

Abstract
Damage to oligodendrocytes caused by glutamate release contributes to mental or physical handicap in periventricular leukomalacia, spinal cord injury, multiple sclerosis and stroke, and has been attributed to activation of AMPA/kainate receptors. However, glutamate also activates unusual NMDA receptors in oligodendrocytes, which can generate an ion influx even at the resting potential in a physiological [Mg2+]. Here we show that the clinically licensed NMDA receptor antagonist memantine blocks oligodendrocyte NMDA receptors at concentrations achieved therapeutically. Simulated ischaemia released glutamate which activated NMDA receptors, as well as AMPA/kainate receptors, on mature and precursor oligodendrocytes. Although blocking AMPA/kainate receptors alone during ischaemia had no effect, combining memantine with an AMPA/kainate receptor blocker, or applying the NMDA blocker MK-801 alone, improved recovery of the action potential in myelinated axons after the ischaemia. These data suggest NMDA receptor blockers as a potentially useful treatment for some white matter diseases and define conditions under which these blockers may be useful therapeutically. Our results highlight the importance of developing new antagonists selective for oligodendrocyte NMDA receptors based on their difference in subunit structure from most neuronal NMDA receptors.
Keywords: Glutamate, white matter, glia, NMDA receptor, ischaemia
Introduction
Glutamate is released in the white matter of the brain in a range of pathological conditions and has been thought to damage oligodendrocytes by activating AMPA/kainate receptors (Volpe, 2001; Matute et al., 2001; Dewar et al., 2003; Park et al., 2004): a concept which has guided therapeutic strategies for preventing excitotoxic damage to these cells. However, glutamate also activates unusual NMDA receptors in oligodendrocytes, which are blocked only weakly by Mg2+ (Káradóttir et al., 2005). These receptors are expressed at all stages of oligodendrocyte development, and in mature oligodendrocytes are preferentially located on the cells' myelinating processes (Káradóttir et al., 2005; Salter & Fern, 2005; Micu et al., 2006). They are activated in conditions of energy deprivation (Káradóttir et al., 2005; Salter & Fern, 2005; Micu et al., 2006), as occurs in periventricular leukomalacia (Volpe, 2001), ischaemia secondary to spinal cord injury (Park et al., 2004) and stroke (Dewar et al., 2003), when glutamate is released from axons and from oligodendrocytes by the reversal of glutamate uptake carriers (Li et al., 1999; Back et al., 2006; Káradóttir & Attwell, 2007). They may also be activated when glutamate is released in active multiple sclerosis lesions (Werner et al., 2001). In ischaemia these NMDA receptors raise the [Ca2+] within myelin and destroy the myelinating processes of oligodendrocytes (Salter & Fern, 2005; Micu et al., 2006), suggesting these receptors as a novel therapeutic target for preventing white matter pathology (Lipton, 2006; Matute, 2006). However, the efficacy of NMDA receptor block for preventing ischaemia-evoked loss of the action potential in myelinated neurons has not been investigated in detail.
Memantine is licensed for therapeutic block of NMDA receptors (Lipton, 2006), raising the possibility that it could also be used to treat NMDA receptor mediated damage occurring to oligodendrocytes in pathological conditions (Salter & Fern, 2005; Micu et al., 2006). However, since oligodendrocyte NMDA receptors are formed from an unusual combination of subunits (probably NR1, NR2C and NR3), different from most neuronal NMDA receptors (Káradóttir et al., 2005), it is uncertain whether memantine will be effective in blocking their activation by glutamate. We therefore tested the effect of memantine on NMDA receptor mediated currents in oligodendrocytes, and investigated whether this drug or another NMDA receptor blocker, MK-801, could help to reduce the loss of action potential propagation occurring in myelinated axons after simulated ischaemia.
