

Abstract
2-Choloroethyl Ethyl Sulfide (CEES) exposure causes inflammatory lung diseases, including acute respiratory distress syndrome (ARDS) and pulmonary fibrosis. This may be associated with oxidative stress, which has been implicated in the desensitization of beta-adrenergic receptors (β-ARs). The objective of this study was to investigate whether lung injury induced by intratracheal CEES exposure (2 mg/kg body weight) causes desensitization of β-ARs. The animals were sacrificed after 7 days and lungs were removed. Lung injury was established by measuring the leakage of iodinated-bovine serum albumin ([125I]-BSA) into lung tissue. Receptor-binding characteristics were determined by measuring the binding of [3H] dihydroalprenolol ([3H] DHA) (0.5–24 nM) to membrane fraction in the presence and absence of DL-propranolol (10 μM). Both high- and low-affinity β-ARs were identified in the lung. Binding capacity was significantly higher in low-affinity site in both control and experimental groups. Although CEES exposure did not change KD and Bmax at the high-affinity site, it significantly decreased both KD and Bmax at low affinity sites. A 20% decrease in β2-AR mRNA level and a 60% decrease in membrane protein levels were observed in the experimental group. Furthermore, there was significantly less stimulation of adenylate cyclase activity by both cholera toxin and isoproterenol in the experimental group in comparison to the control group. Treatment of lungs with 3-isobutyl-1-methylxanthine (IBMX), an inhibitor of phosphodiesterase (PDE) could not abolish the difference between the control group and the experimental group on the stimulation of the adenylate cyclase activity. Thus, our study indicates that CEES-induced lung injury is associated with desensitization of β2-AR.
Keywords: 2-Choloroethyl Ethyl Sulfide (CEES), Lung Injury, β-Adrenergic Receptors, cAMP, Guinea Pig
INTRODUCTION
Mustard gas, an alkylating agent, is an extensively used chemical warfare agent and upon exposure is known to exert local action on eyes, skin, and respiratory tissue followed by impairment of nervous, cardiac, and digestive systems in humans and laboratory animals [1,2]. The upper and lower respiratory tracts may be damaged acutely due to hemorrhagic inflammation after its inhalation and subsequently, a variety of chronic pulmonary sequelae may develop, including acute respiratory distress syndrome (ARDS), chronic bronchitis, and pulmonary fibrosis [3–6]. Yet the exact mechanism is not well understood.
Many inflammatory lung diseases including ARDS are associated with oxidative stress and accumulation of free radicals [7]. Inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), play an important role in the generation of reactive oxygen species (ROS) [8]. Recently, we have reported that guinea pigs exposed intratracheally to a mustard analog, 2-chloroethyl ethyl sulfide (CEES) accumulate high levels of TNF-α, which leads to activation of both acid and neutral sphingomyelinase resulting in high accumulation of ceramides, a second messenger involved in cell apoptosis [9,10]. In addition, CEES activated NF-κB, which rapidly disappeared after 2 h, and because of the disappearance of NF-κB, the cells were damaged continually owing to accumulation of ceramides and activation of several caspases, leading to apoptosis. A subsequent study further revealed that an antioxidant N-acetylcysteine, offers prophylactic protection against the CEES-induced lung injury [11].
Beta-adrenergic receptors (β-ARs) are prototypic members of the G-protein-coupled receptors that regulate the intracellular concentration of important second messenger molecules such as cyclic AMP (cAMP) and play important roles in a variety of cells and organ systems [12,13]. To date, three subtypes of β-ARs (β1, β2, and β3) that differ in their affinities for endogenous catecholamine as well as synthetic ligands have been identified. All three receptors couple to the stimulatory guanine nucleotide binding protein Gs and the agonist binding to the receptor causes activation of Gs-coupled adenylate cyclase, which leads to elevation of intracellular cAMP [14–16].
In the lung, β-ARs especially the β2-ARs play a major role in the secretion of surfactant by the alveolar-type cells [17,18] and clearance of alveolar fluid [19]. Recently, we have reported that isoproterenol, a β2-AR ligand, causes an increase in the secretion of surfactant by stimulating release of Ca2+ from intracellular stores in guinea pig alveolar type II cells [20]. A defect in the β-ARs system in alveolar type II cells has been thought to be a contributing factor in the development of respiratory diseases [21,22]. In addition, oxidative stress has been implicated in the desensitization of β-ARs [23]. Receptor desensitization is a phenomenon in which cellular responsiveness is ultimately diminished when cells are subjected to a continuous stimulus. The important mechanism is the loss of cellular receptors due to desensitization of the β-ARs [24,25]. Desensitization of the β2-AR-adenylate cyclase system characterized by reduction of cAMP production has been observed in expression systems and different cell lines [14–16].
Surfactant defects have been demonstrated in a number of alveolar and airway diseases. In ARDS, deficiencies in surfactant components such as phospholipids, surfactant protein SP-A, and SP-B are evident, and may be caused by proinflammatory cytokines, e.g. TNF-α that decreases surfactant components [26]. Hence, the surfactant defects may additionally play an undefined role in chronic obstructive pulmonary disease (COPD) [27]. Therefore, we can hypothesize that the CEES treatment causes desensitization of β-ARs by up-regulating proinflammatory cytokines; as a result, it causes a defect in surfactant production and secretion, which is very relevant to ARDS.
In order to test this hypothesis, the objective of this study was to investigate whether lung injury induced by CEES exposure causes modification in the structure of alveolar type II cells and desensitization of β-ARs in the lung. Although, both β1 and β2 ARs are present in the lung, there are more β2 than β1 ARs [17,18]. Furthermore, it has been demonstrated by our laboratory [20] and others [28,29] that the release of surfactant is mediated by β2 rather than β1 ARs. Thus, ARDS induced due to exposure to mustard gas could be the result of reduction in responsiveness of the β2-ARs. The prominent mechanism during long-term desensitization of the β-ARs is the loss of cellular receptors [30].
β-ARs gene expression has been shown to be regulated at the transcriptional level by various factors, including steroid hormones such as glucocorticoids [31] and by chronic treatment with β-agonists [32]. It has been reported that down-regulation of β2-AR following long-term agonist exposure is accompanied by a decrease in β2-AR mRNA levels [33]. Thus, in the present study, we also investigated whether CEES exposure causes any change in β2-AR gene expression and its signaling capabilities in the lung.
MATERIALS AND METHODS
Chemicals
2-Chloroethyl ethyl sulfide (CL-CH2CH2–S-CH2CH2, CEES), DL-propranolol, atenolol, and ICI-118, 551 hydrochloride were purchased from Sigma Chemicals (St. Louis, MO). [3H] dihydroalprenolol ([3H] DHA) (40 Ci/mmol) was purchased from NEN™ Life Science products Inc. (Boston, MA), [32P]-α-ATP was obtained from Amersham Pharmacia Biotech (Piscataway, NJ), and [125I]-bovine serum albumin ([125I]-BSA) was purchased from ICN (Aurora, OH).
Animals and the CEES Treatment
Male guinea pigs (Harley strain, 5–6 weeks old, 400 g body weight) were obtained from Harlan Sprague Dawley, Inc. (Indianapolis, IN). Animals were infused intratracheally with single doses of CEES (2.0 mg/kg body weight). Control animals were infused with ethanol as vehicle. The animals were sacrificed after 7 days of CEES exposure.
Lung Injury Study
Lung injury was monitored by studying the leakage of [125I]-BSA from the lung after CEES exposure according to the method of Ward et al. [34] as described by us [10].
