

Abstract
We review the modulatory effects of the catecholamine neurotransmitters noradrenaline and dopamine on prefrontal cortical function. The effects of pharmacologic manipulations of these systems, sometimes in comparison with the indoleamine serotonin (5-HT), on performance on a variety of tasks that tap working memory, attentional-set formation and shifting, reversal learning, and response inhibition are compared in rodents, nonhuman primates, and humans using, in a behavioral context, several techniques ranging from microiontophoresis and single-cell electrophysiological recording to pharmacologic functional magnetic resonance imaging. Dissociable effects of drugs and neurotoxins affecting these monoamine systems suggest new ways of conceptualizing state-dependent fronto-executive functions, with implications for understanding the molecular genetic basis of mental illness and its treatment.
Keywords: prefrontal cortex, noradrenaline, dopamine, serotonin, attention, working memory, reinforcement learning, response inhibition
INTRODUCTION
Researchers still debate the nature of the so-called executive functions of the prefrontal cortex (PFC), which have been defined as the set of control processes that serve to optimize performance on complex cognitive tasks. These executive functions have been variously suggested to be a flexible resource related to Spearman’s general intelligence (g) (Duncan et al. 2000), working memory (Baddeley 1986, Goldman-Rakic 1987), cognitive control (Miller & Cohen 2001), or a toolbox of dissociable mechanisms with some degree of neuroanatomical localization within the prefrontal cortex (e.g., Aron et al. 2004, Robbins 1996). Moreover, investigators are uncovering other features of executive function that include aspects of social cognition, emotional regulation, memory retrieval, and a complex specification of functions of the rostral PFC (Gilbert & Burgess 2008). This review does not attempt to resolve or refine these issues. However, it does emphasize yet another mode of frontal functioning: its capacity for top-down regulatory control over the ascending modulatory systems of the reticular core of the brain (e.g., Amat et al. 2005, Robbins 2005), and in turn, the powerful influences of these neuromodulators on PFC functions. These projections, which include major ascending monoamine [dopamine (DA), norepinephrine (NE), serotonin or 5-hydroxytryptamine (5-HT), histamine, orexin, as well as acetylcholine], have systems that ramify widely to innervate diverse forebrain regions, including the hippocampus, striatum, amygdala, and thalamus, as well as the entire neocortex. These neuromodulatory systems likely adjust signal-to-noise ratios in terminal domains to influence processing there that is presumably appropriate to the state engendered by their conjoint activity (see Sarter et al. 2005 for an excellent review of this approach in terms of the cholinergic coordination of cortical functions). Characterizing these states remains as important a problem as the nature of fronto-executive processes themselves, but they presumably include different levels of arousal. These different levels may reflect fluctuations in the sleep-wake cycle, motivation, mood, and stress, which, while constituting distinct processes, may have a common currency through PFC arousal. Although the extent to which these systems are engaged by these different processes undoubtedly overlaps, with likely interactions between them in forebrain terminal domains, they nevertheless have distinct roles in chemical neuromodulation within the prefrontal cortex. We use the term neuromodulation to mean the set of neurochemical processes that curtail or prolong, augment, or diminish effects of fast signaling in neuronal networks, often conditional on the firing state of the postsynaptic cell. From a neurocomputational perspective, we relate neuromodulation to parameters in equations for artificial neural networks that slowly change the dynamics of the network.
In identifying recent research trends in this area, we have focused on a growing realization that many of these neurotransmitter systems function in both phasic and tonic modes, with considerable implications for understanding their specific functions at the level of the PFC. Elucidation of secondary and tertiary messengers, including common intracellular cascades, has contributed to a better understanding of interactions among the systems. The study of genetic polymorphisms affecting the monoamines has opened up a new approach to studying their role in the PFC, most notably in the case of the catecholamine-o-methyl-transferase polymorphism (COMT). Moreover, the advance of pharmacological functional magnetic resonance imaging (fMRI) and the promise of ligands suitable for evaluating cortical monoaminergic function using positron emission tomography have also provided new evidence. These advances, which have perhaps had greatest influence in human cognitive neuroscience, have been paralleled by superior in vivo voltammetric methodologies for use in experimental animals.