Materials and Methods
Brain slice and optic nerve preparation
Tissue was from P12 rats for patch-clamp studies on corpus callosum (at which age mature myelinating oligodendrocytes are present (Káradóttir et al., 2005)) or P28 rats for studying the effect of simulated ischaemia on the optic nerve compound action potential (an age when myelination is >80% complete (Tennekoon et al., 1977)). Forebrain slices (Káradóttir et al., 2005), 225μm thick, including the corpus callosum, were prepared in solution containing 1mM Na-kynurenate to block glutamate receptors. Optic nerves were isolated and recorded from using suction electrodes (Bolton & Butt 2005). Tissue was superfused at 33±1°C for ischaemia experiments, or at 23±1°C for dose-response studies, with bicarbonate-buffered solution containing (mM) 126 NaCl, 24 NaHCO3, 1 NaH2PO4, 2.5 KCl, 0 MgCl2 (to maximise NMDA receptor mediated currents, for dose-response studies) or 1 MgCl2 (the physiological value, for ischaemia studies), 2.5 CaCl2, 10 glucose, bubbled with 95% O2/5% CO2, pH 7.4. The solution flow rate was ~6 ml/min into a 1.5 ml bath, giving a 15 sec turnover time. To simulate ischaemia we replaced external O2 by N2, and external glucose by 7mM sucrose. In addition, for studies of the effect of ischaemia on patch-clamped oligodendrocytes, we added 2mM iodoacetate to block glycolysis and 25μM antimycin to block oxidative phosphorylation (Allen et al., 2005). Without iodoacetate and antimycin, it took ~3-fold longer for the ischaemia-evoked inward current to develop, probably because in an open chamber O2 can diffuse to the slice allowing glycogen metabolism in mitochondria for longer than would occur in vivo (Allen et al., 2005).
Patch-clamping
White matter oligodendrocytes were whole-cell clamped with pipettes containing a Cs+ (or K+) based solution, comprising (mM) 130 Cs-gluconate (or 130 K-gluconate), 4 NaCl, 4 CsCl, 0.5 CaCl2, 10 HEPES, 10 BAPTA, 4 MgATP, 0.5 Na2GTP, 2 K-Lucifer yellow, pH set to 7.3 with CsOH. Series resistance was ~10-20 MΩ, before 50% compensation. Electrode junction potentials (−10 mV) were compensated. NMDA-evoked currents in corpus callosal oligodendrocytes desensitise, making it hard to distinguish desensitization from the effect of memantine if memantine was applied after starting the application of a high NMDA concentration. We therefore applied memantine and then superimposed NMDA (60μM), and compared the resulting peak NMDA-evoked current in cells pre-exposed to different concentrations of memantine. After recording some slices were fixed in 4% paraformaldehyde, and Lucifer yellow filled cells and antibody labelling were imaged as described previously (Káradóttir et al., 2005; Káradóttir & Attwell, 2006).
Compound action potential
The optic nerve was stimulated using a suction electrode filled with extracellular solution, usually using 0.2 msec, 2-8 volt pulses, at 0.033 Hz (a low stimulus strength was used to avoid activation of the small number (Tennekoon et al., 1977) of unmyelinated axons present at this age), although in some experiments 100 volt pulses were used to produce a nearly maximal response (see Results). The compound action potential was recorded as a current using an Axon Axopatch 200B patch clamp for low stimulus strength experiments, or as a voltage using an Axon Axoclamp 2B amplifier for high stimulus strength experiments) with a second suction electrode at least 2mm from the stimulating electrode. At the end of each experiment TTX (1μM) was applied to obtain the stimulus artefact in the absence of action potentials, which was subtracted from all the records obtained previously. The pre-stimulus baseline of the resulting traces was subtracted, then they were rectified (squared, then square rooted), and the area of the compound action potential was measured out to where the waveform returned to baseline.
Statistics
Data are mean ± s.e.m. P values are from Student's 2-tailed t-tests.
Results
NMDA activates receptors on corpus callosum oligodendrocytes
Mature myelinating oligodendrocytes in the corpus callosum, an area which is thinned in severe periventricular leukomalacia (Maneru et al., 2003) as a result of damage occurring to precursor/immature oligodendrocytes (Craig et al., 2003), were whole-cell clamped and identified by their dye-fill morphology (all cells) and post-recording labelling (n=5) for myelin basic protein (Fig. 1A-C). As reported previously (Káradóttir et al., 2005), NMDA application at −70mV (60μM in 0mM Mg2+ solution) evoked an inward current in these cells (Fig. 2A). To check whether this NMDA current reflects a direct effect of NMDA, or is mediated by NMDA depolarizing neurons and releasing either K+ or a transmitter with receptors on oligodendrocytes, we applied NMDA either in the presence of 1μM tetrodotoxin to block action potentials, or in the presence of blockers of AMPA/kainate, GABAA, glycine and metabotropic glutamate receptors (25μM NBQX, 10μM GABAzine, 5μM strychnine, and 1mM (S)-MCPG). Neither manipulation reduced the NMDA-evoked current in oligodendrocytes. Adding tetrodotoxin did not significantly alter the response (increased by 18%, p=0.74, 6 cells in control solution and 9 cells in TTX, data not shown), and the presence of the receptor blockers also had no effect (increased by 5%, p=0.87, 4 cells in control solution and 5 cells in blockers, data not shown). These results suggest that NMDA acts directly on corpus callosal oligodendrocytes, as shown previously for cerebellar oligodendrocytes (Káradóttir et al., 2005).