Ultrastructural Analysis
Lung samples were fixed with 2.5% glutaraldehyde in 0.1 M Sorensen’s phosphate buffer (pH 7.2) followed by osmium tetroxide in the same buffer as described by us [35]. The specimens were dehydrated through an upgraded ethanol series at room temperature. Preparations were embedded in epon 812. Poststaining was done in a saturated solution of uranyl acetate in 50% ethanol followed by Reynolds’ lead citrate. Sections were examined with a Philips 301 transmissions electron microscope.
β-Adrenergic Receptors Binding Assay
The binding of [3H] DHA to lung membrane fraction was determined as described earlier [18,36]. This was done in a volume of 0.5 mL containing: 0.1 mL membrane protein (50–100 mg), 0.1 mL radioligand (0.125–40 nM), 0.1 mL DL-propranolol (10 mM) (or buffer), and 0.2 mL assay buffer (10 mM Tris-HCl, 1 mM dithiothreitol (DTT), pH 7.6). The reaction was initiated by the addition of membrane protein and terminated after 40 min at room temperature by rapid filtration over a Brandel GF/B filtering manifold, with two 5 mL washes of ice-cold 0.85% NaCl. The specific binding of [3H] DHA was defined as the difference in binding obtained in the presence and absence of propranolol (10 mM). Based on our earlier finding with guinea pig lung [18], two binding sites of β-ARs were identified for β-ARs in this study, one as high-binding sites (KD < 1 nM) and the other as low-binding sites (KD > 4 nM).
In order to characterize these two subtypes, other experiments were carried out in which the incubation medium contained varied concentrations (10−12–10−3 M) of either atenolol (β1-antagonist) or ICI 118,551 (β2-antagonist) at two fixed concentrations of DHA (2 and 8 nM), and IC50 values were determined. IC50 was defined as the concentration of the antagonist producing 50% inhibition of DHA binding. Protein concentration was determined according to modified Lowry’s method [37] using BSA as a standard.
Isolation of RNA and Reverse Transcriptase-Polymerase Chain Reaction
Immediately after sacrifice, lungs were perfused with physiological saline, removed, and flash-frozen in liquid nitrogen and stored at −80°C. Total RNA was extracted using a Qiagen RNAEASY kit. The concentration and purity of the RNA was analyzed in a UV spectrophotometer.
Reverse transcriptase-polymerase chain reaction (RT-PCR) of β-ARs and glyceraldehydes-3-phosphate dehydrogenase (GAPDH) genes were performed using DNAse-treated RNA from each sample, using the One-Step RT-PCR kit (Invitrogen, MD). Both forward and reverse primers were designed based on the sequences from the GenBank Accession No. AJ459814 (cavia porcellus β2-ARs) and U51572 (c. porcellus GAPDH). The RT-PCR products were separated by electrophoresis on a 1% agarose gel, purified using QIAquick PCR purification kit (Qiagen, CA), and sequenced using BidDye-terminators kit (Applied Biosystems, CA). The sequences were analyzed using Applied Biosystems Automated Sequencer (ABI 3700 model).
Northern Blot Hybridization
Total RNA (30 μg) was loaded on a 1.6% agarose gel containing 2.2 M formaldehyde and 0.2 M 3-[N-morpholino]propane sulfonic acid (MOPS) buffer. After 6 h of electrophoresis at 70 V, RNA was transferred overnight onto a Bright Star (Ambion) nylon membrane in 20× sodium chloride–sodium citrate (SSC buffer) and UV cross-linked. Total RNA was DNAse-treated to remove genomic DNA before being reverse-transcribed into cDNA for PCR amplification of the β-AR and GAPDH probes. The primers for the probe are the same used for RT-PCR. After RT-PCR, guinea pig β2-AR cDNA (600 bp) and the GAPDH cDNA (400 bp) were gel-purified using a Qiagen Kit (Cat no. 28104). The fragments were labeled with [α32P] ATP (3,000 Ci/mmol) using DNA polymerase Nick Translation kit (Invitrogen). Following a three-hour prehybridization at 42°C, the filters were hybridized overnight at 42°C with the 32P-labeled β2-AR probe added in ultrahybridization buffer (Ambion). Blots were washed twice in 2× SSC/0.1% sodium dodecyl sulfate (SDS) for 30 min, once with 1× SSC/0.1% SDS at 42°C for 30 min, 0.5× SSC/0.1% SDS at 50°C for 30 min, and finally with 0.1× SSC/0.1% SDS at 55°C for 30 min, and exposed at −80°C to Kodak OMAT Xs film for development. Following autoradiography, blots were stripped and reprobed with a 32P-labeled guinea pig GAPDH cDNA probe, to enable standardization between samples. Band intensities on films were analyzed using Alpha Imager (Inontech) densitometric scan.
Measurement of β2-ARs Level by Western Blot Analysis
Both control and CEES-treated lungs were homogenized in 5 volumes of ice-cold buffer [Manitol (210 mM)/Sucrose(70 mM)/EDTA(1 mM)/CaCl2 (2 mM), pH 7.2] containing 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM DTT, and aprotinin (2 μg/mL) by using a Brinkman Polytron (setting 6–7, 30 s). The homogenates were filtered through a 100 μm Nylon mesh and divided into two parts. One part was used as total homogenate and the other part was then centrifuged at 20,000×g for 20 min at 4°C and washed three times and resuspended in 50 mM Tris-HCl, containing 0.1 mM PMSF, 1 mM DTT, and aprotinin (2 μg/mL), pH 7.4. Protein concentrations of both total homogenate and membrane fraction were measured according to the modified Lowry’s method [37], using BSA as a standard. Protein (50 μg) of total homogenate and membrane protein from each group were resolved by 10% SDS–PAGE. Proteins were transferred onto polyvinylidine difluoride (PVDF) membranes Immobilon-P (Millipore, Bedford, MA), as described by us [38]. Following incubation with β2-specific polyclonal antibodies raised in rabbit (1:500; sc 9042, Santa Cruz Biotechnology Inc., CA), blots were washed and incubated with HRP-conjugated anti-rabbit goat antibody (1:1000; sc 2350, Santa Cruz Biotechnology). Binding of antibodies to the blots was detected with an ECL detection system (Amersham Life Sciences, Arlington Heights, IL) following manufacturer’s instructions. Stripped blots were reprobed with β-actin-specific polyclonal antibodies (1:500; H-196, sc 7120, Santa Cruz Biotechnology) to enable standardization of signals between samples. Band intensities were acquired by Alpha Imager (Inontech).
cAMP Production in the Presence of Isoproterenol, Cholera Toxin, and IBMX
Lung homogenate (100 μg of protein) from both control and CEES-treated groups were incubated in the presence or absence of different concentrations of isoproterenol (6.25 nM–100 μM) at 37°C for 30 min as described by us [39]. The reaction was terminated with 6% trichloroacetic acid and centrifuged at 14,000×g for 15 min. The deproteinized supernatants were washed 10 times with water-saturated diethyl ether and dried by lyophilization and dissolved in water. The levels of cAMP in the extracts were measured by a radio-binding assay described in the instruction material supplied with the Biotrak cAMP assay system (Amersham Life Science).
To determine if there is any difference in the stability of cAMP between control and CEES-treated groups, we measured cAMP production in the presence and absence of 12.5 nM isoproterenol at different times (15–60 min). We also measured cAMP production in the presence or absence of cholera toxin (50 ng/mL) at 60 min.
