin Lab Sci. 2006;36:47–52. [PubMed] [Google Scholar]
- 20.Weiss DJ, Berberich MA, Borok Z, Gail DB, Kolls JK, Penland C, Prockop D. Adult stem sells, lung biology and lung disease. Proc Am Thorac Soc. 2006;3:193–207. doi: 10.1513/pats.200601-013MS. [DOI] [PubMed] [Google Scholar]
- 21.Van Haaften T, Thebald B. Adult bone marrow-derived stem cells for the lung: implications for pediatric lung diseases. Pediatr Res. 2006;59(4):94R–99R. doi: 10.1203/01.pdr.0000203550.50258.5a. [DOI] [PubMed] [Google Scholar]
- 22.Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;179:1855–1863. doi: 10.4049/jimmunol.179.3.1855. [DOI] [PubMed] [Google Scholar]
- 23.Serikov VB, Popov B, Mikhaylov VM, Gupta N, Matthay MA. Evidence of temporary airway epithelial repopulation and rare clonal formation by BM-derived cells following naphthalene injury in mice. Anat Rec. 2007;290:1033–1046. doi: 10.1002/ar.20574. [DOI] [PubMed] [Google Scholar]
- 24.Popov B, Serikov VB, Petrov NS, Izusova TV, Gupta N, Matthay MA. Lung epithelial cells A549 induce endodermal differentiation in mouse mesenchymal BM stem cells by a paracrine mechanism. Tissue Eng. 2007;13:2441–2450. doi: 10.1089/ten.2007.0001. [DOI] [PubMed] [Google Scholar]
- 25.Liang Y, VanZant G, Szilvassy SJ. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood. 2005;106:1479–1487. doi: 10.1182/blood-2004-11-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nat Med. 1996;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- 27.Kim M, Moon H, Spangrude GJ. Major age-related changes of mouse hematopoietic stem/progenitor cells. Ann N Y Acad Sci. 2003;996:195–208. doi: 10.1111/j.1749-6632.2003.tb03247.x. [DOI] [PubMed] [Google Scholar]
- 28.Zander DS, Cogle CR, Theise ND, Crawford JM. Donor-derived type II pneumocytes are rare in the lungs of allogeneic hematopoietic cell transplant recipients. Ann Clin Lab Sci. 2206;36:47–52. [PubMed] [Google Scholar]
- 29.Reynolds SD, Giangreco A, Hong KU, McGrath KE, Ortiz LA, Stripp BR. Airway injury in lung disease pathophysiology: selective depletion of airway stem and progenitor cell pools potentiates lung inflammation and alveolar dysfunction. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1256–L1265. doi: 10.1152/ajplung.00203.2004. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibroblasts as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;170:380–389. doi: 10.4049/jimmunol.171.1.380. [DOI] [PubMed] [Google Scholar]
- 31.London L, Majeski E, Paintlia MK, Harley RA, London SD. Respiratory reovirus 1/L induction of diffuse alveolar damage: A model of Acute Respiratory Distress Syndrome. Exp Mol Pathol. 2002;72:24–36. doi: 10.1006/exmp.2001.2414. [DOI] [PubMed] [Google Scholar]
- 32.Majeski EI, Harley RA, Bellum SC, London SD, London L. Differential role for T cells in development of fibrotic leasions associated with reovirus 1/L-induced bronchiolitis obliterans organizing pneumonia versus acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2003;28:208–217. doi: 10.1165/rcmb.4891. [DOI] [PubMed] [Google Scholar]
- 33.Hashimoto N, Jin H, Liu T, Chensue SW, Phan SH. Bone-marrow-derived progenitor cells in pulmonary fibrosis. JCI. 2004;113:243–252. doi: 10.1172/JCI18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276:71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 35.Prockop DJ, Gregory CA, Spees JL. One strategy for cell and gene therapy: harnessing the power of adult stem cells to repair tissues. Proc Natl Acad Sci U S A. 2003;100:11917–11923. doi: 10.1073/pnas.1834138100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pitt BR, Ortiz LA. Stem cells in lung biology. Am J Physiol Lung Cell Mol Physiol. 2004;286:L621–L622. doi: 10.1152/ajplung.00392.2003. [DOI] [PubMed] [Google Scholar]