

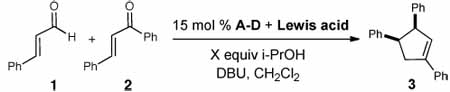
Abstract

A new approach that takes advantage of N-heterocyclic carbene/Lewis acid cooperative catalysis provides access to cis-1,3,4-trisubstituted cyclopentenes from enals and chalcone derivatives with high levels of diastereoselectivity and enantioselectivity. The presence of Ti(OiPr)4 as the Lewis acid allows for efficient substrate pre-organization, which translates into high levels of diastereoselectivity. Additionally, we demonstrate the possibility of controlling the absolute stereochemistry of NHC-catalyzed reactions by employing a catalytic amount of a chiral Lewis acid as the unique source of optically active promoter.
Cooperative catalysis is emerging as a powerful strategy in asymmetric synthesis by providing new modes of activating molecules.1,2 The combination of Lewis base and Lewis acid activation of substrates can lead to new reactivity and selectivity patterns through the pre-organization of starting materials. Cooperative catalysis involving Lewis acids and N-heterocyclic carbenes (NHCs), an important class of Lewis bases,3 has high potential for expanding the field of carbene catalysis.4 An inherent challenge to this idea is the exceptionally strong donor properties of NHCs,5 which have been exploited in the formation of useful late transition metal complexes.6 An intriguing prospect is the combination of early metals which may bind reversibly with NHCs. This scenario might provide an environment in which a Lewis acid and Lewis base operate and activate starting materials simultaneously. The significant potential of this cooperative reactivity mode prompted us to evaluate the feasibility of this concept. In this communication, we report an enantioselective cooperative system that successfully integrates Lewis acid catalysis and carbene catalysis simultaneously, thus providing direct access to previously unobtainable substituted cyclopentenes with excellent levels of enantio- and diastereoselection.
We have been heavily involved in developing N-heterocyclic carbene catalyzed generation of homoenolate equivalents from α,β-unsaturated aldehydes.7,8 In 2006, Nair disclosed the synthesis of (±)-trans-1,3,4-trisubstituted cyclopentenes resulting from the addition of enals to chalcones catalyzed by an achiral carbene.9 Subsequently, Bode reported an enantioselective variant of the Nair process to access cis cyclopentenes, but this process works only with (E)-ethyl 4-oxo-2-butenoate.10 The levels of diastereoselectivity observed in some cases are problematic since the diastereoisomers can be inseparable by standard chromatography.
 |
(1) |
To solve the lack of generality and efficiency to access cis cyclopentenes using carbene catalysis, we chose this reaction as an initial platform to investigate the integration of Lewis acid and carbene catalysis. We hypothesized that the use of a Lewis acid in a cooperative catalysis sense could greatly broaden the substrate scope by activating the conjugate acceptor and providing increased cis diastereoselectivity levels through pre-organization of the substrates (eq 1).
We initiated our studies by combining cinnamaldehyde (1) with chalcone (2) in the presence of 15 mol % achiral triazolium salt A and 1 equiv of various Lewis acids (Table 1). No conversion of the starting material was observed with the metal trifluoromethanesulfonates we examined (entries 2–4). Potentially strong binding of the NHC to the metal might be responsible for the lack of reactivity with these Lewis acids. We hypothesized that the use of Lewis acids with more donating ligands, (e.g., metal alkoxides) might attenuate this interaction. Exploring this possibility, the use of Mg(Ot-Bu)2 afforded a 2:1 mixture of the trans and cis cyclopentenes in 52% yield (entry 5).
Table 1.
Optimization of reaction conditions
 | ||||||
|---|---|---|---|---|---|---|
| entry | azolium |
Lewis acid (equiv) |
X (equiv) |
% yielda | cis:transb | % ee cisc |
| 1 | A | – | – | 73 | 1:3 | – |
| 2 | A | Zn(OTf)2 (1) | – | 0d | – | – |
| 3 | A | Sc(OTf)2 (1) | – | 0d | – | – |
| 4 | A | Mg(OTf)2 (1) | – | 0d | – | – |
| 5 | A | Mg(Ot-Bu)2 (1) | – | 52 | 1:2 | – |
| 6 | A | Ti(Oi-Pr)4 (1) | – | 47 | 20:1 | – |
| 7 | A | Ti(Oi-Pr)4 (1) | 4.0 | 71 | 20:1 | – |
| 8 | A | Ti(Oi-Pr)4 (0.2) | 4.0 | 70 | 5:1 | – |
| 9 | A | Ti(Oi-Pr)4 (0.2) | 0.2 | 75 | 20:1 | – |
| 10 | Be | Ti(Oi-Pr)4 (0.2) | 0.2 | 17f | 2:1 | nd |
| 11 | Ce | Ti(Oi-Pr)4 (0.2) | 0.2 | 68g | 20:1 | 97 |
| 12 | Ce | – | 0.2 | 84 | 1:6 | 94 |
| 13 | De | Ti(Oi-Pr)4 (0.2) | 0.2 | 74h | 20:1 | 99 |
| 14 | De | – | 0.2 | 84 | 1:6 | 99 |
| 15 | De | – | – | 90 | 1:5 | 99 |
Isolated yield.