Furthermore, we studied the effects of 3-isobutyl-1-methylxanthine (IBMX), an inhibitor of phosphodiesterase (PDE) on cAMP production in both control and experimental groups. For this, we incubated homogenates (1 mg protein/mL) with 50 μg IBMX at 37°C for 10 min, and afterwards centrifuged at 105,000 g for 30 min. The pellet was treated with 12.5 nM isoproterenol for 1 h, and analyzed for cAMP levels.
Statistical Analysis
Differences between the control samples and the samples treated with mustard gas were assessed by using ANOVA, and the significance level was set for p ≤ 0.05 using Graph Pad Prism 4 software program.
RESULTS
Effects of CEES Exposure on Lung Injury
Guinea pigs exposed to CEES had a 5-fold increase in lung permeability as measured by leakage of [125I]-BSA (Figure 1). This lung injury was further evident by ultrastructural analysis (photographs not shown because similar observations were reported earlier; see reference 40). Mustard gas exposure caused irregular expansion of alveolar spaces that contained surfactant lamellae and extruded type II cells, indicating disruption of surfactant secretion. Type II cells had irregular outlines and contained large number of lamellar bodies. No protrusion of type II cells was observed in the alveolar spaces in the vehicle-treated control animals.
FIGURE 1.
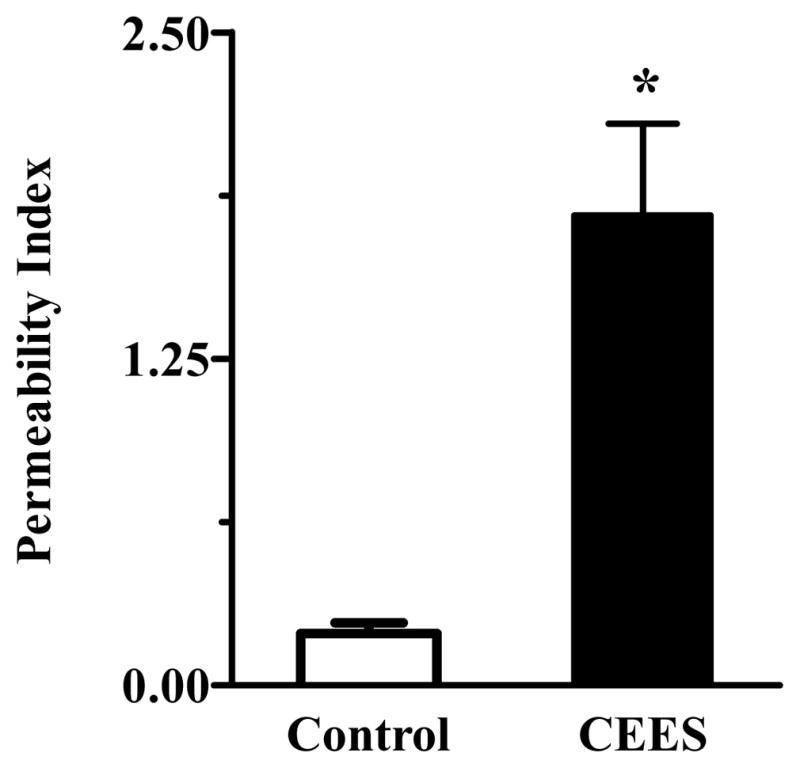
CEES-induced lung injury. Lung injury was expressed as permeability index, which was obtained by dividing total radioactive counts of [125I]-BSA in lung by counts in 1 mL of blood from the same animal (n = 3). Asterisks indicate statistically different compared to control (p < 0.05).
Effects of CEES Exposure on the Binding Characteristics of β-Adrenergic Receptors
The Scatchard plot (Figure 2) by Graph Pad PRISM 4 software program revealed that [3H] DHA binds to whole lung membrane extract at two binding sites with different affinities; high [KD = (0.68 ± 0.17) nM] and low [KD = (6.40 ± 2.1) nM] affinities in control animals. Binding capacity (Bmax) of the low-affinity site [Bmax = (2087 ± 370) fmol/mg protein] was significantly higher than that at the high-affinity site [Bmax = (221.2 ± 56.0) f mol/mg protein], as previously reported by us in guinea pig alveolar type II cells [18]. Lung membranes from CEES-exposed animals also revealed two binding sites with different affinities; KD = (0.62 ± 0.15) nM and KD = (2.85 ± 0.9) nM, respectively. Although CEES exposure did not change KD and Bmax at the high-affinity site, it significantly decreased both KD by 2.2 folds and Bmax by 2.7 folds at the low-affinity site (Table 1).
FIGURE 2.

Representative Scatchard analysis of [3H] DHA binding to β-ARs in guinea pig lung membranes: (A) high-affinity (control), (B) high-affinity (CEES-exposed), (C) low-affinity (control), (D) low-affinity (CEES-exposed). The binding of [3H] DHA to the membrane was measured as described in the “Materials and Methods” section (n = 3). B = Specific binding of ligand and F = Free ligand concentration.
TABLE 1.
Effects of Mustard Gas Exposure on Binding Characteristics of β-Adrenoreceptors
| High Affinity |
Low Affinity |
|||
|---|---|---|---|---|
| Samples | KD(nM) | Bmax(fmol/mg protein) | KD(nM) | Bmax(fmol/mg protein) |
| Control | 0.68 ± 0.17 | 221.2 ± 56.9 | 6.40 ± 2.1 | 2087 ± 370 |
| CEES-exposed | 0.62 ± 0.15 | 203.8 ± 69.0 | 2.85 ± 0.9 | 765.4 ± 164.1* |
Bmax at low-affinity sites is significantly reduced by mustard gas exposure (p = 0.03) (n = 3 for each group).
In order to characterize the high- and low-affinity DHA binding sites in our whole lung membrane preparation, we used subtype-specific antagonists [18,36]. Binding experiments were done in the presence of either atenolol, a β1-specific antagonist or ICI 118,551, a β2-specific antagonist. Both atenolol and ICI 118,551 inhibited the putative binding sites in a dose-dependent manner (Figure 3). The inhibition of DHA binding showed a biphasic distribution by both atenolol and ICI 118,551. Atenolol was more potent in displacing [3H] DHA at a low concentration (2 nM; Figure 3) than at a higher concentration (8 nM) of DHA (Figure 3). With 2 nM and 8 nM concentrations of [3H] DHA, IC50, for atenolol at high-affinity sites were 6 × 10−10 M and 2 × 10−7 M, respectively. On the other hand, at both low and high [3H] DHA concentrations, IC50, for ICI 118,551 for low-affinity sites were 2 × 10−6 M and 18 × 10−6 M, respectively. Hence, β-ARs in guinea pig lung are heterogeneous and contain both β1 (high affinity) and β2 (low affinity) as previously reported by us in alveolar type II cells [18]. Furthermore, the CEES treatment affects both KD and Bmax of β2 subtype, but not β1 subtype of the β-ARs (Table 1).
FIGURE 3.

Inhibition of [3H] DHA binding to the lung membrane by β-antagonists. (■– ■, Atenolol; ▲— ▲, ICI-118, 551). The binding of [3H] DHA to the membrane and IC50 for the two antagonists were measured as described in the “Materials and Methods” section (n = 3).
Effects of CEES Exposure on β2-Adrenergic Receptor Gene Expression
To verify whether a mutation in the coding region of the gene and thus a defect in protein could explain this reduction in Bmax, the β2-AR cDNA from control and CEES-exposed animals were sequenced. The β2-AR cDNA products were obtained by reverse transcription of RNA isolated from the control and CEES-exposed lung tissues, using β2-AR-specific primers. However, when compared by BLAST, no differences in nucleotide sequences between the RT-PCR products (Figure 4) from the control and experimental lungs were observed. The observed sequence had 100% identity with the sequence reported for c. porcellus β2-AR gene in NCBI GenBank (Acccssion No. AJ459814).