Fig. 1.

Identification of corpus callosum oligodendrocytes expressing NMDA receptors. (A-C) Identification of corpus callosal oligodendrocyte. (A) Lucifer yellow (LY, green) fill of cell. (B) Antibody labelling (post-recording) for myelin basic protein (MBP, red). (C) Overlay of A and B (both). (D) Oligodendrocyte filled with Lucifer yellow, labelled post-recording for NR1 subunits of NMDA receptors.
Fig. 2.

Reinforcing this result, antibody directed against the NR1 subunit of NMDA receptors labelled oligodendrocyte processes in cells that had been recorded from (Fig. 1D, n=5) suggesting that, as in cerebellar white matter and optic nerve (Káradóttir et al., 2005; Salter & Fern, 2005; Micu et al., 2006), there are NMDA receptors present on oligodendrocytes.
Memantine blocks oligodendrocyte NMDA receptors at clinically relevant concentrations
We compared NMDA responses in normal solution to NMDA responses evoked after pre-exposure for 1 minute to different concentrations of memantine. Memantine reduced the NMDA-evoked current (Fig. 2A, B). The dependence of current block on memantine concentration could be roughly fitted by a first order inhibition curve with a Km of 54 μM (Fig. 2C), or somewhat better fitted by the sum of two first order inhibition curves (of approximately equal amplitude) with Km = 4.6 and 220 μM.
The data in Fig. 2C underestimate the true IC50 because of the slow penetration of lipophilic memantine into the tissue (Parsons et al., 1999a). In addition, memantine can bind to both open and closed NMDA receptor channels (Blanpied et al., 1997) and in a clinical setting, when it is present in the body for an extended period, it is possible that lower concentrations of memantine would gain access to the receptor channels. We therefore tested the effect of pre-exposing corpus callosum slices to 1μM memantine, a concentration similar to that reached therapeutically in the brain (Parsons et al., 1999a), for 2 hours (compared to exposure for 2 hours to memantine-free solution). This was found to reduce the NMDA-evoked current by 70±7%, which is significantly more (p=0.008) than was found using 1 min pre-exposure to 3μM memantine, and indicates a 100-fold shift of the inhibition curve to lower concentrations with 2 hours pre-exposure to the drug (open circle in Fig. 2C).
The data in Fig. 2 were obtained using Cs+ as the main cation in the internal solution, to reduce the cells' K+ conductance and therefore improve voltage uniformity in the cells. Since, for neuronal NMDA receptors, caesium has been reported to lower (2.6 fold) the affinity of memantine as an NMDA receptor antagonist (Parsons et al., 1999b), we also tested the block of NMDA responses produced by 2 hours pre-incubation with 1μM memantine when the internal solution contained K+ as the main cation. The NMDA-evoked current was reduced by 56±10% which is not significantly different (p=0.49) from the 70±7% reduction observed when the experiment was done with caesium as the main intracellular ion. It is unclear whether this difference with the results of Parsons et al. (1999b) reflects a different effect of Cs+ versus K+ on the oligodendrocyte NMDA receptors compared to neuronal receptors, or a lack of dialysis of the myelinating processes of mature oligodendrocytes by Cs+ introduced via the patch pipette.
Ischaemia activates oligodendrocyte NMDA receptors in corpus callosum
To test whether NMDA receptor block might reduce glutamate-mediated white matter damage in energy deprivation conditions, we whole-cell clamped oligodendrocytes in the corpus callosum during application of solution mimicking ischaemia (and containing a physiological concentration of Mg2+). Ischaemia led to oligodendrocytes generating a slowly developing inward current, part of which was suppressed by the NMDA receptor blocker D-AP5 (used because, unlike memantine, it is rapidly reversible, Fig. 3A) both in mature myelinating oligodendrocytes and in cells with the morphology (Káradóttir et al., 2005) of oligodendrocyte precursors (Fig. 3B). Thus, glutamate is released in conditions of energy deprivation and activates NMDA receptors in mature corpus callosum oligodendrocytes (a potential cause of white matter damage occurring after myelination is complete: Craig et al. (2003)) and in precursor cells (a potential cause of damage associated with periventricular leukomalacia: Maneru et al. (2003); Craig et al. (2003)).