Determined by 1H NMR spectroscopy (500 MHz) of the unpurified reaction.
Enantiomeric excess determined by HPLC.
Starting material was recovered.
10 mol % azolium salt was used.
50% conversion after 48 h.
100% conversion after 48 h.
100% conversion after 12 h.
A further survey of potential weak Lewis acids uncovered that titanium(IV) isopropoxide afforded the desired cyclopentene in moderate yield as a single diastereoisomer (entry 6). In contrast, the reaction without Lewis acid yielded the trans diastereoisomer as the major product (entry 1), illustrating the unique effect of the Lewis acid on the outcome of the reaction. To increase the yield from 47%, we hypothesized that the use of an additive might be beneficial for catalyst turnover. After surveying different alcohols, we observed the addition of 2-propanol improved both the rate and the yield to 71% (entry 7). However, performing the reaction with 20 mol % Ti(Oi-Pr)4 with 4 equiv of 2-propanol led to diminished diastereoselectivity levels (entry 8). Excess 2-propanol might have a detrimental effect on substrate pre-organization by competing for coordination sites on the titanium, thus favoring the non-Lewis acid-catalyzed pathway. In order to minimize this effect, the amount of 2-propanol was reduced to 0.2 equiv. This change resulted in a clean conversion to afford the cis cyclopentene in good yield and with excellent diastereocontrol (20:1 dr, entry 9).
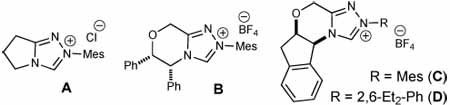
With optimized conditions employing titanium(IV) and Lewis base catalyst identified, we turned our attention to rendering this reaction enantioselective. The use of chiral triazolium salt B7c led to a sluggish reaction with low levels of diastereoselectivity for the product (Table 1, entry 10). However, full conversion of the cinnamaldehyde could be achieved with (R,R)-aminoindanol-derived triazolium salt C after 48 hours. This catalyst in the presence of 20 mol % Ti(Oi-Pr)4 afforded the cis cyclopentene with excellent diastereo- and enantioselectivity (entry 11). A survey of different N-aryl substituents in place of a mesityl unit (Mes, Me3C6H2) on the triazolium pre-catalyst identified that 2,6-diethylphenyl (D) led to higher reaction rates. With 10 mol % D, we could obtain 100% conversion of the aldehyde in 25% of the time compared to reactions with azolium C (12 hours, entry 13). The origins of this rate enhancement and the generality of this observation with regard to carbene catalyzed reactions are currently under investigation. Importantly, NHCs C and D in the absence of Ti(IV), with or without i-PrOH, provide predominantly the originally observed trans products (Table 1, entries 12,14, 15).
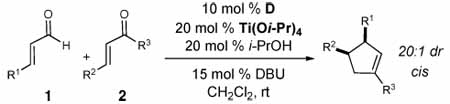
A variety of β-substituted enals were then evaluated as homoenolate substrates with chalcone (Table 2, entries 2–9). Both electron-donating and electron-widthdrawing substituents are accomodated on the aromatic ring and excellent levels of enantioselectivity were obtained.11 The highest yields were observed for enals bearing an electron-donating group at the para position of the aryl ring (e.g., entry 6). Expanding the scope to enals with an alkyl β-substituent (n-hexenal) resulted in moderate yield, but only with achiral triazolium salt A (62% entry 9).
Table 2.
