

Abstract
Oxygen-atom transfer reactions of metalloporphyrin species play an important role in biochemical and synthetic oxidation reactions. An emerging theme in this chemistry is that spin-state changes can play important roles, and a ‘two-state’ reactivity model has been extensively applied especially in iron-porphyrin systems. Herein we explore the gas phase oxygen-atom transfer chemistry of meso-tetrakis(pentafluorophenyl)porphyrin (TPFPP) chromium complexes, as well as some other tetradentate macrocyclic ligands. Electrospray ionization in concert with Fourier transform ion cyclotron resonance (FT-ICR) spectrometry has been used to characterize and observe reactivity of the ionic species [(TPFPP)CrIII]+ (1) and [(TPFPP)CrVO]+ (2). These are an attractive system to examine the effects of spin state change on oxygen atom transfer because the d1 CrV species are doublets while the CrIII complexes have quartet ground states with high-lying doublet excited states. In the gas phase, [(TPFPP)CrIII]+ forms adducts with a variety of neutral donors but O-atom transfer is only observed for NO2. Pyridine N-oxide adducts of 1 do yield 2 upon collision induced dissociation (CID), but the ethylene oxide, DMSO, and TEMPO analogs do not. [(TPFPP)CrVO]+ is shown by its reactivity and by CID experiments to be a terminal metal-oxo with a single vacant coordination site. It also displays limited reaction chemistry, being deoxygenated only by the very potent reductant P(OMe)3. In general, [(TPFPP)CrVO]+ species are much less reactive than the Fe and Mn analogs. Thermochemical analysis of the reactions points towards the involvement of spin issues in the lower observed reactivity of the chromium complexes.
Introduction
The role of spin states in reactions of transition metal complexes is being increasingly discussed, specifically how reactions are affected when a change in spin state occurs between reactants and products.1 This fundamental question appears to be particularly important in the oxidative chemistry of metalloporphyrin-containing enzymes and complexes. Heme-containing monooxygenase enzymes, such as cytochromes P450, nitric oxide synthases and peroxidases, mediate myriad transformations of biological importance.2–4 In particular, the reaction chemistry of the cytochromes P450 has been intensively studied, both in enzymatic systems and using small molecule models, because this enzyme superfamily is primarily responsible for processing xenobiotic substances in vivo. In addition, the enzymes and models accomplish very interesting reaction chemistry, including the hydroxylation of alkanes and epoxidation of olefins. The mechanisms of these reactions have therefore been of much interest, and have been the subject of some controversy.5
For all these reasons, there have been many computational studies of metalloporphyrin oxidation reactions and their mechanisms. Shaik, Schwarz and their coworkers have proposed that multiple spin states play an important role in these reactions, with many of them involving what they have called “two-state reactivity (TSR).”6,7 They state, “A thermal reaction which involves spin crossover along the reaction coordinate from reactants to products needs to be described in terms of two-state reactivity if product formation arises from an interplay of spin inversion and the respective barrier heights on both spin surfaces.”7 Perhaps the clearest experimental evidence for the TSR model comes from the gas phase reactions of FeO+ or OCrO+ studied by Schwarz and coworkers.8 The interpretation of the experiments and computations for the iron-porphyrin species in oxidation reactions is however complicated by the multiplicity of accessible spin states. The ‘Compound I’ intermediate, two oxidation levels above FeIII, is most simply described as an FeIV-oxo (ferryl) species coupled to a porphyrin radical cation and can have closely spaced doublet, quartet, and sextet states.9 The FeIII state can similarly have accessible doublet, quartet and sextet states.6 Experimental and computational studies also indicate that spin-state effects play important roles in the reactions of oxomanganese porphyrin complexes,10 other metal-oxo complexes,11 and in oxygen atom abstractions by M(OCtBu3)3 [M = V, Nb(PMe3), and Ta].12
The chromium(V)-oxo/chromium(III) redox couple could provide a more direct test of the importance of spin state changes on oxygen atom transfer reactions because there is one predominant spin state present at each oxidation level in such a system (eq 1). CrV has a d1 configuration and therefore is an obligate doublet state.13 A number of CrV oxo species have
| (1) |
been prepared, including with porphyrin, salen, and other ligands.14 Some of these compounds undergo oxygen atom transfer reactions such as epoxidation, as found by Groves,15 Kochi,16 and others.17 CrIII coordination complexes essentially always have quartet ground states (except for organometallic species with μ-acid ligands).18,19 In octahedral symmetry, the ground state for CrIII is 4A2g and the lowest lying excited state is 2E.20 Since both states have a t2g3 configuration, the energy of the 2E state is much less dependent in the nature of the ligands than most ligand-field excited states, and is at ~13,000 cm−1 (37 kcal mol−1) for Cr(NH3)63+.20 When X and XO are closed shell (singlet) species, oxygen atom transfer from doublet ground state CrVO to a quartet ground state CrIII is thus formally spin forbidden. Herein we report our investigations of the gas phase reactivity of ligated chromium(III) and chromium(V)oxo complexes with a variety of oxygen atom donors and acceptors, using electrospray ionization (ESI) coupled to mass spectrometry (MS).
Earlier studies based on Fourier Transform ion cyclotron resonance (FT-ICR) mass spectrometry combined with ESI have provided valuable information on functional models of the Compound I intermediates of monooxygenase enzymes, namely the [(TPFPP)•+FeIVO]+ and [(TPFPP)MnVO]+ (TPFPP = meso-tetrakis(pentafluorophenyl)porphyrinato dianion) complexes.21 These species have been obtained as naked ions and their reactivity has been investigated in an environment lacking any proximal or distal ligand, solvent or counterion. In this way it could be ascertained that the [(TPFPP)MnVO]+ complex is responsible for catalytic activity while a strong trans axial ligand (an additional oxygen) can switch off the reactivity, an outcome consistent with barriers due to TSR effects.21c By gas phase synthesis using ozone as oxidant, the naked core of Compound I, [(PPIX)•+FeIVO]+ (PPIX = protoporphyrin IX dianion) has been obtained for the first time.22 The dilute gas phase has ensured that these species have a lifetime long enough to reveal both structural details and elementary steps of the catalytic activity. High valent manganese(V)-oxo complexes of non-heme ligands have also been examined in the gas phase by Plattner, showing that ESI-tandem MS techniques is a convenient approach for the study of the coordination chemistry of highly reactive solution-phase species.23 On these premises an attempt to answer the spin-forbidden question for oxochromium complexes was undertaken.