A general principle that has emerged in the past decade in considering the functions of the chemical modulatory inputs to the PFC has been that of the Yerkes-Dodson, inverted U-shaped function linking the efficiency of behavioral performance to the level of activity in the ascending monoaminergic (mainly DA- and NE-ergic) systems. The inverted U dose response has been demonstrated with pharmacological agents in both animals (e.g., Granon et al. 2000, Zahrt et al. 1997) and humans (Gibbs & D’Esposito 2006), and also in relationship to COMT genotype. Thus, substitutions of methionine (met) for valine (val) weaken COMT catabolic activity and shift the inverted U leftward both in normal human subjects and in patients (Egan et al. 2001, Williams-Gray et al. 2007).
Further complications have related to heterogeneity of function within the PFC and the fact that the Yerkes-Dodson relationships may posit different U-shaped functions depending on the nature of the task; therefore, a level of monoaminegic function optimal for one may be sub- or supraoptimal for another. The most obvious functional divisions are between orbitofrontal and dorsolateral regions of the primate PFC, the former associated especially with emotional decision-making and the latter with working memory. But these crude functional divisions are now being fractionated further: The role of component subregions is slowly being elucidated, as is the conceivably top-down role of regions such as the anterior PFC, which highlights potentially hierarchical or heterarchical modes of organization. The growing sophistication of cognitive-behavioral and computational approaches to PFC function has also presented what sometimes appear to be competing accounts of the contributions of the ascending modulatory systems, which require resolution. What is increasingly evident is that PFC executive functions must be considered in terms of a state dependency that is mainly provided by the chemical modulatory systems. In simple terms, the executive process required to respond optimally to stress (e.g., during a battle) may differ dramatically from those required to cope with bereavement or to stay awake during a boring, though important, lecture.
CATECHOLAMINES ARE RELEASED IN PFC ACCORDING TO AROUSAL STATE
The chemical arousal systems projecting to the PFC probably all affect fronto-executive cognition via their modulatory effects. However, most of our detailed knowledge on patterns of neuronal firing and terminal release in PFC has focused on the catecholamines. Release of NE and DA is low during periods of drowsiness, moderate during conditions of alert interest, and high during uncontrollable stress. Recordings from NE and DA cell bodies have shown that under optimal, alert conditions, NE and DA neurons have relatively low levels of tonic firing but fire to stimuli that are relevant and/or predict reward, respectively (Aston-Jones et al. 2000, Berridge & Waterhouse 2003, Schultz 1998). Under conditions of psychological stress, the amygdala also activates norepinephrine (NE) and DA cells at source and increases catecholamine release in the PFC (Goldstein et al. 1996).
The orexin (hypocretin) neurons in the hypothalamus have key arousing effects needed for the brain to enter the waking state (Saper et al. 2001). The excitatory orexins project to the NE cell bodies of the locus coeruleus (Horvath et al. 1999) and to the thalamus and cortex, including the PFC (Moore et al. 2001). In rats, orexins excite the same thalamocortical synapses in PFC as does nicotine, and they improve attentional function (Lambe et al. 2005). The antinarcolepsy drug, modafinil, may act via the orexin system and has improved PFC functions in human subjects (Turner et al. 2003), suggesting that this will be an important area for future research. Orexins appear to activate the subgroup of DA neurons in the ventral tegmental area that project to the PFC and the nucleus accumbens shell (Vittoz et al. 2008), suggesting a highly specific and coordinated arousal system.