FIGURE 4.
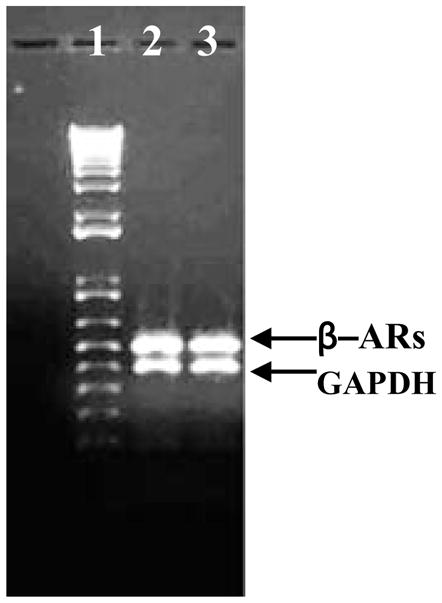
RT-PCR products of β-ARs gene of lung; 1 = kb+ DNA ladder; 2 = Control; 3 = CEES-exposed.
In order to evaluate whether the CEES treatment exerts an inhibitory effect on β2-AR gene expression, Northern blot analysis was performed. In both control and CEES-exposed lungs, we consistently detected a distinct band at 2.2 kb as expected for the expressed β2-AR message (Figure 5A). However, densitometry evaluation and comparison after normalization with the housekeeping gene GAPDH, indicates only a 20% reduction in β2 gene expression in CEES-exposed lungs (Figure 5B). However, this minimal reduction was not statistically significant.
FIGURE 5.
Effect of mustard gas on guinea pig lung β2-AR receptor gene expression. (A) A representative autoradiogram of Northern blot analysis, (B) bar graph of normalized densitometric values (n = 3).
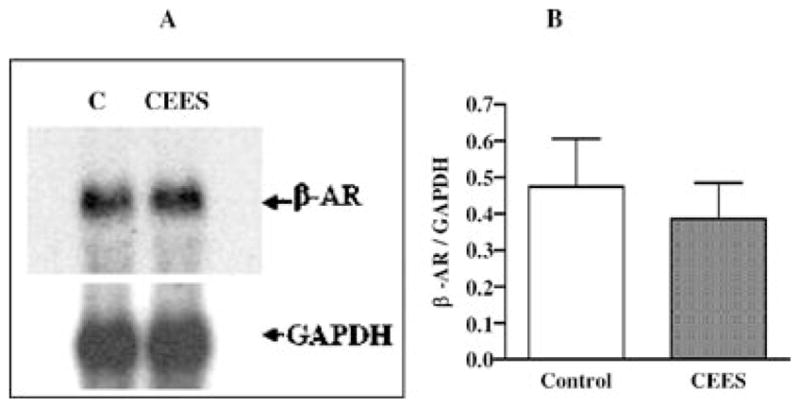
To further characterize whether the CEES treatment has any effect on translation of β2-AR mRNA, both whole lung homogenate and membrane isolated from both control and CEES-exposed lungs were immunodetected by using β2-AR-specific polyclonal antibody by Western blot (Figure 6). The protein level of β2-AR after normalization with β-actin levels indicates a 25% reduction in total lung homogenate (Figure 6A) and 60% reduction in the membrane (Figure 6B) from CEES-treated animals compared to the corresponding control. Furthermore, the CEES-induced decrease in β2-AR level was more in the membrane fraction than that in whole homogenate fraction. This indicates that the CEES treatment causes rapid internalization and degradation of β2-AR in the lung.
FIGURE 6.

Effect of mustard gas on guinea pig lung β2-AR receptor levels. (A) Homogenate (n = 3), (B) membrane (n = 3).
Effects of CEES Exposure on cAMP Production
Binding of agonists to the β2-AR stimulates adenylate cyclase activity and converts ATP to cAMP [12,13]. In order to assess whether the decrease in the expression of β2-ARs by CEES exposure causes a decreased response to β-agonists due to desensitization of β2-ARs, we measured the effects of CEES exposure on isoproterenol-induced cAMP production. Figure 7A shows a dose-dependent production of cAMP in response to isoproterenol treatment for 30 min of lung homogenate in both control and CEES-treated groups. The maximal induction was at 12.5 nM in both control (150 fmol/mg) and experimental lungs (212.5 fmol/mg). Thus, the stimulation was 29.4% less in the CEES-exposed group in comparison to the control group. The same degree of less stimulation by isoproterenol was observed at all dose levels in the experimental group than in the control group. There was a significant decrease (p < 0.001) in endogenous cAMP level in a time-dependent manner when incubated with optimum dose of isoproterenol (12.5 nM) in the CEES-exposed lungs compared to the control lungs (Figure 7B).
FIGURE 7.

Effects of mustard gas exposure on cAMP production in guinea pig lung. (A) Dose-response curve in the presence and absence of isoproterenol (n = 3), (B) time-response curve in the presence and absence of isoproterenol (n = 3), (C) response in the presence of isoproterol and IBMX singly or in combination (n = 3).
The cAMP production in response to cholera toxin, a direct activator of adenylate cyclase, was significantly less in the CEES-exposed group compared to the control group. For example, the stimulation was 21.1-fold for control group whereas it was 16-fold for the experimental group (Table 2). Hence, it indicates that the CEES treatment not only affects the agonist-mediated cellular response, but also stimulated degradation of cAMP. This is also evident from the results obtained from the study with IBMX (Figure 7C). The cAMP level was significantly lower in the CEES-exposed group than in the control group in the presence of both isoproterenol and IBMX.
TABLE 2.
Intracellular Level of cAMP in Choleratoxin-Treated Guinea Pig Lung: Role of the CEES Treatment
| cAMP (fmol/mg of Protein) |
||
|---|---|---|
| Addition | Vehicle | CEES |
| Endogenous | 98.20 ± 2.65 | 35.62 ± 0.67 |
| Cholera toxin (50 ng/mL) | 2074.51 + 6.77 | 1050.34 ± 6.87 |
The values are mean ± SEM of four samples.
In addition to the morphological changes in alveolar Type II cells that correlate with functional compromise, our binding studies and gene expression analysis strongly indicate that CEES exposure indeed down-regulates β-AR, especially the β2 subtype in the lung at both expression and functional levels.
DISCUSSION
Our recent studies with the guinea pig as an experimental model indicated that within 1 h of CEES exposure, there is induction of TNF-α [9] that initiates a series of signaling events leading to cellular apoptosis [10]. In this study, we investigated whether CEES exposure induced lung injury by modulating the binding characteristics and/or the expression of the β-ARs in lung. In fact, our results clearly show that the CEES treatment not only causes lung injury, ultrastructural modification of alveolar type II cells, and decrease in binding capacity of β-ARs, but also inhibits β2-AR gene expression as well as induces desensitization, internalization, and degradation of the β2-ARs.
We have previously reported the presence of high-and low-binding sites of β-ARs in the guinea pig lung alveolar Type II cells [18]. It was also established in that study that the high-affinity site is the β1 subtype and the low-affinity site is the β2 receptor subtype. The widely used β-AR ligand [3H] DHA binding to both control and experimental lung membrane preparations showed saturable characteristics. Binding characteristics of both the β1 and β2 subtypes in the whole lung membrane preparations in the current study is in good agreement with the previous observations [18] with alveolar type II cells from guinea pig. The β-AR ligand [3H] DHA exhibited two binding sites with different affinities, in both control and experimental lungs. The binding studies with control and experimental lungs we report here clearly indicate the presence of two β-AR subtypes and prominent reduction in binding capacity of the β2 subtype due to CEES exposure.