Substrate scope
 | |||||
|---|---|---|---|---|---|
| entry | R1 | R2 | R3 | % yielda | % ee cisb |
| 1 | Ph | Ph | Ph | 74 (3) | 99 |
| 2 | 2-naphthyl | Ph | Ph | 76 (4) | 99 |
| 3 | 4-Br-C6H4 | Ph | Ph | 67 (5) | 98 |
| 4 | 4-Cl-C6H4 | Ph | Ph | 65 (6) | 99 |
| 5 | 4-F-C6H4 | Ph | Ph | 62 (7) | 99 |
| 6 | 4-MeO-C6H4 | Ph | Ph | 80 (8) | 99 |
| 7 | 2-MeO-C6H4 | Ph | Ph | 54 (9) | 99 |
| 8 | 2-furyl | Ph | Ph | 73 (10) | 99 |
| 9c | n-propyl | Ph | Ph | 62 (11) | – |
| 10d | Ph | 4-Br-C6H4 | 4-Cl-C6H4 | 70 (12) | 99 |
| 11 | Ph | 4-Cl-C6H4 | 4-Cl-C6H4 | 67 (13) | 99 |
| 12 | Ph | 2-Cl-C6H4 | 4-Cl-C6H4 | 82 (14) | 99 |
| 13 | Ph | 4-F-C6H4 | 4-Cl-C6H4 | 70 (15) | 99 |
| 14d | Ph | 3-NO2-C6H4 | 4-Cl-C6H4 | 65 (16) | 99 |
| 15d | Ph | 4-Tol | 4-Cl-C6H4 | 60 (17) | 98 |
| 16d | Ph | 4-MeO-C6H4 | 4-Cl-C6H4 | 62 (18) | 99 |
| 17 | Ph | 2-furyl | 4-Cl-C6H4 | 67 (19) | 98 |
| 18 | Ph | 2-thienyl | Ph | 50 (20) | 98 |
| 19d | Ph | 4-pyridyl | Ph | 78 (21) | 99 |
| 20e | Ph | Ph | 4-Br-C6H4 | 81 (22) | 99 |
| 21d | Ph | Ph | 2-furyl | 75 (23) | 99 |
Isolated yield.
Enantiomeric excess determined by HPLC. Dr determined by 1H NMR (500 MHz) of the unpurified reaction.
15 mol % azolium salt A was used.
50 mol % Ti(Oi-Pr)4.
30 mol % Ti(Oi-Pr)4.
The scope of the chalcone substrate was also examined (Table 2, entries 10–21). Aromatic and heteroaromatic substituents at the β-position of the chalcone are well accommodated, although electron-donating substituents were less reactive and required an increase of Ti(Oi-Pr)4 (entries 15–16). Reactions involving substrates bearing heteroatoms that could potentially interact with the Lewis acid were also conducted with 50 mol% of Ti(Oi-Pr)4 to ensure good diastereoselectivity levels (entries 14, 19, 21). Importantly, the trans diastereoisomer was not detected in any case (as measured by 1H NMR).
Our current understanding of the reaction pathway is shown in Scheme 1. Initial coordination of the α,β-unsaturated aldehyde to the titanium Lewis acid promotes the formation of the extended Breslow intermediate I. The subsequent coordination of the chalcone to this carbene-aldehyde-titanium(IV) intermediate a) activates the enone towards conjugate addition, and b) situates the homoenolate in close proximity to the β-carbon of the enone as shown in II. The ensuing conjugate addition involving chalcone reacting in the s-cis conformation12 would then generate bisenolate III. The protonation and tautomerization of this chalcone carbonyl titanium enolate and a resulting intramolecular aldol reaction affords intermediate IV.13 A final acylation and decarboxylation cascade affords the cyclopentene. We postulate that 2-propanol might accelerate the acylation step from III to regenerate the carbene catalyst and Ti(Oi-Pr)4 by facilitating the disassociation of the tertiary alkoxide.
Scheme 1.

Proposed Catalytic Pathway
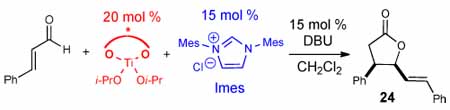
The efficiency and unique selectivity pattern observed with this new metal-NHC cooperative catalytic reaction prompted us to evaluate the possibility of combining chiral Lewis acid catalysis with achiral carbenes. An additional source of chirality in NHC-catalyzed reactions would be highly valuable and offers great potential for future methodology development. We selected the dimerization of enals via NHC-catalyzed homoenolates as a model reaction to probe the feasibility of this new strategy. While the NHC-catalyzed synthesis of γ-butyrolactones has been the object of many publications,14 a general enantioselective version of this reaction has not yet emerged.15
With the successful integration of Ti(IV) and NHC catalysis in the synthesis of cyclopentenes, we commenced the investigation of chiral titanium alkoxides as Lewis acids with NHC catalysis as a way to access optically active γ-butyrolactones. Starting with 20 mol % (S)-Ti-BINOL and 15 mol % Imes-derived carbene led to a low conversion of the starting material (Table 3, entry 1). However, with (R,R)-Ti-TADDOL16 as the Lewis acid and 15 mol % Imes only the cis diastereoisomer was detected with 75% yield and 38% ee (entries 2). Importantly, the observed enantiomeric excess originates from the influence of the chiral Lewis acid and implicates Ti(IV) catalysis is involved in the key bond forming event. By reducing the temperature, the cis γ-butyrolactone could be obtained in 60% yield and 60% ee, the highest levels of diastereo- and enantioselectivity achieved to date for this transformation (entry 3).