Classical neurochemical evidence has indicated that medial PFC (mPFC) DA levels in the rat may be affected in a protracted way, especially by aversive stimuli such as electric foot shock (Thierry et al. 1976). Investigators have found similar results for a variety of stressors, including handling, forced swimming, tail pinch, social defeat, conditioned aversive stimuli, and pharmacological anxiogenesis (see review by Seamans & Robbins 2009). These increases can be countermanded by chronic treatment with antidepressant drugs without affecting the corticosteroid response per se, which suggests that they arise as emotional (or arousing) concomitants of stress rather than reflecting the stress itself (Dazzi et al. 2001).
Appetitive stimuli also enhance activity in the mesocortical DA system, although to a lesser degree and for a more transient period than do aversive stimuli (see Seamans & Robbins 2009 for a review). Furthermore, such activity can be evoked in several types of cognitive demand situations, including learning and extinction, (e.g., Feenstra et al. 2001), reversal learning (van der Meulen et al. 2007), working memory (Phillips et al. 2004, Rossetti & Carboni 2005, Watanabe et al. 1997), sustained attention (Dalley et al. 2002) or attentional set-shifting (Stefani & Moghaddam 2006), and intertemporal choice during reward discounting (Winstanley et al. 2006). Without discussing any of these particular situations in detail, we can make a few general points. First, many of these studies provided controls to show that the PFC DA activation could be over and above that provided by the passive receipt of food rewards, suggesting indeed that the cognitive demands were key. Second, some of the individual variation in PFC neurochemistry was consistent with mesocortical DA being associated with specific traits (e.g., impulsivity; Dalley et al. 2002). Third, some evidence indicates differential recruitment of PFC DA according to the sector of the PFC monitored; in the study by Winstanley et al. (2006), DA in the orbital frontal cortex (OFC) was more associated with choice of large delayed rewards than was mPFC DA (which contrasted with the regional pattern of 5-HT activation). Finally, these data must be considered together with pharmacological findings of effects, for example, of specific DA receptor agonists. An elegant integration was achieved in the study by Phillips et al. (2004) on working memory in which knowledge of the fluctuations in PFC DA in the delays imposed during a spatial working memory task for rats enabled understanding of the contrasting effects of a D1 receptor agonist to improve, but also to impair, memory as a function of the retention interval. Specifically, both the D1 agonist-induced improvements in low baseline performance afforded by long delays and the deficits in performance after brief delays (Floresco & Phillips 2001) could be related to relatively low and high levels of PFC DA respectively at those retention intervals, as measured using in vivo dialysis.
Differing levels of NE release engage qualitatively different noradrenergic receptors (Arnsten 2000). Moderate levels of NE release engage high affinity α2a-adrenergic receptors (α2a-ARs), whereas higher levels of NE release during stress engage lower-affinity α1-adrenoceptors (α1-ARs) and β-adrenoceptors (β-ARs). As described below, these receptors have opposing effects on PFC function: Stimulation of postsynaptic α2a-ARs improves, whereas stimulation of α 1-ARs and (β1-ARs impairs, PFC function. Thus, arousal state can alter our cognitive abilities through powerful catecholamine actions in PFC. A parallel situation exists with respect to DA receptors; D1 and D2 receptors may be stimulated optimally at different levels of DA presynaptic activity, which may improve some aspects of cognition and hinder others (see below and reviews Floresco & Magyar 2006, Seamans & Robbins 2009).