It has been reported that CEES acts as a mutagen. In humans, several single nucleotide polymorphisms that affect the function of β2-AR have been identified [40]. Since we did not observe any change in nucleotide sequences between the RT-PCR products from the control and experimental lungs, the reduction in binding due to CEES exposure is not due to any mutation. However, the data related to the gene expression (Figure 5) and protein level (Figure 6) indicate that CEES exposure causes reduction of the β2-ARs expression, and also induces internalization of β2-AR. We observed an overall 20% reduction in β2 receptor message levels in the lung tissues exposed to CEES.
In recent years, substantial evidence has been accumulated in support of the concept that cytokines are key regulators of the cascade of cellular events associated with airway inflammation in asthma and COPD [42–45]. Moreover, there is increasing evidence that cytokines may contribute to the β-AR responsiveness that is observed in asthma [45]. Cells in the airway wall also show that there is a close relationship between increased expression of IL-β, TNF-α, and IL-5 with reduced capacity of airway smooth muscle to generate cAMP or to relax in response to β-agonists. A decrease in β-AR-mediated relaxation of airway smooth muscle caused by IL-1β and/or TNF-α has also been described in guinea pigs, rats, rabbits, and dogs [46,47]. Finally, administration of specific cytokines has been shown to induce impairment of β-AR-mediated airway relaxation in the isolated guinea pig trachea [48]. Furthermore, we previously reported that the CEES treatment causes increased TNF-α in both lung tissue and macrophage [9,10]. Hence, we can postulate that CEES may affect both transcription and translation of β-ARs by stimulating inflammatory cytokines, such as TNF-α.
The protein level of β2-ARs significantly decreased in membrane fraction (Figure 6B; 60%) more than that in total homogenate (Figure 6A; 25%) isolated from CEES-treated lungs (but not from control lung) control. A significant difference of protein level between homogenate and membrane fraction indicates that the CEES treatment increases uncoupling of β2-ARs from Gs, and as a result internalization of β2-ARs. Then internalized β2-ARs may be either reactivated and recycled to the plasma membrane following dephosphorylation in intracellular vesicles or degraded in lysosomes. Both the above-mentioned processes can contribute to a reduction in the β2-ARs level in the plasma membrane of the guinea pig lung. Mounting evidence suggests that the physiological function of the various subtypes of adrenergic receptors is controlled by phosphorylation/dephosphorylation reactions. Different types of kinases appear to be involved. On the other hand, phosphorylation reactions may operate in a classical feedback regulatory sense. Thus cAMP-dependent kinase, once activated by a β-agonist, can feedback-regulate the function of the receptors by phosphorylating and desensitizing them. There may also be “cross-talk” between systems. Lefkowitz and Caron [49] reported that the cAMP-dependent protein kinases could phosphorylate the alpha 1 adrenergic receptor in vitro. Moreover, protein kinases C, when stimulated by phorbols, are able to phosphorylate and desensitize the β-ARs. Thus, one transmembrane-signaling system can regulate the function of another, which indicates that receptor-mediated signals are tightly regulated by feedback inhibition and act to prevent signal overload and reset the receptor to a changing environment. It has also been shown that short-term regulation i.e. uncoupling of β-ARs involves receptor phosphorylation and uncoupling of the receptor from the G protein Gs [50].
Chronic exposure to ligand leads to reduced receptor number, i.e. down-regulation, which results from a combination of receptor internalization, and degradation and decreased mRNA abundance. It has also been reported that regulation of β-ARs is species- and subtype-specific, with a rank order of β2-ARs> β1-ARs> β3-ARs, and most tissues express more than one subtype [50]. Several reports have shown that oxidative stress modulates differentially various receptor responses in guinea pig and β-AR response is more susceptible than muscarinic receptor response [51–53]. Our observation is in parallel with previous reports of a reduction in β-AR mRNA after chronic in vivo β-agonist treatment [30,32,54]. Nishikawa et al. [54] reported that β2-AR mRNA was significantly reduced as early as one day and persisted at day 7 after isoproterenol infusion. The mechanism by which CEES exerts its effect on the β2-AR mRNA levels is not known.
We previously reported that the CEES treatment causes not only increase of TNF-α, but also oxidative stress in guinea pig lung [9–11]. CEES may affect transcription, translation, and degradation of β2-ARs as a result of desensitization of β-AR by increasing production of TNF-α, generation of ROS, and endogenous catecholamine. Zang et al. [55] have shown that oxygen radicals play an important role in the nonvagal component of the noncholinergic bronchoconstriction in guinea pigs in vivo. Morken et al. [56] have shown that there is an immediate and sustained systemic elevation of catecholamines including epinephrine, which may set the stage for development of neutrophils-mediated acute lung injury. Furthermore, it has been reported that stress and hypoxia of anaphylaxis cause catecholamine release, loss of choline-containing phospholipids, and thereby ARDS [57]. A decrease in both β2-AR receptor and mRNA levels in response to chronic stimulation by β-AR agonists has been reported [33]. In addition, the β2-AR gene contains cAMP and glucocorticoid receptor response elements (CRE and GRE), and studies indicate regulation of β2-receptor by cAMP (and/or corticosteroids) at the gene expression level [33,58]. CRE-binding protein is also believed to maintain the basal transcription of β2-receptor gene [8,32]. Reduced cAMP regulatory element binding (CREB) activity has been suggested to result in reduced β2 mRNA in rat and guinea pig lungs following prolonged exposure to β-agonist [32]. Endogenous catecholamines may increase due to CEES-induced stress and trauma, and thereby modulate basal transcription of β2-AR gene.
The early signaling event in classical G-protein-coupled receptor signal transduction pathway induced upon β2-ARs binding to endogenous catecholamines is the activation of adenylate cyclase activity [30,59]. Our results with in vitro treatment of isoproterenol show a dose-dependent decrease of β-agonist-induced activation of adenylate cyclase activity as expressed in terms of cAMP production, in CEES-treated lungs in comparison to control lungs (Figure 7A). The potent activator of adenylate cyclase, cholera toxin causes 50% reduced stimulation of adenylate cyclase in CEES-treated lungs than control lungs (Table 2). This seems to suggest that the CEES treatment may cause β-ARs ligand-mediated desensitization in guinea pig lung. The β-AR ligand-mediated desensitization has been demonstrated in several in vitro systems, including ventricular strips, adipocytes, and a variety of cell lines [60]. The CEES treatment causes a 2.8-fold decrease in endogenous cAMP compared to the levels in control lungs. Moreover, the fold stimulation from basal level is higher than control, but stability of cAMP is significantly decreased (p < 0.001) in CEES-treated lungs than in control lungs (Figure 7B). This indicates that the CEES treatment not only affects the β2-AR responsiveness to agonists, but also increases the rapid degradation of cAMP. It has been reported that the cyclic nucleotides are synthesized by adenylate cyclase or guanylate cyclase and degraded by PDEs [61]. Desensitization may also involve activation of Gi and inhibition of adenylate cyclase [62]. It has been shown that protein kinase A (PKA) phosphorylation of β-AR increases the affinity for Gi, thus effectively switching signaling pathways Gs to Gi [61]. Hence, the CEES treatment may modulate directly or indirectly multiple events of β2-ARs signaling.