Experimental
Materials
5,10,15,20-tetrakis(pentafluorophenyl)porphyrinato chromium(III) chloride, (TPFPP)CrIIICl (1-Cl) was purchased from Frontier Scientific (Logan, UT, USA). 5,10,15,20-meso-tetraphenylporphyrin (TPP, Strem) was purified by the method of Adler.24 (TPP)CrIIICl was prepared from TPP and anhydrous CrIICl2 (Strem) according to the literature.25 3,3′-5,5′-tetra-tert-butyl-salen, and salphen were prepared by condensation of the appropriate di-amine and benzaldehyde (both from Sigma), and recrystallized from ethanol (J.T. Baker).26 (salen)CrIIICl complexes were synthesized from CrIICl2 according to known procedures.27 The corresponding triflate salts were prepared by addition of stoichiometric AgOTf (Strem) and filtration. Iodosylbenzene (C6H5IO) was prepared according to the literature procedure28 and stored at −20°C. Pyridine-N-oxide (pyO) was synthesized by reaction of pyridine with a stoichiometric amount of m-chloroperbenzoic acid (m-CPBA) in MeOH at room temperature. All the solvents were analytical grade. H218O (95%) and all other chemicals were research grade products obtained from commercial sources (Sigma-Aldrich; Matheson Gas Products Inc.) and used as received.
Instrumental
Experiments were performed either with a Bruker BioApex 4.7 T Fourier Transform ion cyclotron resonance (FT-ICR) mass spectrometer, or with a Triple Quadrupole Linear Ion Trap mass spectrometer (Applied Biosystems 2000 Q TRAP). The first instrument is equipped with an Apollo I electrospray ionization (ESI) source. Ions are led into a cylindrical “infinity” cell where neutrals may be admitted by way of either pulsed valves or leak valves. The second device consists of an ESI source combined with a tandem mass spectrometer (QHQ, quadrupole-hexapole-quadrupole configuration).
Generation of oxochromium(TPFPP)+ [(TPFPP)CrV(O)]+ ions
Two routes were followed in order to generate the [(TPFPP)CrV(O)]+ ions of interest. In the first one, a 10 μM solution of 1-Cl in methanol was submitted to ESI, and the so formed [(TPFPP)CrIII]+ ions (1) were exposed to a stream of NO2 in the ionic path driving the ions into the FT-ICR cell. This procedure leads to incorporation of an O-atom into 1, presumably resulting in formation of [(TPFPP)CrV(O)]+ (2). In the second method, iodosylbenzene (C6H5IO, 0.044 mg, 2 × 10−4 mol) was added to 1-Cl (10 μM) in methanol (2 mL) at room temperature, yielding a red colored solution, stable for several hours at room temperature. When submitted to ESI, this solution delivered the same species described as [(TPFPP)CrV(O)]+ (2). In both cases, the mass spectra showed two prominent species, both characterized by the same characteristic isotopic pattern. The first one is assigned to ion 1, [(TPFPP)CrIII]+, centered at m/z = 1024, while the second one is centered at m/z = 1040, consistent with ion 2 [(TPFPP)CrV(O)]+.
Reactions of [(TPFPP)CrV(O)]+
The solutions were continuously sprayed at a 2 μL min−1 flow rate controlled by a syringe pump. A countercurrent flow of heated dry gas (nitrogen at 120 °C) was used to desolvate the ions. After an accumulation interval of 0.6 s in a rf-only hexapole, the ion population was pulsed into the ICR cell placed within a 4.7 T superconducting magnet. In the cell, maintained at room temperature (ca. 300 K), the neutral reagent was leaked at a constant pressure in the range of 0.5–10 × 10−8 mbar. The pressure readings, obtained from a cold-cathode ionization gauge (IKR Pfeiffer Balzers S.pA., Milan, Italy), were calibrated by using the rate constant k = 11.7 × 10−10 cm3 molecule−1 s−1 for the reference reaction CH4 +• + CH4 → CH5+ + CH3• and weighted by using individual response factors.29 The ion of interest was isolated by ejection routines as commonly performed in FT-ICR mass spectrometry and allowed to react with the neutral leaked by a needle valve. To avoid unintended off-resonance excitation of the reactant ion, the whole isotopic cluster corresponding to ion 1 or 2, as desired, was isolated and allowed to react with the selected neutral. Pseudo-first-order rate constants were obtained from the semilogarithmic decrease of the reactant ion abundance over time at each selected pressure and were divided by the concentration of the neutral species to attain second-order rate constants (kexp). The rate constants were also expressed as percentages (efficiencies) of the collision rate constant (kcoll) calculated by the parameterized trajectory theory.30 Whereas the reproducibility of kexp, reported as the average of usually three experiments run at different neutral pressures, was good, the estimated error in their absolute values (±30%) is mainly due to uncertainty in pressure measurements.
Collision-induced dissociation (CID) experiments
For CID performed in FT-ICR, the mass-selected reagent ions were accelerated in the presence of argon gas pulsed into the cell (peak pressure ca 4 × 10−6 mbar) for 1 s. In ESI-MS/MS effected in the Q TRAP, the ions of interest were isolated using Q1, accelerated at variable collision energy (CE) using N2 as a collision gas (normally at 2 × 10−4 mbar) in the hexapole H and detected together with their ionic fragments in Q2. The energy dependence of the dissociation process has been analyzed yielding the phenomenological appearance energies (Eapp) for the fragmentation processes under study.
Results
We have primarily focused on the tetrakis(pentafluorophenyl)porphyrin (TPFPP) complexes of chromium because this ligand has proven very amenable to ESI-FT-ICR studies with the related iron21a,b and manganese21c complexes. Studies with other porphyrin and related tetradentate ligands are described after the TPFPP results.
I. Generation of [(TPFPP)CrV(O)]+
Treatment of chromium(III)-porphyrin chloride complexes (P)CrCl with oxygen atom donors are reported to give CrIV-oxo and/or CrV–oxo compounds, depending on the reaction conditions (in the presence of limited oxidant, CrV and CrIII comproportionate to CrIV).14,31 With the TPFPP ligand, there has been a brief comment about the formation of (TPFPP)CrIV(O) from (TPFPP)CrII and O2, but no mention of the CrV-oxo complex.31 We find that treatment of (TPFPP)CrIIICl (abbreviated 1-Cl) with iodosylbenzene (C6H5IO) in methanol solution gives substantial amounts of [(TPFPP)CrV(O)]+ (2), as indicated by electrospray ionization (ESI) mass spectrometry of these mixtures (Scheme 1). The same product is formed by the gas-phase reaction of [(TPFPP)CrIII]+ (1) and NO2 in the ICR (Scheme 1). Other reactions of 1 are described below.
Scheme 1.
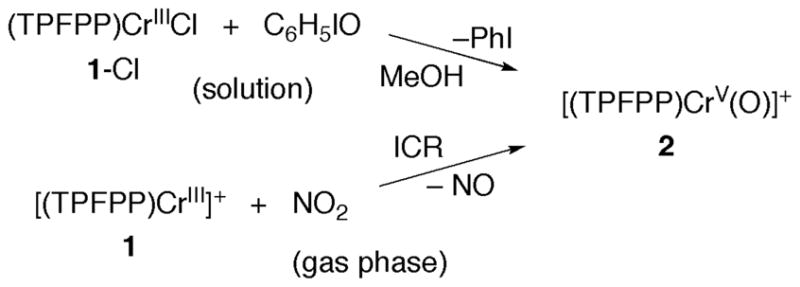
Generation of (TPFPP)CrV(O)+ (2).