MECHANISMS OF OPTIMAL REGULATION OF WORKING MEMORY IN PFC BY CATECHOLAMINES
Brozoski et al. (1979) were the first to show that catecholamines are essential to dlPFC spatial working memory function. Although this landmark study focused on DA actions, investigators now agree that both NE and DA have essential, probably complementary, actions that affect cognition in the PFC. Blockade of either NE α2a-ARs or DA D1 receptors (D1-Rs) in the dlPFC markedly impairs spatial working memory performance (Li & Mei 1994, Sawaguchi & Goldman-Rakic 1991). Results from electrophysiological studies in monkeys performing working memory tasks are consistent with these behavioral findings: Blockade of α2a-ARs (Li et al. 1999) or D1-Rs (Sawaguchi & Goldman-Rakic 1994, Williams & Goldman-Rakic 1995) erodes the spatially tuned, persistent firing of dlPFC neurons. As described below and in Figure 1, NE stimulation of postsynaptic α2a-ARs strengthens iso-directional networks (i.e., increases signals within the network), whereas optimal DA stimulation of D1-Rs weakens network connections from contra-directional inputs (i.e., decreases noise within the network). These gating actions likely occur in dendritic spines by regulating cAMP influences on HCN channels (hyperpolarization-activated cyclic nucleotide gated cation channels, which pass the h-current when in the open state).
Figure 1.

The effects of NE and DA on the task-related firing of PFC neurons in a monkey performing a working memory task. NE stimulation of α2a-AR increases firing during the delay period for the preferred direction of the neuron (left panel), whereas DA stimulation of D1-R suppresses firing for the nonpreferred directions of the neuron (right panel) compared with control conditions. Thus, optimal levels of NE and DA have complementary enhancing effects on PFC physiology during spatial working memory. Higher levels of D1-R stimulation suppress all firing (not shown). Adapted from Wang et al. 2007 and Vijayraghavan et al. 2007.
HCN channels are localized on the heads and necks of dendritic spines near incoming synapses in the superficial layers of monkey PFC, the layers that form the cortical-cortical networks (Wang et al. 2007). When the HCN channels are opened in the presence of cAMP, nearby synaptic inputs are shunted, likely owing to a reduction in membrane resistance. Thus, activation of cAMP-HCN signaling weakens the network inputs onto that spine compartment. α2a-ARs are localized next to HCN channels on spines and are thus ideally positioned to modulate the local concentration of cAMP near the channels via Gi inhibition of cAMP production. Electrophysiological studies have shown that α2a-AR stimulation increases network firing for preferred directions, and this improvement can be reversed by manipulations that increase or mimic cAMP (Wang et al. 2007). Conversely, blockade of α2a-ARs induces network collapse that can be rescued by blocking HCN channels. Similar results have been observed at the behavioral level, where viral knockdown of HCN channels or pharmacological blockade of these channels in the rat prelimbic PFC improves spatial working memory performance (Ramos et al. 2005, Wang et al. 2007). These beneficial actions are reversed by agents that increase cAMP signaling at both the cellular and the behavioral levels.
Information cannot be represented in the PFC by a single neuron increasing its firing rate: Neuronal activity must be spatially tuned and accurately timed to convey accurate representations of spatial position. Spatial tuning of PFC networks is created by GABAergic inhibition (Rao et al. 1999,2000) and DAD1-R stimulation (Vijayraghavan et al. 2007). D1-Rs and α2a-ARs appear to be on different dendritic spines (C. Paspalas and A.F.T. Arnsten, unpublished data), which suggests that they modulate different sets of network inputs. Optimal D1-R stimulation tunes PFC microcircuits by suppressing cell responses to nonpreferred stimuli [e.g., spatial directions for which the microcircuit is not tuned (Vijayraghavan et al. 2007)]. Conversely, blocking D1-R increases firing to nonpreferred inputs and makes the neuron noisy or less tuned. D1-R stimulation may dynamically regulate the breadth of network tuning on the basis of task demands (Vijayraghavan et al. 2007).
Catecholamines have additional actions in PFC, including fundamental excitatory effects (Seamans et al. 2001, Young & Yang 2004) and traditional effects on neuroplasticity (Runyan & Dash 2005). However, their very powerful effects on working memory are most evident in PFC networks engaged in working memory operations.
EXCESSIVE CATECHOLAMINE RELEASE DURING STRESS IMPAIRS PFC FUNCTION
In contrast to the essential effects of moderate levels of catecholamines, very high levels of catecholamine release in the PFC during stress exposure markedly impair working memory function through network collapse and suppression of PFC cell firing.