Fivefold increases in leakage of [125I]-BSA (Figure 1) and altered alveolar Type II cell morphology [41] indicate that lung injury is induced and surfactant secretion may be compromised following CEES exposure. In healthy and diseased animals/or humans, oxidative stress has been shown to compromise host defense (e.g. excess mortality from bacterial infections in rodents) and cause lung injury and inflammation (e.g., alveolar protein leak, neutrophils influx, etc). The impaired host defense may arise from increased alveolar macrophage (AM) apoptosis and polymorphonucleus phagocytosis and respiratory burst, whereas inflammation appears to arise from increased NF-κB activity, leading to the up-regulation of cytokines and adhesion molecules in endothelial and epithelial cells [63,64]. cAMP and PKA have been implicated in cell migration and wound repair. It has been shown that isoproterenol increased the migration of cells to speed up closing of mechanically and enzymatically induced wounds of submerged monolayers of bovine bronchial and human airway epithelial cells [65,66]. Dumasius et al. [67] have demonstrated that alveolar β2-AR overexpression improves β2-AR function and maximally up-regulates β-agonist responsive active Na+ transport by improving responsiveness to endogenous catecholamines, and as a result accelerates clearance of the alveolar fluid. Previously, we have reported that the NF-κ B level increases within 1 h of the CEES treatment, leading to the up-regulation of cytokines and causing oxidative stress.
Here we report that the CEES treatment results in down-regulation of β2-AR and causes lung injury and inflammation as evident by alveolar leakages. In summary, the CEES treatment causes desensitization of β2-AR by (a) up-regulating TNFα; (b) increasing generation of ROS; (c) enhancing infiltration of inflammatory cells, and probably (d) increasing stress-related endogenous catecholamine. Therefore, the CEES treatment may not only cause agonist-stimulated cAMP-dependent receptor phosphorylation, but also cAMP-independent receptor phosphorylation, and as a consequence, it will reduce responsiveness of the β2-ARs. Furthermore, the CEES treatment directly or indirectly stimulates PDEs activity (Figure 7B), which not only could cause the loss in responsiveness of the β2-ARs, but also could stimulate degradation of camp and affect the repairing of CEES-induced lung injury and inflammation as evident by leakage of [125I]-BSA (Figure 1). Furthermore, the CEES treatment causes down-regulation of mRNA (Figure 5) following an increase in TNFα and ROS and causes desensitization of β2-ARs, which is relevant to ARDS, since β2-ARs are responsible for surfactant secretion by alveolar type II cells.
Acknowledgments
Contract Grant Sponsor: US Army.
Contract Grant Number: W81XWH-06-2-0044.
Contract Grant Sponsor: NIH.
Contract Grant Number: 5T32HL-07751-09.
References
- 1.Smith MG, Stone W, Crawford K, Ward P, Till GO, Das SK. A promising new treatment for mustard gas with the potential to substantially reduce the threat posed by chemical, biological and radiological agents. Jane’s Chem-Bio Web. 2003:1–5. [Google Scholar]
- 2.Papirmeister B, Fenster AJ, Robinson SI, Ford RD. Sulfur mustard injury: Description of lesions and resulting incapacitations. In: Fenster AJ, editor. Medical defense against mustard gas. Toxic mechanisms and pharmacological implications. Boca Raton, FL: CRC; 1991. pp. 13–42. [Google Scholar]
- 3.Worwser U. Toxicology of mustard gas. Trends Pharmacol Sci. 1991;12:164–167. doi: 10.1016/0165-6147(91)90534-y. [DOI] [PubMed] [Google Scholar]
- 4.Momeni AZ, Enshaelh S, Meghdadi SM, Amindjavaheri M. Skin manifestation of mustard gas. Clinical study of 535 patients exposed to mustard gas. Arch Dermatol. 1992;128:775–780. [PubMed] [Google Scholar]
- 5.Calvet JH, Jarrean PH, Levame M, D’ortho MP, Lorino H, Harf A, Mavier IM. Acute and chronic respiratory effects of sulfur mustard intoxication in guinea pigs. J Appl Physiol. 1994;76:681–688. doi: 10.1152/jappl.1994.76.2.681. [DOI] [PubMed] [Google Scholar]
- 6.Emad A, Rezaian GR. The diversity of the effects of sulfur mustard gas inhalation on respiratory system 10 years after a single, heavy exposure: Analysis of 197 cases. Chest. 1997;112:734–738. doi: 10.1378/chest.112.3.734. [DOI] [PubMed] [Google Scholar]
- 7.Leff JA, Parsons PE, Day CE, Taniguchi N, Jochum M, Fritz H, Moore FA, Moore EE, McCord JM, Repine JE. Serum antioxidants as predictors of adult respiratory distress syndrome in patients with sepsis. Lancet. 1003;341:777–780. doi: 10.1016/0140-6736(93)90558-x. [DOI] [PubMed] [Google Scholar]
- 8.Lloyd SS, Cheng AK, Taylor FB, McCay JEG. Free radicals and septic shock in primates: The role of tumor necrosis factor. Free Radic Biol Med. 1993;14:233–242. doi: 10.1016/0891-5849(93)90020-u. [DOI] [PubMed] [Google Scholar]
- 9.Rajaratnam VS, Das SK. Array of cytokine induction in early lung injury response to 2-chloroethyl ethyl sulfide, a mustard gas analog. FASEB J. 2005;19:A852. [Google Scholar]
- 10.Chatterjee D, Mukherjee S, Smith MG, Das SK. Signal transduction events in lung injury induced by 2-chloroethyl ethyl sulfide (CEES), a mustard analog. J Biochem Mol Toxicol. 2003;17:1–8. doi: 10.1002/jbt.10068. [DOI] [PubMed] [Google Scholar]
- 11.Das SK, Mukherjee S, Smith MG, Chatterjee D. Prophylactic protection by N-acetylcysteine against the pulmonary injury induced by 2-chloroethyl ethyl sulfide, a mustard analogue. J Biochem Mol Toxicol. 2003;17:177–184. doi: 10.1002/jbt.10076. [DOI] [PubMed] [Google Scholar]
- 12.Liggett SB. Update on current concepts of the molecular basis of beta-2 adrenergic receptor signaling. J Allergy Clin Immunol. 2002;110:S223–S227. doi: 10.1067/mai.2002.129945. [DOI] [PubMed] [Google Scholar]
- 13.Hein L, Kobilka BK. Adrenergic receptor signal transduction and regulation. Neuropharmacology. 1995;34:357–366. doi: 10.1016/0028-3908(95)00018-2. [DOI] [PubMed] [Google Scholar]
- 14.Collins S, Caron MG, Lefkowitz RJ. From ligand binding to gene expression: New insight into the regulation of G-protein-coupled receptors. Trends Biochem Sci. 1992;17:37–40. doi: 10.1016/0968-0004(92)90425-9. [DOI] [PubMed] [Google Scholar]
- 15.Brady AE, Limbird LE. G protein-coupled receptor interacting proteins: Emerging roles in localization and signal transduction. Cell signal. 2002;14:297–309. doi: 10.1016/s0898-6568(01)00239-x. [DOI] [PubMed] [Google Scholar]