Figure 1 shows a high-resolution ESI-FT-ICR mass spectrum of 2 generated in solution from PhIO + [(TPFPP)CrIII]+ (1), showing ions at m/z = 1039.9806 (calculated value 1039.9778). Complex 1 is also seen in such mass spectra, at m/z = 1023.9857 (calculated value 1023.9829). If no oxidizable species are added to the solution, 2 is remarkably inert towards side reactions, as has been found for iron-oxo cations containing porphyrin ligands with electron-withdrawing groups.21b When the oxidation of 1-Cl with iodosylbenzene is effected in the presence of excess H218O (95%-18O content) the ion at m/z = 1040 is shifted to m/z = 1042 (2-18O). This indicates that compound 2 contains an added oxygen atom, likely a formal chromium(V)-oxo complex (Figure 1), as discussed in further detail below.
Figure 1.

ESI (+) FT-ICR mass spectrum of a methanol solution of (TPFPP)CrIIICl (1-Cl, 10 μM) and a ten-fold excess of iodosylbenzene, showing [(TPFPP)CrIII]+ (1) and [(TPFPP)CrIII(O)]+ (2). Inset: enlargement of the isotopic cluster at m/z 1040.
II. Gas-Phase Reactions of [(TPFPP)CrV(O)]+
The nature and reactivity of [(TPFPP)CrV(O)]+ (2), has been assayed by FT-ICR mass spectrometry. In all cases, there is no difference between 2 formed either in solution with PhIO or in the ICR with NO2. When mass-selected and exposed in the ICR cell to a stationary concentration of potential reducing agents (L), 2 proved to be quite unreactive. No change was observed on exposure to NO, NO2, Me2S, β-pinene, pyridine or thioanisole. This indicates that reactions with these substrates are disfavored either by a positive free energy change,32 or by rate constants less than kexp < 10−13 cm3 molecule−1 s−1. Reactions do occur with γ-picoline (p-CH3C5H4N) and trimethylphosphite (P(OMe)3) to give addition products [(TPFPP)CrOL)]+ (2-L) and, for P(OMe)3, the bis-addition product [(TPFPP)CrOL2]+ (2-L2). These adduct ions likely owe their stabilization to the dissipation of any excess energy released in the association process by radiative and/or collisional cooling.33 The bimolecular rate constants (kexp) and the relative efficiencies (Φ) are summarized in Table 1. Note that the abbreviations used in this paper indicate how the ions were made, in this case “2-L” and “2-L2” indicating that the complexes were made from 2 and L. The chemical structure of these ions is discussed below.
Table 1.
Gas phase reactivity of [(TPFPP)CrV(O)]+ ions (2)a with substrates (L) in the gas phase.
| L | Productb | kexpc | Φ (efficiency, %)d |
|---|---|---|---|
| NO | NR | – | – |
| NO2 | NR | – | – |
| Me2S | NR | – | – |
| β-pinene | NR | – | – |
| pyridine | NR | – | – |
| γ-picoline | [(TPFPP)Cr(O)(γ-picoline)]+ | 1.2 | 8.7 |
| P(OMe3) | [(TPFPP)Cr(O)P(OMe)3]+e | 3.9 | 35 |
[(TPFPP)CrV(O)]+ ions were produced either in solution, by treatment of 1-Cl with C6H5IO, or in the gas phase by reaction of 1 with NO2.
NR = no observed reaction.
Second-order rate constants in units of 10−10 cm3 molecule−1 s−1. The estimated error is ± 30%, while the internal consistency of the data is within ± 10%.
Reaction efficiency, Φ = kexp/kcoll × 100. Collision rate constants (kcoll) evaluated with the parameterized trajectory theory.30
In a consecutive second addition step the [TPFPP)Cr(O)(P(OMe)3)2]+ ion is formed.
When assayed by low-energy collision-induced dissociation (CID), the primary adduct [(TPFPP)Cr(O)(γ-picoline)]+ yields ion 2 and γ-picoline as dissociation products. This suggests that γ-picoline has simply bound to 2 as a ligand to chromium, namely that this material is a picoline-ligated CrV-oxo complex, 2-γ-picoline (eq 2). In contrast, the CID spectra of [(TPFPP)CrOP(OMe)3]+ (2-P(OMe)3, formed as shown in eq 3) display the release of 1 by
| (2) |
| (3) |
concomitant loss of OP(OMe)3 (Figure 2). The CID spectra show no significant fragmentation of the phosphite ligand in 2-P(OMe)3, irrespective of the origin of 2 (either from the solution or the gas-phase synthesis). These products imply a net transfer of oxygen from 2 to P(OMe)3. The same fragment ion 1 is observed when the CID experiment is performed on labeled [(TPFPP)Cr18OP(OMe)3]+, from 2-18O + P(OMe)3, indicating loss of 18OP(OMe)3. These results support a CrIII–trimethylphosphate coordination structure for the ion, [(TPFPP)CrIII(OP(OMe)3)]+. The observed reactivity with P(OMe)3 also shows that 2 is a high-valent chromium oxo complex. Alternative structures that could be conceived for the elemental composition of the sampled ion 2, for example isomers bearing an O-atom on the porphyrin ligand, would not account for the observed net O-transfer to P(OMe)3.
Figure 2.

CID spectrum of mass-selected [(TPFPP)CrIII(OP(OMe)3)]+ (m/z = 1164) prepared in a FT-ICR mass spectrometer by ion-molecule reactions of 2 (a) and 2-18O (b) with P(OMe)3 and by electrospray ionization of a 1-Cl + OP(OMe)3 mixture in acetonitrile (c).
Compound 2-P(OMe)3 also undergoes a subsequent addition process to give [(TPFPP)CrIII{OP(OMe)3}{P(OMe)3}]+. This is evidence that the complex has a vacant sixth coordination site. In the presence of Me2S, 2-P(OMe)3 undergoes a second addition step, yielding what must be a six-coordinate species with OP(OMe)3 and Me2S ligands, [(TPFPP)CrIII{OP(OMe)3}(Me2S)]+ (eq 4).
| (4) |
The reactivity of 2-P(OMe)3 has been compared with that of the isobaric species 1-OP(OMe)3 generated by electrospray of in situ mixtures of 1-Cl + OP(OMe)3. Interestingly, this cation 1-OP(OMe)3 is also able to undergo addition of one molecule of Me2S with a reaction efficiency similar to that of reaction 4. Further analogy is found when comparing the CID behavior of the complex at m/z 1226 obtained by the gas phase addition of Me2S to either complex 2-P(OMe)3 or 1-OP(OMe)3 formed in solution. In both cases, CID of these mixed adducts release 1-Me2S by loss of neutral OP(OMe)3. This is further indication that the m/z 1226 ion is best described as a [(TPFPP)CrIII]+ ion bound to OP(OMe)3 and Me2S axial ligands.