Table 3.
Catalysis with chiral Lewis acid and achiral azolium
 | |||||
|---|---|---|---|---|---|
| entry | Temp. (°C) | % yielda | cis:transb | %eec | |
| 1 | (S)-BINOL | 23 | 10 | 4:1 | nd |
| 2 | (R,R)-TADDOL | 23 | 75 | >20:1 | 38 |
| 3 | (R,R)-TADDOLd | −20 | 60 | >20:1 | 60 |
Isolated yield.
Diastereomeric ratio determined by 1H NMR (500 MHz) of the unpurified reaction.
Enantiomeric excess determined by HPLC.
20 mol% i-PrOH was used as an additive.
In summary, a highly enantioselective and diastereoselective cyclopentene forming reaction that employs catalytic amounts of Lewis acids and N-heterocyclic carbenes has been developed. This cooperative catalysis process integrating titanium(IV) and triazolium-derived NHCs allows the synthesis of cis cyclopentenes with a broader substrate scope and higher stereoselectivity than previously published methods. In addition, we have discovered that cooperative catalysis with chiral titanium Lewis acids in the presence of achiral NHCs can promote the formation of cis γ-butyrolactones with promising levels of enantioselectivity. These studies in general lay the foundation for the discovery of new productive combinations of Lewis acids and carbenes. While reversible binding between the Lewis acids and N-heterocyclic carbenes used in these studies is possible, this aspect is not detrimental and each catalyst operates in concert successfully. Additionally, the use of two catalysts allows for either or both promoter to be optically active, thereby providing new opportunities to control the absolute stereochemistry in these processes. This new cooperative carbene catalysis is a powerful strategy for asymmetric synthesis and studies pursuing its development are ongoing.
Supplementary Material
Acknowledgements
Financial support for this work has been provided by the NIGMS (GM73072), the Sloan Foundation, Amgen, AstraZeneca, and GlaxoSmithKline. We thank Wacker Chemical and FMCLithium for reagent support and John M. Roberts (NU) for assistance with X-ray crystallography. FQRNT (Fonds Québécois de la Recherche sur la Nature et les Technologies) is also gratefully acknowledged for a postdoctoral fellowship (B.C.-D).
Footnotes
Supporting Information Available: Experimental procedures and spectral data for new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent selected examples, see: Yamagiwa N, Qin H, Matsunaga S, Shibasaki M. J. Amd. Chem. Soc. 2005;127:13419–13427. doi: 10.1021/ja054066b. Hu W, Xu X, Zhou J, Liu W-J, Huang H, Hu J, Yiang L, Gong L-Z. J. Am. Chem. Soc. 2008;130:7782–7783. doi: 10.1021/ja801755z. Li C, Villa-Marcos B, Xiao J. J. Am. Chem. Soc. 2009;131:6967–6969. doi: 10.1021/ja9021683. Yang T, Ferrali A, Sladojevich F, Campbell L, Dixon DJ. J. Am. Chem. Soc. 2009;131:9140–9141. doi: 10.1021/ja9004859.
- 2.For reviews, see: Ma J-A, Cahard D. Angew. Chem., Int. Ed. 2004;43:4566–4583. doi: 10.1002/anie.200300635. Kanai M, Kato N, Ichikawa E, Shibasaki M. Synlett. 2005:1491–1508. Paull DH, Abraham CJ, Scerba MT, Alden-Danforth E, Lectka T. Acc. Chem. Res. 2008;41:655–663. doi: 10.1021/ar700261a.