- 16.Penn RB, Benovic JL. Desensitization of G protein-coupled receptors. In: Conn PM, editor. Handbook of physiology. Vol. 1. New York: Oxford University Press; 1998. pp. 125–164. [Google Scholar]
- 17.Brown LA, Longmore WJ. Adrenergic and cholinergic regulation of lung surfactant secretion in the isolated perfused rat lung and in the alveolar type II cell in culture. J Biol Chem. 1981;256:66–72. [PubMed] [Google Scholar]
- 18.Das SK, Sikpi MO, Skolnick P. Heterogeneity of beta adrenoreceptors in guinea pig alveolar type II cells. Biochem Biophys Res Commun. 1987;142:898–903. doi: 10.1016/0006-291x(87)91498-7. [DOI] [PubMed] [Google Scholar]
- 19.McGraw DW, Fukuda N, James PF, Fobes SL, Woo AL, Lingrel JB, Witte DP, Matthay MA, Liggett SB. Targeted transgenic exppession of beta (2)-adrenergic receptors to type II cells increases alveolar fluid clearance. Am J Physiol Lung Cell Mol Physiol. 2001;281:L895–903. doi: 10.1152/ajplung.2001.281.4.L895. [DOI] [PubMed] [Google Scholar]
- 20.Das SK, Mukherjee S. Role of peripheral benzodiazepines receptors on secretion of surfactant in guinea pig alveolar type II cells. Biosci Rep. 1999;19:461–471. doi: 10.1023/a:1020272508250. [DOI] [PubMed] [Google Scholar]
- 21.Peterson JW, Luich KM, Goldie RG. The role of beta-adrenoreceptors in hyperreactivity. In: Morley J, editor. Perspectives in asthma. London: Academic Press; 1982. pp. 245–268. [Google Scholar]
- 22.Pauwels R. Bronchial adrenergic receptors and asthma, tachyphylaxis and its prevention. Allerg Immunol (Paris) 1988;20:261–265. [PubMed] [Google Scholar]
- 23.Persad S, Elimban V, Kaila J, Dhalla NS. Biphasic alterations in cardiac Beta-adrenoceptor signal transduction mechanism due to oxyradicals. J Pharmacol Exp Ther. 1997;282:1623–1631. [PubMed] [Google Scholar]
- 24.Ferguson SSG. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 25.Stadel JM, Nambi P, Shorr RGL, Sawer DF, Caron MG, Lefkowitz RJ. Catecholamine-induced desensitization of Turkey erythrocyte adenylate cyclase is associated with phosphorylation of the β-adrenergic receptor. Proc Natl Acad Sci USA. 1983;80:3173–3177. doi: 10.1073/pnas.80.11.3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Profita M, Chiappara G, Mirabella F, Di Giorgi R, Chimenti L, Costanzo G, Riccobono L, Bellia V, Bousquet J, Vignola AM. Effect of cilomilast (Ariflo) on TNFα, IL-8 and GM-CSF release by airway cells of patients with COPD. Thorax. 2003;58:573–579. doi: 10.1136/thorax.58.7.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall M, Glumoff V, Ramet M. Surfactant in respiratory distress syndrome and lung injury. Comp Biochem Physiol. 2001;129:287–294. doi: 10.1016/s1095-6433(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 28.Dobbs LG, Mason RJ. Pulmonary alveolar type II cells isolated from rats: Release of phosphatidylcholine in response to β-adrenergic stimulation. J Clin Invest. 1979;63:378–387. doi: 10.1172/JCI109313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whitsett JA, Manton MA, Darovec-Beckerman C, Adams KG, Moore JJ. Beta-adrenergic receptors in the developing rabbit lung. Am J Physiol. 1981;240:E351–357. doi: 10.1152/ajpendo.1981.240.4.E351. [DOI] [PubMed] [Google Scholar]
- 30.Mills HE. Implications of feedback regulation of beta-adrenergic signaling. J Anim Sci. 2001;80 (E Suppl 1):E30–E35. [Google Scholar]
- 31.Davies AO, Lefkowitz RJ. Regulation of β-adrenergic receptors by steroid hormones. Annu Rev Physiol. 1984;46:119–130. doi: 10.1146/annurev.ph.46.030184.001003. [DOI] [PubMed] [Google Scholar]
- 32.Mak JCW, Nishikawa M, Shirasaki H, Miyayasu K, Barnes PJ. Protective effects of glucocorticoids on down regulation of pulmonary β2-Adrenergic receptors in vivo. J Clin Invest. 1995;96:99–106. doi: 10.1172/JCI118084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hadcock JR, Malbon CC. Regulation of β-adrenergic receptors by permissive hormones; glucocorticoids increase steady state levels of receptor mRNA. Proc Natl Acad Sci USA. 1988;85:8415–8419. doi: 10.1073/pnas.85.22.8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward PA, Till GO, Kunel R, Beaucham C. Evidence for role of hydroxyl radical in complement and neutrophil-dependent tissue injury. J Clin Invest. 1983;72:789–801. doi: 10.1172/JCI111050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherjee S, Nayyar T, Chytil F, Das SK. Mainstream and sidestream cigarette smoke exposure increases retinol in guinea pig lungs. Free Radic Biol Med. 1995;18:507–514. doi: 10.1016/0891-5849(94)00161-c. [DOI] [PubMed] [Google Scholar]
- 36.Das SK, Mukherjee S, Banerjee D. β-adrenoreceptors of multiple affinities in a clonal capillary endothelial cell line and its functional implication. Mol Cell Biochem. 1994;140:49–54. doi: 10.1007/BF00928365. [DOI] [PubMed] [Google Scholar]
- 37.Peterson GL. A simplification of the protein assay method of Lowry et al. Which is more generally applicable. Anal Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 38.Das SK, Tsao FHC, Mukherjee S. Mainstream and sidestream cigarette smoke exposure increases Ca2+-dependent phospholipid binding proteins in guinea pig alveolar type II cell. Mol Cell Biochem. 2002;231:37–42. doi: 10.1023/a:1014468521403. [DOI] [PubMed] [Google Scholar]
- 39.Mukherjee S, Coaxum SD, Maleque M, Das SK. Effects of oxidized low-density lipoprotein on nitric oxide synthetase and protein kinase C activities in bovine endothelial cells. Cell Mol Biol. 2001;47:1051–1058. [PubMed] [Google Scholar]
- 40.Belfer I, Buzas B, Evans C, Hipp H, Phillips G, Taubman J, Lorincz I, Lipsky RH, Enoch MA, Max MB, Goldman D. Haplotype structure of the beta adrenergic receptor genes in US Caucasians and African Americans. Eur J Hum Genet. 2004;13:341–351. doi: 10.1038/sj.ejhg.5201313. [DOI] [PubMed] [Google Scholar]
- 41.Sinha Roy S, Mukherjee S, Kabir S, Rajaratnam V, Smith M, Das SK. Inhibition of cholinephosphotransferase activity in lung injury induced by 2-chloroethyl ethyl sulfide, a mustard analog. J Biochem Mol Toxicol. 2005;19:289–297. doi: 10.1002/jbt.20092. [DOI] [PubMed] [Google Scholar]
- 42.Watson ML, Smith D, Bourne AD, Thompson RC, West-wick J. Cytokines contribute to airway dysfunction in antigen-challenged guinea pigs: Inhibition of airway hyperreactivity, pulmonary eosinophil accumulation and tumor necrosis factor by pretreatment with an interleukin-1 receptor antagonist. Am J Respir Cell Mol Biol. 1993;8:365–369. doi: 10.1165/ajrcmb/8.4.365. [DOI] [PubMed] [Google Scholar]