Likewise, when 2 is exposed to a gaseous mixture of P(OMe)3 and OP(OMe)3, only the primary addition of the former ligand exhibits a second addition step, whereas OP(OMe)3 displayed the addition of a just one molecule to ion 2, to form 2-OP(OMe)3. This result is inconsistent with the presence of distinct oxo and P(OMe)3 axial ligands within 2-P(OMe)3. Rather, it implies that an O-coupling occurs upon reaction of 2 with P(OMe)3, such that a sixth coordination site within 2-P(OMe)3 is still vacant and may further coordinate a second ligand molecule. However, when the neutral ligand is not a good oxygen atom acceptor, when L = OP(OMe)3 or γ-picoline, a six-coordinate addition complex is formed with 2 in which the oxo group and L are distinct axial ligands. Consistently, these complexes do not display any tendency to give a second addition step.
As a further comparison, separately prepared 2-P(OMe)3 and 1-OP(OMe)3 were each reacted with a 70:30 mixture of P(OMe)3 and OP(OMe)3. Once again, 2-P(OMe)3 and 1-OP(OMe)3 display the same reactivity. Reversible addition reactions are observed, which attain the same equilibrium when starting from either reactant ion (Figure 3). One may thus infer that a sixth coordination site with indistinguishable binding properties is present in both 2-P(OMe)3 and 1-OP(OMe)3 under these experimental conditions, and therefore that 2-P(OMe)3 and 1-OP(OMe)3 are actually the same species.
Figure 3.

Time dependence of relative ion abundances when either 2-P(OMe)3 or 1-OP(OMe)3 ions are allowed to react with a 70:30 mixture of P(OMe)3, and OP(OMe)3 at 7 × 10–8 mbar in the FT-ICR cell. The reactant ion is seen to undergo addition of either one P(OMe)3 or one OP(OMe)3 molecule.
III. Reactions of [(TPFPP)CrIII]+ (1) with Oxygen Atom Donors and CID experiments
[(TPFPP)CrIII]+ (1) reacts with a variety of small molecules to form addition products [(TPFPP)CrIII(L)]+ and, in some cases, [(TPFPP)CrIII(L)2]+, as summarized in Table 2. Some of these ions are formed by electrospraying an equimolar solution (5 μM) of 1-Cl and L in MeOH, while others have been formed in the ICR by gas-phase ion-molecule reactions. Many of the ligands are potential oxygen atom donors, such as ethylene oxide which can be deoxygenated to ethylene, but in only two cases was the oxo complex 2 formed. These are the two reactions described above, the solution reaction of 1-Cl + iodosylbenzene and the gas-phase reaction of 1 + NO2 (Scheme 1). The latter reaction is characterized by a notable efficiency of 22 % (note d in Table 2). The potential oxygen atom donors XO are given in the Table in increasing order of X–O bond dissociation enthalpy (BDE), as discussed below.
Table 2.
Products from [(TPFPP)CrIII]+ (1) + ligands and their behavior upon CID.
| synthesisa | L | Product | CID reaction | Eappb |
|---|---|---|---|---|
| Sc | PhIO | 2 | - | - |
| S | γ-picoline-O | +L | 1-L → 2 + γ-picoline (90%) | 0.48 |
| 1-L → 1 + γ-picoline-O (10%) | 0.58 | |||
| S,G | γ-picoline | +L | 1-L → 1 + γ-picoline | 0.55 |
| S | py-O | +L | 1-L → 2 + py (75%) | 0.41 |
| 1-L → 1 + py-O (25%) | 0.38 | |||
| S,G | py | +L | 1-L → 1 + py | 0.56 |
| G | NO2 | 2d | –c | – |
| G | C2H4O | +L, +2L | 1-L → 1 + L | –e |
| 1-L2 → 1-L + L | –e | |||
| S | DMSO | +L | 1-L → 1 + L | 0.43 |
| G | Me2S | +L, +2L | 1-L → 1 + L | –e |
| 1-L2 → 1-L + L | –e | |||
| S,G | TEMPO | +L | 1-L → TEMPO+ + [(TPFPP)CrII] | –e |
| S | OPPh3 | +L, + 2L | 1-L → 1 + L | 0.56 |
| S,G | OP(OMe)3 | +L, + 2L | 1-L → 1 + L | 0.58 |
| 1-L2 → 1-L + L | ||||
| S,G | P(OMe)3 | + L | 1-L → 1 + L | 0.31 |
S = prepared in solution from 5 μM 1-Cl + 5 μM LO in MeOH, followed by electrospray; G = formed in the gas phase from 1 + L.
Phenomenological appearance energies in kcal mol−1.
The so-formed ion 2 is stable towards CID in the energy range explored.
Efficiency 22%.
Not available.
To understand the nature of the ligand adducts, a number of them were analyzed by collision induced dissociation, CID. Upon CID the adducts of ethylene oxide, DMSO (Me2SO), OPPh3, and OP(OMe)3 all dissociated to give the neutral ligand and 1. A representative CID spectrum of the mass-selected ion 1-OP(OMe)3, showing the loss of intact trimethylphosphate, is presented in Figure 2(c). In contrast, pyridine N-oxide and γ-picoline N-oxide undergo CID to yield the oxo complex 2 and (presumably) the deoxygenated ligand. The phenomenological appearance energies for these CID processes are 0.41 and 0.48 eV, respectively. The adduct of 1 with TEMPO (2,2′-6,6′-tetramethylpiperidin-1-oxyl), dissociates into TEMPO+ and, presumably, neutral (TPFPP)CrII, as the only product observed.
IV. Reactivity of other [(L4)CrV(O)]+ and [(L4)CrIII]+ cations
Several chromium complexes with tetradentate macrocyclic ligands L4 other than TPFPP have been tested for potential oxygen transfer reactivity using ESI-FT-ICR. These have involved L4 = 5,10,15,20-meso-tetraphenyl-porphyrin (TPP), 3,3′-5,5′-tetra-tert-butyl-salen (salen), and N,N′-phenylenebis(salicylideneimine) (salphen); see Table S1 in the Supporting Information. As in the TPFPP case, addition of ligands to [(L4)CrIII]+ results in sequential formation of the mono- and bis-adducts, without formation of the corresponding CrV-oxo complex. The one exception is the reaction of [(tBu4salen)CrIII]+ with NO2 to give [(tBu4salen)CrV(O)]+, the product of a net O-transfer. However, neither [(salphen)CrIII]+ nor [(TPP)CrIII]+ react with NO2.
The [L4Cr(O)]+ complexes (see Supporting Information) showed no reactivity with potential reductants including Me2S, P(OMe)3, γ-picoline, NO, p-Me-C6H4SMe and β-pinene. The lack of reactions with P(OMe)3 or alkenes contrasts with the reactivity of these oxo complexes in solution, such as the reported epoxidations.15,16 We have also found that, in solution, the salen complex is very reactive with phosphines, rapidly forming Cr(III) products.34 A distinct effect by donor ligands such as (OPEt3) is also reported to enhance the epoxidation rates and yields displayed by (Salen)CrV(O) complexes in solution.16a The reaction of [(tBu4salen)CrV(O)(OPPh3)]+ probed in the gas phase with a powerful reductant such as p-CH3C6H4SCH3 did not lead to any enhanced reactivity, however, and this complex remains unreactive just as [(tBu4salen)CrV(O)]+ (See Table S1).