Fig. 3.

NMDA receptor mediated ischaemia-evoked inward current in corpus callosal oligodendrocytes. (A) Membrane current at −70mV in a mature oligodendrocyte during application of ischaemia solution. D-AP5 (200μM) blocks a component of the ischaemia-evoked current. (B) Mean current (±s.e.m.) blocked by D-AP5 (~6 mins after the start of ischaemia) in oligodendrocyte precursors (Prec) and mature myelinating oligodendrocytes (Mature). Numbers of cells shown on bars.
NMDA receptor block reduces loss of the myelinated nerve action potential in ischaemia
To assess whether activation of oligodendrocyte NMDA receptors blocks action potential propagation in pathological conditions, we studied the optic nerve: a myelinated fibre tract that contains no neuronal somata, and which suffers glutamate-mediated damage in ischaemia and multiple sclerosis. Using the optic nerve avoids confounding effects of neuronal NMDA receptors since NMDA receptors have not been reported on axons. The compound action potential, reflecting the activity of all the stimulated axons in the nerve, was recorded and, after subtracting the stimulus artefact (Fig. 4A), was quantified from its area (see Materials and Methods). In the absence of glutamate receptor blockers, 20 mins ischaemia reduced the compound action potential area (which was stable for over an hour in normal solution) by over 80%, with no recovery 30 mins after returning to normal solution containing oxygen and glucose (Figs. 4A; 5A). Part of this irreversible decline probably reflects axonal damage caused, for example, by a rise of axonal [Ca2+]i caused by reversal of Na/Ca exchange when ion gradients run down in ischaemia (Stys, 2005). However, the effect of glutamate receptor blockers (see below) indicates that the decline is partly due to glutamate-mediated damage to oligodendrocytes, and is preventable.
Fig. 4.

Ischaemia-evoked changes in optic nerve compound action potential (CAP). (A) Top row, traces from left are: CAP in control solution, after 20 mins ischaemia, 30 mins after returning to control solution, and the stimulus artefact recorded subsequently in 1μM TTX. Bottom row shows same traces with stimulus artefact trace subtracted. (B-G) Artefact-subtracted responses with 25μM NBQX (B), 300μM memantine (C), NBQX + memantine (D), 50μM MK-801 (E) or NBQX + MK-801 (F) present during ischaemia, and (G) NBQX present during ischaemia + 1 μM memantine pre-incubated 2 hours before ischemia and also present during ischaemia.
Fig. 5.
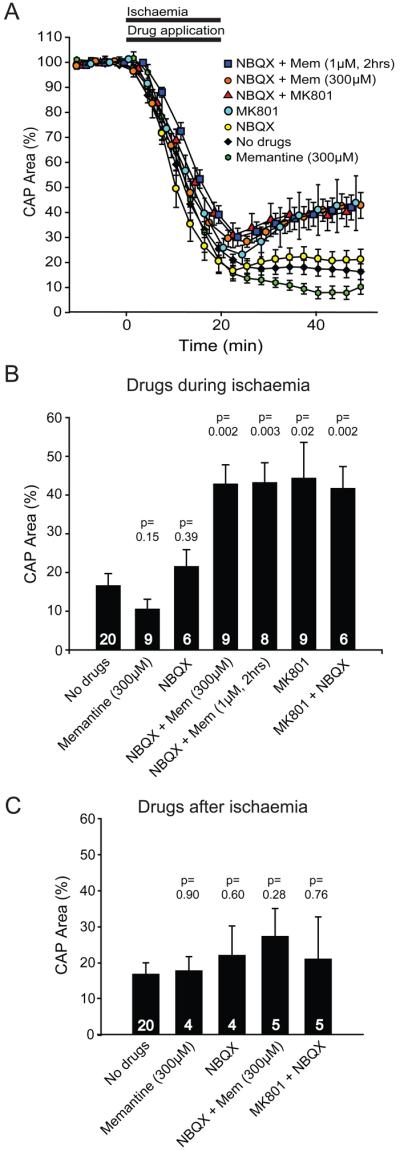
Effect of glutamate receptor blockers on ischaemia-evoked changes in optic nerve compound action potential (CAP). (A) Normalized area of rectified CAP before, during and after ischaemia (from 0-20 mins), using ischaemia solutions containing no glutamate receptor blockers (No drugs), 25μM NBQX, 300μM memantine (Mem), NBQX + Mem, 50μM MK-801, or NBQX + MK-801 (drugs were applied during ischaemia as shown by the bar), and also using solutions containing 1μM Mem from 2 hours before the ischaemia solution was applied (1μM Mem was present for the whole experiment, with NBQX present during ischaemia). (B) Normalised CAP area after 30 mins in normal solution, following 20 mins application of ischaemia solution containing different glutamate receptor blockers. Number of nerves studied is shown on each bar. (C) Normalised CAP area after 30 mins in normal solution containing the glutamate receptor blockers indicated, which was preceded by 20 mins of ischaemia solution lacking blockers. P values in (B) and (C) are for comparison with ‘No drugs’.