- 3.Denmark SE, Beutner GL. Angew. Chem. Int. Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]
- 4.(a) Zeitler K. Angew. Chem., Int. Ed. 2005;44:7506–7510. doi: 10.1002/anie.200502617. [DOI] [PubMed] [Google Scholar]; (b) Marion N, Diez-Gonzalez S, Nolan SP. Angew. Chem. Int. Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; (c) Enders D, Niemeier O, Henseler A. Chem. Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]
- 5.(a) Regitz M. Angew. Chem., Int. Ed. Engl. 1996;35:725–728. [Google Scholar]; (b) Arduengo AJ. Acc. Chem. Res. 1999;32:913–921. [Google Scholar]; (c) Bourissou D, Guerret O, Gabbai FP, Bertrand G. Chem. Rev. 2000;100:39–92. doi: 10.1021/cr940472u. [DOI] [PubMed] [Google Scholar]; (d) Herrman WA. Angew. Chem., Int. Ed. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 6.(a) Burgess K, Perry MC. Tetrahedron: Asymmetry. 2003;14:951–961. [Google Scholar]; (b) Nolan SP, editor. N-Heterocyclic Carbenes in Synthesis. Weinheim, Germany: Wiley-VCH; 2006. [Google Scholar]; (c) Glorius F. Top. Organomet. Chem. 2007;21:1–20. [Google Scholar]; (d) Hahn FE, Jahnke MC. Angew. Chem., Int. Ed. 2008;47:3122–3172. doi: 10.1002/anie.200703883. [DOI] [PubMed] [Google Scholar]
- 7.(a) Chan A, Scheidt KA. Org. Lett. 2005;7:905–908. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]; (b) Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew. Chem., Int. Ed. 2007;46:3107–3110. doi: 10.1002/anie.200605235. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chan A, Scheidt KA. J. Am. Chem. Soc. 2007;129:5334–5335. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wadamoto M, Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2007;129:10098–10099. doi: 10.1021/ja073987e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chan A, Scheidt KA. J. Am. Chem. Soc. 2008;130:2740–2741. doi: 10.1021/ja711130p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Phillips EM, Reynolds TE, Scheidt KA. J. Am. Chem. Soc. 2008;130:2416–2417. doi: 10.1021/ja710521m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Maki BE, Chan A, Scheidt KA. Synthesis. 2008:1306–1315. doi: 10.1055/s-2008-1072516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burstein C, Glorius F. Angew. Chem., Int. Ed. 2004;43:6205–6208. doi: 10.1002/anie.200461572. Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b. Nair V, Babu BP, Vellalath S, Varghese V, Raveendran AE, Suresh E. Org. Lett. 2009;11:2507–2510. doi: 10.1021/ol900571x. For a review of NHC-generated homoenolate methodology, see: Nair V, Vellalath S, Babu BP. Chem. Soc. Rev. 2008;37:2691–2698. doi: 10.1039/b719083m.
- 9.Nair V, Vellalath S, Poonoth S, Suresh E. J. Am. Chem. Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- 10.Chiang P-C, Kaeobamrung J, Bode JW. J. Am. Chem. Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]
- 11.The absolute and relative stereochemistry was determined by X-ray crystallography and assigned by analogy (see Supporting Information). The use of (E)-ethyl 4-oxo-2-butenoate affords only low conversion with our reaction (<20%).
- 12.Patel D, Liddle ST, Mungur SA, Rodden M, Blake AJ, Arnold PL. J. Chem. Soc., Chem. Commun. 2006:1124–1126. doi: 10.1039/b514406j. [DOI] [PubMed] [Google Scholar]
- 13.An alternative mechanistic rationale invoking a benzoin/oxy-Cope rearrangement sequence has been proposed for cyclopentene formation using only NHC catalysis (not with NHC/Lewis acid cooperative catalysis), see reference 10. Our data support a more direct homoenolate addition depicted in Scheme 1.
- 14.(a) Sohn SS, Rosen EL, Bode JW. J. Am. Chem. Soc. 2004;126:14370–14371. doi: 10.1021/ja044714b. [DOI] [PubMed] [Google Scholar]; (b) Nair V, Vellalath S, Poonoth M, Mohan R, Suresh E. Org. Lett. 2006;8:507–509. doi: 10.1021/ol052926n. [DOI] [PubMed] [Google Scholar]; (c) Nair V, Vellalath S, Poonoth M, Suresh E, Viji S. Synthesis. 2007:3195–3200. [Google Scholar]; (d) Hirano K, Piel I, Glorius F. Adv. Synth. Catal. 2008;350:984–988. [Google Scholar]
- 15.For a recent report combining cinnamaldehyde with α-keto esters in the presence of a chiral carbene, see Y, Zhao Z-A, He H, You S-L. Adv. Synth. Catal. 2008;350:1885–1890. For an NHC-catalyzed γ-butyrolactone formation with 2,2,2-trifluoroacetophenone in up to 66% ee (23% yield), see, Matsuoka Y, Ishida Y, Saigo K. Tetrahedron Lett. 2008;49:2985–2989.
- 16.2,2-Dimethyl-α,α,α',α'-tetraphenyl-1,3-dioxolane-4,5-dimethanolato-titanium diisopropoxide. For a review on TADDOL, see: Seebach D, Beck AK, Heckel A. Angew. Chem. Int. Ed. 2001;40:92–158.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.