Discussion
The gas phase reactivity of several porphyrin- and salen-ligated complexes of chromium have been explored using gas phase ICR-MS. Most of the work has focused on the tetrakis(pentafluorophenyl) porphyrin (TPFPP) complexes because of their higher reactivity. The results, particularly the reactions with P(OMe)3 and CID studies of the products, show that complex 2 is the metal-oxo cation, [(TPFPP)CrV≡O]+. For example, the addition of P(OMe)3 to 2 gives an adduct that loses OP(OMe)3 on collisional activation. This adduct is essentially identical to the complex formed from [(TPFPP)CrIII]+ (1) and OP(OMe)3; for instance, both species bind P(OMe)3 and OP(OMe)3 with the same equilibrium constants. This assignment is similar to chromium(V)-terminal oxo complexes made in solution with porphyrin and salen supporting ligands.15–17
Net oxygen atom transfers have been observed, from NO2 to 1, and from 2 to P(OMe)3. However, 2 shows no observable reactivity with NO, NO2, Me2S, β-pinene or pyridine. This chromium-oxo cation is thus much less reactive than the analogous iron cation, [(TPFPP•+)FeIVO]+, which reacts rapidly with all of these substrates.21a The difference in reactivity is consistent with the generally much higher oxidizing power of high-valent iron versus chromium compounds as observed in solution.35
I. Thermochemistry of oxygen atom transfer
The oxygen atom donors/acceptors XO/X used in this study have a wide range of homolytic bond dissociation enthalpies (BDEs, ΔH° for XO → X + O), as shown in Table 3.
Table 3.
. Homolytic X-O bond dissociation enthalpies (BDEs).a
| Reaction | ΔH° (kcal mol−1)b |
|---|---|
| PhIO → PhI + O | ~26c |
| γ-picoline-O → γ-picoline + O | 62.6 |
| pyO → py + O | 63.3 |
| NO2 → NO + O | 73.2 |
| C2H4O → C2H4 + O | 84.7 |
| Me2SO → Me2S + O | 86.8 |
| Me2NO• → Me2N• + O | 86d |
| H2NO• → H2N• + O | 91d |
| Ph3PO → Ph3P + O | 139 |
| (EtO)3PO → (EtO)3P + O | 148 |
Data from 41 unless otherwise noted.
Derived from solution data or calculated gas phase heats of formation.
Estimated from data in reference 42.
The reaction of NO2 with [(TPFPP)CrIII]+ (1) to give [(TPFPP)CrV≡O]+ (2) and (presumably) NO (eq 5) is a particularly simple reaction that provides a lower limit for the CrV≡O bond strength. This reaction proceeds with a moderately high efficiency (22% at room temperature, Table 2, footnote d) consistent with a negative value of ΔG°.32,36 Indeed, definitely exoergic reactions with ΔG° less than −10 kcal mol−1 and negligible intrinsic barriers (such as proton transfer processes) proceed with high efficiency (>70%).32 We may then consider that if ΔG° for the reaction of 1 with NO2 is < 0, the bond dissociation free energy (BDFE) should be higher than that of NO2, which has a BDFE of 64 kcal mol−1 (see Supporting Information).37 Since the entropy change for reaction (5) is likely to be small,38 we can conclude that the more commonly discussed bond dissociation enthalpy (BDE) of CrV≡O should be greater than that of NO2, BDE = 73 kcal mol−1.39 For comparison, the Cr–O BDE in the gas-phase diatomic CrO+ has been measured to be 86 ± 3 kcal mol−1.40
| (5) |
Rough upper limits to the CrV≡O bond strength can be derived from the reduction of 2 by P(OMe)3 and by the absence of O-atom transfer from TEMPO (2,2′-6,6′-tetramethylpiperidin-1-oxyl) to chromium (eq 6), with the assumption that the lack of reaction has a thermochemical rather than kinetic origin. The analyses of these reactions are more complicated than that of eq 5
 |
(6) |
because the ligand, OP(OMe)3 or TEMPO, is bound to the CrIII so that the enthalpy of binding plays a role. The Cr–OP(OMe)3 binding energy is estimated to be near 13 ± 5 kcal mol−1 based on the CID experiments at variable collision energy (Table 2). The Cr-O bond strength in 1-TEMPO is taken as equal to the Ti–O BDE in Cp2TiCl(TEMPO), 25 kcal mol−1.43 This should be an upper limit because Cr is less oxophilic than Ti and because CrIV is more oxidizing than TiIV [these compounds are formally M4+–(TEMPO−)]. In addition, the CrV sides of these reactions are likely to be entropically favorable by ca. 10 kcal mol−1 (ΔS° ~ 34 cal K−1 mol−1).44 The reactions at the top of Scheme 2, with X = OP(OMe)3 or TEMPO, should be at least 10 kcal mol−1 enthalpically unfavorable. Finally, the BDEs of OP(OMe)3 and TEMPO are taken to be the same as those of OP(OEt)3 and Me2NO•, respectively (Table 3). With these various values, Scheme 2 implies that the CrV≡O BDE must be less than ca. 143 kcal mol−1 to account for the observed reaction of 2 with P(OMe)3. Similarly, the lack of deoxygenation of TEMPO• by 1 suggests that BDE(CrV≡O) < ~101 kcal mol−1. These thermochemical considerations are consistent with the estimated BDE for CrV≡O in ion 2 to be higher than 73 kcal mol−1 based on the reaction of 1 with NO2.
Scheme 2.

Thermochemical analysis of the lack of oxygen atom transfer from 1-OP(OMe)3 or 1-(TEMPO).
II. Effect of spin on the oxygen atom transfer reactions
It is surprising that none of the XO or X reagents with BDEs between NO2 and OP(OMe)3 (73.2 and ca. 148 kcal mol−1, Table 3) are observed to interconvert 1 and 2. The loss of ethylene from 1•C2H4O should be only slightly less favorable than the loss of NO from the putative 1•NO2 intermediate in the NO2 reaction. However, not only does this process not occur, but 2 is unable to release an O atom to an activated olefin such as β-pinene. Similarly, the 1-DMSO adduct does not fragment into 2 and Me2S when activated to by CID, displaying no evidence for an O-atom transfer from sulfur to 1. Once again the reverse reaction is not observed either, because 2 appears to be unreactive towards gaseous Me2S. The pyridine N-oxides py-O and γ-picoline-O are among the strongest oxidizing agents in the set of species that were considered (Table 3). Both pyridine N-oxide complexes 1•Opy and 1•O-γ-picoline share similar Eapp values upon CID at variable energy, leading to either 2 (75–90%) or 1 (25-10%) in comparable amounts. This finding of only partial oxidation of 1 by relatively powerful oxidizing agents, stronger than NO2 which does efficiently oxidize 1 in the gas phase, suggests that kinetic effects play an important role in the partitioning of the O-atom between 1 and the pyridine nitrogen, and, more generally, in O-transfer reactions involving 1 and 2.