Including the AMPA/kainate receptor blocker NBQX (25μM) in the ischaemia solution did not reduce the suppression of the compound action potential (Figs. 4B; 5A, B), and a similar lack of improvement was obtained in the presence of 300μM memantine alone (Figs. 4C; 5A, B). However, when NBQX and memantine were present together during ischaemia, the compound action potential started to recover when the ischaemia solution was removed (Figs. 4D; 5A), and reached over 40% of its original area 30 mins after returning to non-ischaemic solution (Fig. 5B). Applying the classical NMDA receptor blocker MK-801 (50μM), either alone or with NBQX, led to a similar recovery (Figs. 4E, F; 5A, B), confirming the potential of NMDA receptor block to promote the recovery of myelinated axon function after ischaemia.
To mimic better the effect of a clinically relevant exposure time and concentration of memantine, we also measured recovery of the compound action potential from ischaemia in nerves that were pre-exposed to 1μM memantine for 2 hours before the ischaemia. Pre-exposure to this clinically-relevant (Parsons et al., 1999a) concentration, combined with NBQX during the ischaemia, led to a recovery of the compound action potential similar to that seen for short-term application of 300μM memantine with NBQX (Figs. 4G, 5A, B). Thus, clinically attainable levels of memantine in the brain or spinal cord could reduce damage to white matter oligodendrocytes.
In neurons, NMDA receptor blockers can prevent ischaemia-evoked damage even when applied after the ischaemic insult (Gill et al., 1988). However, when memantine or MK-801, together with NBQX, were applied to the optic nerve after, rather than during, ischaemia they were no longer significantly protective (Fig. 5C).
To test whether the lack of protective effect of blocking AMPA/kainate receptors, which disagrees with previous work (see Discussion), could reflect either a slow penetration of NBQX into the nerve (although the similarly sized TTX abolished the action potential rapidly), or our use of a low stimulus strength to activate only low threshold myelinated fibres, we tested the effect of pre-incubating with NBQX for 2 hours, while using a large stimulus that produced an almost saturating response. NBQX again had no protective effect (p=0.7, Fig 6), while including MK-801 with the NBQX greatly improved recovery after the ischaemia (p=0.0001).
Fig. 6.

Effect of pre-incubation with glutamate receptor blockers on ischaemia-evoked changes in optic nerve compound action potential (CAP). (A) Normalized area of rectified CAP before, during and after ischaemia, using solutions containing no glutamate receptor blockers (No drugs), pre-incubation with 25μM NBQX from 2 hours before ischaemia until the end of ischaemia, or pre-incubation with 25μM NBQX and 50μM MK-801 from 2 hours before ischaemia until the end of ischaemia. These experiments used strong stimulation that evoked a near maximal response. (B) Normalised CAP area after 30 mins in normal solution, following the period of ischaemia. Number of nerves studied is shown on each bar. P values are compared with ‘No drugs’.
Discussion
The presence of NMDA receptors in CNS white matter was suspected over 10 years ago (Matute & Miledi, 1993), but later papers reported only AMPA/kainate receptors in oligodendrocytes, and the expression of NMDA receptors by oligodendrocytes has only recently been documented (Káradóttir et al., 2005; Salter & Fern, 2005; Micu et al., 2006). Consequently, the idea that oligodendrocytes express only AMPA/kainate receptors and not NMDA receptors has guided previous therapeutic strategies for preventing excitotoxic damage to these cells. Blocking glutamate's action on AMPA/kainate receptors is reported to help to preserve the function of myelinated axons in ischaemia (Li et al., 1999; Tekkök & Goldberg, 2001; Wrathall et al., 1997; Follett et al., 2000) and in a model of multiple sclerosis (Pitt et al., 2000; Smith et al., 2000). Surprisingly, in our experiments the AMPA/kainate blocker NBQX alone had no significant effect on the recovery of the optic nerve compound action potential after ischaemia. However blocking NMDA receptors, either alone using MK-801, or together with block of AMPA/kainate receptors when using memantine, significantly improved recovery (Fig. 5).