- 43.Hakonarson H, Herrick DJ, Gonzalez P, Grunstein MM. Mechanism of cytokine-induced modulation of β-adrenoceptor responsiveness in airway smooth muscle. J Clin Invest. 1996;97:2593–2600. doi: 10.1172/JCI118708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore PE, Lahiri T, Laporte JD, Church T, Panettieri RA, Jr, Shore SA. Signal transduction in smooth muscle selected contribution: Synergism between TNFα and IL-1β in airway smooth muscle cells: Implications for β-adrenergic responsiveness. J Appl Physiol. 2001;91:1467–1474. doi: 10.1152/jappl.2001.91.3.1467. [DOI] [PubMed] [Google Scholar]
- 45.Hegab AE, Sakamoto T, Saitoh W, Massoud HH, Massoud HM, Hassanein KM, Sekizawa K. Polymorphisms of IL4, IL13 and ADR2 gene in COPD. Chest. 2004;126:1832–1839. doi: 10.1378/chest.126.6.1832. [DOI] [PubMed] [Google Scholar]
- 46.Wills-Krap M, Uchida Y, Lee JY, Jinot J, Hirata A, Hirata F. Organ culture with proinflammatory cytokines reproduces impairment of the beta-adrenoreceptor-mediated relaxation in tracheas of a guinea pig antigen model. Am J Respir Cell Mol Biol. 1993;8:153–159. doi: 10.1165/ajrcmb/8.2.153. [DOI] [PubMed] [Google Scholar]
- 47.Koto H, Mak JC, Haddad EB, Xu WB, Salmon M, Barnes PJ, Chung KR. Mechanisms of impaired beta-adrenoceptor-induced airway relaxation by interleukin-1 beta in vivo in the rat. J Clin Invest. 1996;98:1780–1787. doi: 10.1172/JCI118977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boskabady MH, Teymoory S. The influence of epithelium on the responsiveness of guinea-pig trachea to -adrenergic agonist and antagonist. Med Sci Monit. 2003;9(9):BR336–342. [PubMed] [Google Scholar]
- 49.Lefkowitz RJ, Caron MG. Regulation of adrenergic receptor function by phosphorylation. Curr Top Cell Regul. 1986;28:209–231. doi: 10.1016/b978-0-12-152828-7.50007-x. [DOI] [PubMed] [Google Scholar]
- 50.Mills SE. Implications of feedback regulation of beta-adrenergic signaling. J Anim Sci. 2003;80(E Suupl 1):E30–E35. [Google Scholar]
- 51.Van Hoof IH, Van Bree L, Bast A. Changes in receptor function by oxidative stress in guinea pig tracheal smooth muscle. Cent Eur J Public Health. 1996;4(Suppl):3–5. [PubMed] [Google Scholar]
- 52.Doelman CJ, Leurs R, Oosterom WC, Bast A. Mineral dust exposure and free radical-mediated lung damage. Exp Lung Res. 1990;16:41–55. doi: 10.3109/01902149009064698. [DOI] [PubMed] [Google Scholar]
- 53.Doelman CJ, De Vlieger JF, Sprong RC, Bast A. Oxidative stress and receptor responses in guinea-pig tracheal tissue. Agents Actions Suppl. 1990;31:143–145. doi: 10.1007/978-3-0348-7379-6_19. [DOI] [PubMed] [Google Scholar]
- 54.Nishikawa M, Mak JCW, Shirasaki H, Harding SE, Barnes PJ. Differential down-regulation of pulmonary β1- and β2-adrenergic receptor messenger RNA with prolonged in vivo infusion of isoprenaline. Eur J Pharmacol Mol Pharmacol. 1993;247:131–138. doi: 10.1016/0922-4106(93)90070-p. [DOI] [PubMed] [Google Scholar]
- 55.Zhang HQ, Tai HH, Lai YL. Oxygen radicals in the nonvagal component of cholinergic airway constriction. Respir Physiol. 1996;104:213–220. doi: 10.1016/0034-5687(96)00004-7. [DOI] [PubMed] [Google Scholar]
- 56.Morken JJ, Warren KU, Xie Y, Rodriguez JL, Lyte M. Epinephrine as a mediator of pulmonary neutrophils sequestration. Shock. 2002;18:46–50. doi: 10.1097/00024382-200207000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Goadby P. Release of adrenaline by anaphylaxis in the guinea pig: Its effect on lung surfactant. J Pharm Pharmacol. 1975;27:254–261. doi: 10.1111/j.2042-7158.1975.tb10695.x. [DOI] [PubMed] [Google Scholar]
- 58.Collins S, Altschmied J, Herbsman O, Caron MG, Mellon PL, Lefkowitz RJ. A cAMP response element in the β2-adrenergic receptor gene confers transcriptional autoregultion by cAMP. J Biol Chem. 1990;265:19330–19335. [PubMed] [Google Scholar]
- 59.Tran TM, Friedman J, Qunaibi F, Baameur F, Moore RH, Clark RB. Characterization of agonist stimulation of cAMP-dependent protein kinase and G-protein-coupled receptor kinase phosphorylation of the β2-adrenergic receptor using phosphoserine-specific antibodies. Mol Pharmacol. 2004;65:106–206. doi: 10.1124/mol.65.1.196. [DOI] [PubMed] [Google Scholar]
- 60.Penn RB, Benovic JL. Regulation of G protein coupled receptors. In: Conn PM, editor. Handbook of physiology. Oxford: Oxford University Press; 1998. pp. 125–164. [Google Scholar]
- 61.Raeburn D, Adrenier C. Isoenzyme-selective cyclic nucleotide phospho-diesterase inhibitors: Effects on airways smooth muscle. Int J Biochem Cell Biol. 1995;27:29–37. doi: 10.1016/1357-2725(94)00060-3. [DOI] [PubMed] [Google Scholar]
- 62.Tepe NM, Liggett SB. Functional receptor coupling to Gi a mechanism of an agonist-promoted desensitization of the beta2-adrenergic receptor. J Recept Signal Transduct Res. 2000;20:75–85. doi: 10.3109/10799890009150038. [DOI] [PubMed] [Google Scholar]
- 63.Lakshminarayanan V, Drab-Weiss EA, Roeback KA. Hydrogen peroxide and tumor necrosis factor-α induce differential binding of the redox-responsive transcription factors AP-1 and NF-κB to the interleukin–8 promoters in endothelial and epithelial cells. J Biol Chem. 1996;273:32670–32678. doi: 10.1074/jbc.273.49.32670. [DOI] [PubMed] [Google Scholar]
- 64.Rahman I, MacNee W. Role of transcription factors in inflammatory lung diseases. Thorax. 1998;53:601–612. doi: 10.1136/thx.53.7.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spurzem JR, Gupta J, Veys T, Kneifl KR, Rennard SI, Wyatt TA. Activation of protein kinase A accelerates bovine bronchial epithelial cell migration. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1108–L1116. doi: 10.1152/ajplung.00148.2001. [DOI] [PubMed] [Google Scholar]
- 66.Nishimura K, Tamaoki J, Isono K, Aoshiba K, Nagai A. Beta-adrenergic receptor-mediated growth of human airway epithelial cell lines. Eur Respir J. 2002;20:353–358. doi: 10.1183/09031936.02.01352001. [DOI] [PubMed] [Google Scholar]
- 67.Dumasius V, Sznajder JL, Azzam ZS, Boja J, Mutlu GM, Maron MB, Factor P. Beta (2)-adrenergic receptor overexpression increases alveolar fluid clearance and responsiveness to endogenous catecholamines in rats. Circ Res. 2001;89:907–914. doi: 10.1161/hh2201.100204. [DOI] [PubMed] [Google Scholar]