As noted above, the interconversion of 1 + XO with 2 + X is formally spin-forbidden with closed-shell (singlet) reagents X and XO. All but two of the X/XO reagents used here are closed-shell species, including ethylene oxide/ethylene, pyO/py, Me2SO/Me2S, and OP(OMe)3/P(OMe)3. The two exceptions are NO2 and TEMPO, which are both free radicals with an unpaired electron and are therefore doublet states. Reaction of quartet 119 with doublet NO2 can therefore give doublet 2 + doublet NO in a spin-allowed reaction, as shown in eq 7 (the orbitals underneath the reagents show only the unpaired electrons). Reaction 7 formally involves transfer of a triplet oxygen atom NO2 to CrIII. This analysis is consistent with the measured efficiency of 22% for reaction 7.
 |
(7) |
TEMPO can react with 1 in a similar spin-allowed fashion.45 Therefore the absence of O-atom transfer from TEMPO to chromium (eq 6) is likely due to the reaction being endoergic, rather than due to a spin-based spin barrier. The thermodynamic values given above support this conclusion: the Cr≡O BDE is indicated to be only a little larger than that the N–O BDE in NO2 (73 kcal mol−1), and the N–O BDE in TEMPO is estimated as being significantly stronger, close to that in Me2NO•, namely 86 kcal mol−1 (Table 3). Hence, the reaction of 1 with TEMPO is likely to be quite unfavorable based on thermodynamic considerations.
The reactivity pattern of 1 and 2 discussed above appears to be a case where the spin state change between CrIII and CrV affects the reaction chemistry. The most striking feature of these chromium complexes is their low reactivity in either reaction direction. One notable exception is the facile and spin-allowed reaction of 1 + NO2. The other exception is the reaction of 2 + P(OMe)3 to give (TPFPP)CrIII(OP(OMe)3)+. This reaction is so highly exoergic that it could proceed in a spin allowed fashion to initially form the 2E excited state of the CrIII product. The P(OMe)3 reaction is >60 kcal mol−1 downhill, using the conservative upper limit on BDE(CrV≡O) from the lack of deoxygenation of TEMPO. This is much larger than the estimate of the energy of the 2E state by analogy with [CrIII(NH3)6]3+, ~37 kcal mol−1 (as noted above). Attempts in solution to observe emission from such a potential 2E intermediate (chemi-luminescence), potentially generated thermally upon reaction of the (salen)CrV(O)+ derivatives with phosphines, have unfortunately not been successful.34 Consistent with the suggestion that the P(OMe)3 reaction could proceed via a doublet CrIII excited state, this reaction has a very similar rate constant as the analogous reaction of gas phase (TPFPP•+)FeIVO with trimethylphosphite. The iron reaction proceeds with somewhat higher efficiency and proceeds with full oxygen atom transfer to give [(TPFPP)FeIII]+, while 2 forms the CrIII adduct (TPFPP)CrIII(OP(OMe)3)+, perhaps because of the more substitution-inert character of Cr(III) complexes.
Conclusions
High-valent chromium(V)oxo complexes have been obtained from chromium(III) precursors both by an oxidation reaction in solution, and by a gas phase reaction using NO2 as an oxygen-atom donor. Among several sampled species, the [(TPFPP)CrVO]+ ion (2) from the oxidation of the [(TPFPP)CrIII]+ (1) has been the most thoroughly studied. The presence of a CrVO unit in 2 has been probed by a variety of processes in the gas phase, including O-atom transfer to potential reductants and the assessment of association equilibria. These tools allow us to establish that the complex formed in solution using PhIO and then transferred to the gas phase by ESI, and the species obtained by the gas phase reaction of 1 with NO2, are actually the same chromium(V)oxo cation [(TPFPP)CrVO]+ (2). Gas phase FT-ICR mass spectrometry has allowed us to investigate the gas phase reactivity of these cationic Cr species.
The major purpose of this study was aimed at exploring the role of spin state constraints on the O-transfer reactivity connecting [(TPFPP)CrIII]+ (1) and [(TPFPP)CrVO]+ (2). To this end, gas phase reactivity data have been collected regarding both the bimolecular kinetics for the reaction with selected gaseous neutrals, and the fragmentation patterns observed when adducts of 1 and selected ligands undergo CID at variable energy. The reactivity behavior of the 1/2 couple has been interpreted with reference to the thermodynamic parameters of the reactions, namely to the X-O bond dissociation free energies of the XO/X neutral reaction partners. The gas phase reaction of 1 with NO2 sets an approximate value for the CrV≡O BDE of 2. The efficiency of this reaction and the absence of the reverse reaction (2 + NO) indicate a CrV≡O BDE somewhat larger than the O–NO BDE. In agreement with this evaluation, no O-atom transfer is observed with TEMPO, whose N-O BDE is definitely higher. Both NO2 and TEMPO are free radicals with one unpaired spin, and therefore they can transfer an oxygen atom to 1 to give 2 in a spin allowed process.
Oxygen atom transfer reactions of the 1/2 couple with closed-shell XO/X neutrals, however, are formally spin forbidden. In these cases, the O-atom transfer reactivity is found to be abnormally low. Compound 2 does not oxidize olefins, sulfides, or pyridines, and the corresponding epoxides, sulfoxides, and pyridine N-oxides are not deoxygenated by 1. Thus there are substantial kinetic impediments to these reactions, and it seems likely that this is due to the requirement of a spin change from doublet 2 to quartet 1. Of the various closed-shell reagents explored, only trimethylphosphite reacts with 2 to give a CrIII product ([(TPFPP)CrIII(O(P(OMe)3]+ (1-OP(OMe)3), albeit with a relatively modest efficiency of 35%. P(OMe)3 has a very high affinity for an oxygen atom, with an estimated (MeO)3P–O BDE of >140 kcal mol−1, so that its reaction with 2 could proceed in a spin-allowed fashion to initial make the 2E excited state of the CrIII product. Thus all of the data are consistent with spin-forbidden reactions of 1 and 2 being inhibited while thermodynamically favorable spin-allowed reactions occur with reasonable efficiency.
As a final remark, the relative reactivity of [(TPFPP)Metal(O)]+ complexes in the gas phase may now be compared with regard to the oxygen atom transfer reaction with olefins. While the iron complex reacts with 2-butenes with ca. 1% efficiency21a and the manganese complex with 0.8 to 8% efficiency,21c depending on the configuration at the double bond, the corresponding chromium complex is unreactive, even with a highly activated olefin as β-pinene. Dissecting the thermodynamic and kinetic contribution to the observed reactivity behavior is not straightforward. Moreover the mechanism of the alkene epoxidation by compound I and model species involves intermediates of different nature and presents TSR and multistate reactivity scenarios.6 A more complete analysis of these issues will require computational examination, as well as more experimental studies.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the Università di Roma “La Sapienza” and the Italian Ministero dell’Università e della Ricerca to SF and MEC, and from the U.S. National Institutes of Health (GM50422) to JMM.