Interestingly, the NMDA receptor blocker ketamine was previously reported to improve recovery of the compound action potential after anoxia of the optic nerve (Ransom et al., 1990), although at the time this was not attributed to an action on NMDA receptors. Similarly, a previous study on an animal model for multiple sclerosis also found a beneficial effect of memantine (Wallström et al., 1996): this was attributed to an action on neuronal NMDA receptors but, now that it is known that oligodendrocytes express NMDA receptors, a more parsimonious explanation is that memantine directly protects the oligodendrocytes from glutamate which is released in this condition (Werner et al., 2001; Pitt et al., 2000; Smith et al., 2000). Nevertheless, Tekkök et al. (2007) recently reported no protective effect of blocking NMDA receptors when optic nerves were oxygen and glucose deprived. This difference from our data may stem from the methodological difference that they used mice whereas we used rats.
Although our experiments focus on preventing NMDA receptor mediated damage to myelin and loss of the action potential, the NMDA receptors on oligodendrocyte precursors have properties similar to those on mature myelinating oligodendrocytes (including a weak Mg2+ block that allows a glutamate evoked Ca2+ influx at the resting potential (Káradóttir et al., 2005)) and so NMDA receptor block may also reduce loss of precursors in periventricular leukomalacia.
We assume that the NMDA receptors which need to be blocked to reduce the loss of action potential propagation in myelinated axons during ischaemia are those on oligodendrocytes, for three reasons. First, as we show in Fig. 1, these receptors are present on the myelinating processes of oligodendrocytes, where an ion influx into the small intracellular space is likely to disrupt the structure of myelin and thus abolish propagation of the action potential. Second, we have shown that, in the corpus callosum, oligodendrocyte currents evoked by NMDA are not blocked by TTX, and so do not reflect NMDA depolarizing neurons and evoking the release of K+ or other neurotransmitters which then generate a current in oligodendrocytes. Also, the oligodendrocyte NMDA-evoked currents are unaffected by block of AMPA/kainate, GABAA, glycine or metabotropic glutamatergic receptors, demonstrating that the NMDA-evoked current was not a secondary response to the activation of any of these receptor types by released glutamate, GABA or glycine. Third, these receptors are activated in ischaemia (Fig. 3; Káradóttir et al., 2005), and in other brain areas their activation in ischaemia is known to cause disruption of the structure of myelin (Salter & Fern, 2005; Micu et al., 2006). Nevertheless, astrocytes (at least in grey matter) are also reported to express NMDA receptors (Schipke et al., 2001; Lalo et al., 2006), as well as non-NMDA receptors (García-Barcina & Matute, 1996), and we cannot rule out the possibility that it is activation of these receptors (rather than those on the myelinating processes of oligodendrocytes) during ischaemia which somehow abolishes action potential propagation in myelinated axons.
Our data specify two important constraints on how NMDA receptor block can be used to protect white matter in pathological conditions. First, it is important that oligodendrocyte NMDA receptors are blocked during the period when glutamate is released pathologically, such as during ischaemia occurring secondary to spinal cord injury, and during the elevation of extracellular glutamate concentration that occurs in active multiple sclerosis lesions. This requirement probably stems from the fact that disruption of myelin structure occurs relatively quickly (~30 mins) when glutamate is released (Salter & Fern, 2005; Micu et al., 2006). Second, MK-801 alone protected optic nerve oligodendrocytes against ischaemia, while memantine was only effective if AMPA/kainate receptors were also blocked and so clinical application of memantine to treat white matter damage would require combining it with a clinically approved AMPA/kainate receptor blocker such as topiramate (Follett et al., 2004). A possible explanation for this difference is that the block of NMDA receptors by memantine is much more voltage-dependent (Frankiewicz et al., 1996) than block by MK-801, so that when the glutamate concentration rises in ischaemia, activation of oligodendrocyte AMPA/kainate receptors and consequent depolarization of the cell could reduce the block of NMDA receptors by memantine, but have much less effect on the block by MK-801.
Even blocking NMDA and AMPA/kainate receptors with memantine and NBQX during ischemia provided only 40% recovery of the compound action potential after ischaemia, probably because some of the loss of the action potential reflects damage to axons (Stys, 2005; McCarran & Goldberg, 2007) rather than to their ensheathing oligodendrocytes. Combining block of glutamate receptors on oligodendrocytes with block of voltage-gated calcium channels on axons (Ouardouz et al., 2003) might therefore further enhance action potential recovery after ischaemia.
Although memantine is already licensed for clinical use, if it is employed to block oligodendrocyte NMDA receptors there may be side-effects caused by its block of neuronal NMDA receptors. Thus, our results suggest that attention should be given to developing NMDA blockers that are specific for the receptors on oligodendrocytes. Producing such blockers may be feasible, since oligodendrocyte NMDA receptors have a subunit structure different from that of most neuronal NMDA receptors (Káradóttir et al., 2005; Salter & Fern, 2005; Micu et al., 2006).
Acknowledgements
Supported by the Wellcome Trust. We thank Robert Fern, Alasdair Gibb, Peter Mobbs and Angus Silver for comments on the manuscript, and Lucia Sivilotti & David Colquhoun for the loan of equipment.
References
- Allen NJ, Káradóttir R, Attwell D. A preferential role for glycolysis in preventing the anoxic depolarization of rat hippocampal area CA1 pyramidal cells. J Neurosci. 2005;25:848–859. doi: 10.1523/JNEUROSCI.4157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Craig A, Kayton RJ, Luo NL, Meshul CK, Allcock N, Fern R. Hypoxia-ischemia preferentially triggers glutamate depletion from oligodendroglia and axons in perinatal cerebral white matter. J Cereb Blood Flow Metab. 2007;27:334–347. doi: 10.1038/sj.jcbfm.9600344. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping block of NMDA-activated responses by amantadine and memantine. J Neurophysiol. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- Bolton S, Butt AM. The optic nerve: a model for axon-glia interactions. J Pharm Toxicol Meth. 2005;51:221–233. doi: 10.1016/j.vascn.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Craig A, Ling Luo N, Beardsley DJ, Wingate-Pearse N, Walker DW, Hohimer AR, Back SA. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp Neurol. 2003;181:231–240. doi: 10.1016/s0014-4886(03)00032-3. [DOI] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP. Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab. 2003;23:263–274. doi: 10.1097/01.WCB.0000053472.41007.F9. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: a protective role for topiramate. J Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankiewicz T, Potier B, Bashir ZI, Collingridge GL, Parsons CG. Effects of memantine and MK-801 on NMDA-induced currents in cultured neurones and on synaptic transmission and LTP in area CA1 of rat hippocampal slices. Brit J Pharmacol. 1996;117:689–697. doi: 10.1111/j.1476-5381.1996.tb15245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Barcina JM, Matute C. Expression of kainate-selective glutamate receptor subunits in glial cells of the adult bovine white matter. Eur J Neurosci. 1996;8:2379–2387. doi: 10.1111/j.1460-9568.1996.tb01201.x. [DOI] [PubMed] [Google Scholar]
- Gill R, Foster AC, Woodruff GN. MK-801 is neuroprotective in gerbils when given during the post-ischaemic period. Neuroscience. 1988;25:847–855. doi: 10.1016/0306-4522(88)90040-1. [DOI] [PubMed] [Google Scholar]
- Káradóttir R, Cavelier P, Bergersen L, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, Attwell D. Combining patch-clamping of cells in brain slices with immunocytochemical labelling to define cell type and developmental stage. Nature Protocols. 2006;1:1977–1986. doi: 10.1038/nprot.2006.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Káradóttir R, Attwell D. Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience. 2007;14:1426–1438. doi: 10.1016/j.neuroscience.2006.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Kirchhoff F, North RA, Verhkratasky A. NMDA receptors mediate neuron-glia signaling in mouse cortical astrocytes. J Neurosci. 2006;26:2673–2683. doi: 10.1523/JNEUROSCI.4689-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mealing GA, Morley P, Stys PK. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci. 1999;19:RC16. doi: 10.1523/JNEUROSCI.19-14-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA. NMDA receptors, glial cells and clinical medicine. Neuron. 2006;50:9–11. doi: 10.1016/j.neuron.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Maneru C, Junque C, Salgado-Pineda P, Serra-Grabulosa JM, Bartres-Faz D, Ramirez-Ruiz B, Bargallo N, Tallada M, Botet F. Corpus callosum atrophy in adolescents with antecedents of moderate perinatal asphyxia. Brain Inj. 2003;17:1003–1009. doi: 10.1080/0269905031000110454. [DOI] [PubMed] [Google Scholar]