Footnotes
Supporting Information Available: Derivation of hydroxylamine BDE and BDFE, analysis of reactions with TEMPO, Table S1. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Maria Elisa Crestoni, Email: mariaelisa.crestoni@uniroma1.it.
James M. Mayer, Email: mayer@chem.washington.edu.
References
- 1.(a) Harvey JN. Chem Phys Phys Chem. 2007;9:331–343. doi: 10.1039/b614390c. [DOI] [PubMed] [Google Scholar]; (b) Poli R, Harvey JN. Chem Soc Rev. 2003;32:1–8. doi: 10.1039/b200675h. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ortiz de Montellano PR. Cytochrome P-450: Structure, Mechanism and Biochemistry. 3. Plenum Press; New York: 2005. [Google Scholar]; (b) Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]; (c) Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]; (d) Ortiz de Montellano, De Voss JJ. Nat Prod Rep. 2002;19:477–493. doi: 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- 3.(a) Griffith OW, Stuehr DJ. Annu Rev Physiol. 1995;57:707–734. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]; (b) Woodward JJ, Chang MM, Martin NI, Marletta MA. J Am Chem Soc. 2009;131:297–305. doi: 10.1021/ja807299t. [DOI] [PubMed] [Google Scholar]
- 4.(a) Dunford HB. Heme Peroxidases. Wiley; New York: 1999. [Google Scholar]; (b) Davies MJ, Hakwins CL, Pattison DI, Rees MD. Antioxidants and Redox Signaling. 2008;10:1199–1234. doi: 10.1089/ars.2007.1927. [DOI] [PubMed] [Google Scholar]
- 5.(a) Groves JT, McClusky GA, White RE, Coon MJ. Biochem Biophys Res Commun. 1978;81:154–160. doi: 10.1016/0006-291x(78)91643-1. [DOI] [PubMed] [Google Scholar]; (b) Auclair K, Hu Z, Little DM, Ortiz de Montellano PR, Groves JT. J Am Chem Soc. 2002;124:6020–6027. doi: 10.1021/ja025608h. [DOI] [PubMed] [Google Scholar]; (c) Newcomb M, Toy PH. Acc Chem Res. 2000;33:449–455. doi: 10.1021/ar960058b. [DOI] [PubMed] [Google Scholar]; (d) Newcomb M, Shen R, Choi SY, Toy PT, Hollenberg PF, Vaz ADN, Coon MJ. J Am Chem Soc. 2000;122:2677–2686. [Google Scholar]; (e) Wang Q, Sheng X, Horner JH, Newcomb M. J Am Chem Soc. 2009;131:10629–10636. doi: 10.1021/ja9031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]; (b) Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Chem Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]; (c) Shaik S, Hirao H, Kumar D. Acc Chem Res. 2007;40:532–542. doi: 10.1021/ar600042c. [DOI] [PubMed] [Google Scholar]; (d) Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. Chem Rev. 2010;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 7.Schröder D, Shaik S, Schwarz H. Acc Chem Res. 2000;33:139–145. doi: 10.1021/ar990028j. [DOI] [PubMed] [Google Scholar]
- 8.(a) Schröder D, Fiedler A, Ryan MF, Schwarz H. J Phys Chem. 1994;98:68–70. [Google Scholar]; (b) Fiedler A, Kretzschmar I, Schröder D, Shaik S. J Am Chem Soc. 1996;118:9941–9952. [Google Scholar]
- 9.Lai W, Chen H, Shaik S. J Phys Chem B. 2009;113:7912–7917. doi: 10.1021/jp902288q. [DOI] [PubMed] [Google Scholar]
- 10.cf., De Angelis F, Jin N, Car R, Groves JT. Inorg Chem. 2006;45:4268–4276. doi: 10.1021/ic060306s.Song WJ, Seo MS, George SD, Ohta T, Song R, Kang M-J, Takehiko Tosha T, Kitagawa T, Solomon EI, Wonwoo Nam W. J Am Chem Soc. 2007;129:1268–1277. doi: 10.1021/ja066460v.
- 11.cf., Strassner T, Houk KN. Org Lett. 1999;1:419–421. doi: 10.1021/ol990064i.Cavallo L, Jacobsen H. Eur J Inorg Chem. 2003:892–902.Abashkin YG, Burt SK. Org Lett. 2004;6:59–62. doi: 10.1021/ol036051t.(d) Reference 23
- 12.Veige AS, Slaughter LM, Lobkovsky EB, Wolczanski PT, Matsunaga N, Decker SA, Cundari TR. Inorg Chem. 2003;42:6204–6224. doi: 10.1021/ic0300114. [DOI] [PubMed] [Google Scholar]
- 13.CrV is not nearly as oxidizing as FeV so CrIV/ligand radical cation states are very high in energy and do not need to be considered for these species (especially for the electron deficient fluorinated porphyrin used here). See, for example, references 13b and 15–17. Penner-Hahn JE, Benfatto M, Hedman B, Takahashi T, Doniach S, Groves JT, Hodgson KO. Inorg Chem. 1986;25:2255–2259.
- 14.cf., references 15, 16, 17, 31 and Rong C, Anson FC. Inorg Chem. 1994;33:1064–1070.
- 15.(a) Groves JT, Kruper WJ., Jr J Am Chem Soc. 1979;101:7613–7615. [Google Scholar]; (b) Groves JT, Haushalter RC. J Chem Soc, Chem Commun. 1981:1165–1166. [Google Scholar]; (c) Groves JT, Kruper WJ, Haushalter RC, Butler WM. Inorg Chem. 1982;21:1363–1368. [Google Scholar]
- 16.(a) Samsel EG, Srinivasan K, Kochi JK. J Am Chem Soc. 1985;107:7606–7617. [Google Scholar]; (b) Srinivasan K, Kochi JK. Inorg Chem. 1985;24:4671–4679. [Google Scholar]
- 17.cf., Traylor TG, Miksztal AR. J Am Chem Soc. 1989;111:7443–7448.Garrison JM, Bruice TC. J Am Chem Soc. 1989;111:191–198.Creager SE, Murray RW. Inorg Chem. 1985;24:3824–3828.Venkataramanan SN, Premsingh S, Rajagopal S, Pitchumani K. J Org Chem. 2003;68:7460–7470. doi: 10.1021/jo034558b.
- 18.Greenwood NN, Earnshaw A. Chemistry of the Elements. 2. Butterworth-Heinemann; Oxford: 1997. pp. 1027–1031. [Google Scholar]
- 19.Balch AL, Latos-Grażyński L, Noll BC, Olmstead MM, Zovinka EP. Inorg Chem. 1992;31:1148–1151. [Google Scholar]
- 20.Figgis BN, Hitchman MA. Ligand Field Theory and Its Applications. Wiley-VCH; New York: 2000. especially p. 136, 207, and 220 which show that E(2E) ≈ 20 B for an octahedral d3 complex and that B for V(NH3)63+ = 657 cm−1. [Google Scholar]
- 21.(a) Crestoni ME, Fornarini S. Inorg Chem. 2005;44:5379–5387. doi: 10.1021/ic048595c. [DOI] [PubMed] [Google Scholar]; (b) Chiavarino B, Cipollini R, Crestoni ME, Fornarini S, Lanucara F, Lapi A. J Am Chem Soc. 2008;130:3208–3217. doi: 10.1021/ja077286t. [DOI] [PubMed] [Google Scholar]; (c) Crestoni ME, Fornarini S, Lanucara F. Chem Eur J. 2009;15:7863–7866. doi: 10.1002/chem.200901361. [DOI] [PubMed] [Google Scholar]
- 22.Crestoni ME, Fornarini S. Inorg Chem. 2007;46:9018–9020. doi: 10.1021/ic700698d. [DOI] [PubMed] [Google Scholar]
- 23.(a) Feichtinger D, Plattner DA. J Chem Soc, Perkin Trans 2. 2000:1023–1028. [Google Scholar]; (b) Feichtinger D, Plattner DA. Chem Eur J. 2001;7:591–599. doi: 10.1002/1521-3765(20010202)7:3<591::aid-chem591>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]; (c) Plattner DA. Int J Mass Spectrom. 2001;207:125–144. [Google Scholar]
- 24.Adler AD, Longo FR, Finavells JD, Goldmacher JA, Korsakoff L. J Org Chem. 1967;32:476–476. [Google Scholar]
- 25.(a) Summerville DA, Jones RD, Hoffman BM, Basolo F. J Am Chem Soc. 1977;99:8195–8202. doi: 10.1021/ja00467a012. [DOI] [PubMed] [Google Scholar]; (b) Garrison JM, Bruice TC. J Am Chem Soc. 1989;111:191–198. [Google Scholar]; (c) Traylor TG, Miksztal AR. J Am Chem Soc. 1989;111:7443–7448. [Google Scholar]
- 26.Pfeiffer P, Breith E, Lübbe E, Tsumaki T. Justus Liebigs Ann Chem. 1933;503:84–103. [Google Scholar]
- 27.Coggon P, McPhail AT, Mabbs FE, Richards A, Thornley AS. J Chem Soc A. 1970;1970:3296–3303. [Google Scholar]
- 28.Saltzman H, Sharefkin JG. Org Synth. Vol. 5. Wiley; New York: 1973. p. 658. Coll. [Google Scholar]
- 29.(a) Meot-Ner M. In: Gas Phase Ion Chemistry. Bowers MT, editor. Vol. 1 Academic Press; New York: 1979. [Google Scholar]; (b) Bartmess JE, Gerogiadis RM. Vacuum. 1983;33:149–153. [Google Scholar]
- 30.Su T, Chesnavich WJ. J Chem Phys. 1982;76:5183–5185. [Google Scholar]
- 31.Liston DJ, West BO. Inorg Chem. 1985;24:1568–1576. [Google Scholar]
- 32.Bouchoux G, Salpin JY, Leblanc D. Int J Mass Spectrom Ion Processes. 1996;153:37–48. [Google Scholar]
- 33.(a) Angelelli F, Chiavarino B, Crestoni ME, Fornarini S. J Am Soc Mass Spectrom. 2005;16:589–598. doi: 10.1016/j.jasms.2005.01.011. [DOI] [PubMed] [Google Scholar]; (b) Dunbar RC. Mass Spectrom Rev. 2004;23:127–158. doi: 10.1002/mas.10074. [DOI] [PubMed] [Google Scholar]
- 34.Warren JJ, Mayer JM. unpublished results [Google Scholar]
- 35.Sheldon RA, editor. Metalloporphyrins in Catalytic Oxidations. Marcel-Dekker; New York: 1994. [Google Scholar]
- 36.(a) Meot-Ner M. J Am Chem Soc. 1982;104:5–10. [Google Scholar]; (b) Bucher H-H, Grutzmacher H-F. Int, J Mass Spectrom Ion Processes. 1991;109:95–104. [Google Scholar]; (c) Baciocchi E, Bietti M, Chiavarino B, Crestoni ME, Fornarini S. Chem Eur J. 2002;8:532–537. doi: 10.1002/1521-3765(20020118)8:2<532::AID-CHEM532>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 37.Afeefy HY, Liebman JF, Stein SE. Neutral Thermochemical Data. In: Linstrom PJ, Mallard WG, editors. NIST Chemistry WebBook, NIST Standard References Database Number 69. National Institutes of Standards and Technology; Gaithersburg MD: [accessed 9 October 2009]. p. 20899. http://webbook.nist.gov/chemistry/ [Google Scholar]
- 38.Gas phase A + B → C + D reactions typically have small entropy changes. In this case, the S° values for NO2 and NO (57.37, 50.37 cal K−1 mol−1) differ only by 7 cal K−1 mol−1.37 For a discussion of entropies in hydrogen-atom transfer reactions, including cases where ΔS° ≠ 0, see: Mader EA, Manner VW, Markle TF, Wu A, Franz JA, Mayer JM. J Am Chem Soc. 2009;131:4335–4345. doi: 10.1021/ja8081846.Mader EA, Davidson ER, Mayer JM. J Am Chem Soc. 2007;129:5153–5166. doi: 10.1021/ja0686918.
- 39.For XO → X + O, when S°(XO) = S°(X) the gas phase BDFE will be smaller than the gas phase BDE by TS°(O) = (298 K)(38.49 cal K−1 mol−1) = 11.5 kcal mol−1.37
- 40.Schröder D, Schwarz H. Angew Chem Int Ed Engl. 1995;34:1973–1195. [Google Scholar]
- 41.Acree WE, Jr, Pilcher G, Ribeiro da Silva MDMC. J Phys Chem Ref Data. 2005;34:553–572. [Google Scholar]
- 42.Deubel DV. J Am Chem Soc. 2004;126:996–997. doi: 10.1021/ja039234j.The BDE(PhI–O) of 26 kcal is estimated from the solution thermodynamic oxygen-transfer potentials (TOP) in this reference, assuming constant solvation effects on the XO/X couples.
- 43.Huang KW, Han JH, Cole AP, Musgrave CB, Waymouth RM. J Am Chem Soc. 2005;127:3807–3816. doi: 10.1021/ja044512f. [DOI] [PubMed] [Google Scholar]
- 44.Conry RR, Mayer JM. Inorg Chem. 1990;29:4862–4867. [Google Scholar]
- 45.The initial adduct 1-TEMPO is probably best described as CrIV-(TEMPO−) and is probably a spin triplet. This species can then dissociate a doublet tetramethylpipridinyl radical to give 2.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.