- Matute C. Oligodendrocyte NMDA receptors: a novel therapeutic target. Trends Molec Med. 2006;12:289–292. doi: 10.1016/j.molmed.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Matute C, Miledi R. Neurotransmitter receptors and voltage-dependent Ca2+ channels encoded by mRNA from the adult corpus callosum. Proc Natl Acad Sci USA. 1993;90:3270–3274. doi: 10.1073/pnas.90.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C, Alberdi E, Domercq M, Perez-Cerda F, Perez-Samartin A, Sanchez-Gomez MV. The link between excitotoxic oligodendroglial death and demyelinating diseases. Trends Neurosci. 2001;24:224–230. doi: 10.1016/s0166-2236(00)01746-x. [DOI] [PubMed] [Google Scholar]
- McCarran WJ, Goldberg MP. White matter axon vulnerability to AMPA/kainate receptor-mediated ischemic injury is developmentally regulated. J Neurosci. 2007;27:4220–4229. doi: 10.1523/JNEUROSCI.5542-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Nikolaeva MA, Coderre E, Zamponi GW, McRory JE, Trapp BD, Yin X, Wang W, Woulfe J, Stys PK. Depolarization-induced Ca2+ release in ischemic spinal cord white matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron. 2003;40:53–63. doi: 10.1016/j.neuron.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Park E, Velumian AA, Fehlings MG. The role of excitotoxicity in secondary mechanisms of spinal cord injury: a review with an emphasis on the implications for white matter degeneration. J Neurotrauma. 2004;21:754–774. doi: 10.1089/0897715041269641. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist - a review of preclinical data. Neuropharmacology. 1999a;38:735–767. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Bartmann A, Spielmanns P, Frankiewicz T, Hesselink M, Eilbacher B, Quack G. Amino-alkyl-cyclohexanes are novel uncompetitive NMDA receptor antagonists with strong voltage-dependency and fast blocking kinetics: in vitro and in vivo characterization. Neuropharmacology. 1999b;38:85–108. doi: 10.1016/s0028-3908(98)00161-0. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nature Medicine. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Waxman SG, Davis PK. Anoxic injury of CNS white matter: protective effect of ketamine. Neurology. 1990:1399–1403. doi: 10.1212/wnl.40.9.1399. [DOI] [PubMed] [Google Scholar]
- Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- Schipke CG, Ohlemeyer C, Matyaash M, Nolte C, Kettenmann H, Kirchhoff F. Astrocytes of the mouse neocortex express functional N-methyl-D-aspartate receptors. FASEB J. 2001;15:1270–1272. doi: 10.1096/fj.00-0439fje. [DOI] [PubMed] [Google Scholar]
- Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nature Medicine. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci. 2005;233:3–13. doi: 10.1016/j.jns.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Tekkök SB, Goldberg MP. AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkök SB, Ye Z, Ransom BR. Excitotoxic mechanism of ischemic injury in myelinated white matter. J Cereb Blood Flow Metab. 2007 doi: 10.1038/sj.jcbfm.9600455. doi: 10.1038/sj.jcbfm.9600455. [DOI] [PubMed] [Google Scholar]
- Tennekoon GI, Cohen SR, Price DL, McKhann GM. Myelinogenesis in optic nerve: a morphological, autoradiographic and biochemical analysis. J Cell Biol. 1977;72:604–616. doi: 10.1083/jcb.72.3.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Wallström E, Diener P, Ljungdahl A, Khademi M, Nilsson CG, Olsson T. Memantine abrogates neurological deficits, but not CNS inflammation, in Lewis rat experimental autoimmune encephalomyelitis. J Neurol Sci. 1996;137:89–96. doi: 10.1016/0022-510x(95)00339-4. [DOI] [PubMed] [Google Scholar]
- Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169–180. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- Wrathall JR, Teng YD, Marriott R. Delayed antagonism of AMPA/kainate receptors reduces long-term functional deficits resulting from spinal cord trauma. Exp Neurol. 1997;145:565–573. doi: 10.1006/exnr.1997.6506. [DOI] [PubMed] [Google Scholar]