High levels of D1-R stimulation in the PFC suppress both preferred and nonpreferred inputs, inducing network collapse (Vijayraghavan et al. 2007) and impairing working memory (Zahrt et al. 1997). These detrimental actions occur through increased levels of cAMP (Vijayraghavan et al. 2007), likely through opening of HCN channels (N.J. Gamo, M. Wang, and A.F.T. Arnsten, unpublished observations). High levels of NE stimulation of β1-AR may also contribute to excessive cAMP signaling and PFC cognitive dysfunction (Ramos et al. 2005).
High levels of NE released during stress impair working memory and neuronal firing by stimulating α1-ARs (Birnbaum et al. 2004). NE α1-AR stimulation impairs PFC function by activating the phosphatidylinositol (PI) cascade, leading to intracellular Ca2+ release and activation of protein kinase C (Birnbaum et al. 2004). Ca2+-dependent kinases negatively influence PFC cognitive functions. Activation of Ca2+-calmodulin-dependent kinase II and inhibition of the Ca2+-activated phosphatase, calcineurin, impair spatial working memory in the rat prelimbic PFC (Runyan et al. 2005). These actions may be mediated, in part, by the opening of small-conductance Ca2+-gated potassium channels (SK channels), which suppress neuronal firing (Brennan et al. 2008, Hagenston et al. 2008). Protein kinase C is cooperatively activated by Ca2+ and DAG (diacyl glycerol). Activation of protein kinase C suppresses delay-related firing of PFC neurons in monkeys performing a working memory task and impairs spatial working memory performance in both rats and monkeys (Birnbaum et al. 2004, Runyan et al. 2005). Conversely, inhibition of protein kinase C signaling in PFC protects working memory performance from the detrimental effects of stress. As described below, these findings are of great relevance to mental illness because this pathway is the target of genetic insults as well as of many medications used to treat bipolar disorder and schizophrenia (Manji & Lenox 1999).
TOP-DOWN PFC CONTROL OF MONOAMINE FUNCTION
PFC neurons project down to the NE, DA, and 5-HT cell bodies in the brain stem and likely regulate optimal firing (Arnsten & Goldman-Rakic 1984, Jodo et al. 1998, Sara & Herve-Minvielle 1995). This anatomical organization provides an intriguing opportunity for PFC to recruit monoamine systems in specific circumstances; for example, in the case of Amat et al. (2005), 5-HT neuron activity in the dorsal raphé was influenced by the perception of mPFC’s instrumental control over environmental contingencies, thus enabling a coping strategy to ameliorate the stress. Moreover, Giorgi et al. (2003) showed that tail pinch or pharmacological stressors increased PFC DA levels of Roman high-avoidance but not low-avoidance rats, implying that the rise in PFC DA levels may be related to the recruitment of a more effective coping strategy rather than the stress response per se, suggesting that PFC DA is also recruited in part to mediate the coping strategy.
ROLE OF CATECHOLAMINES IN COGNITIVE FLEXIBILITY AND INHIBITORY CONTROL
Although the catecholamine systems modulate working memory mechanisms within the PFC, they also play important roles in other executive functions, including attentional selection and inhibitory response control. The noradrenergic system has been implicated in sustained attention in its phasic mode and in distractibility in its tonic mode in nonhuman primates performing a go/no-go visual attentional task (Aston-Jones & Cohen 2005). Single-unit recordings of the locus coeruleus showed that optimal performance on a go/no-go visual target-detection paradigm in rhesus monkeys was accompanied by phasic firing of the NE cells. However, when performance was suboptimal for example as a result of anxiety or agitation, the firing of the locus coeruleus cells was generally at a high tonic level. Aston-Jones & Cohen (2005) speculated that this mode of firing is well suited to situations in which the animal responds to novel stimuli or explores new contingencies. These findings are probably consistent with those from rodents in analogous tests of sustained visual attention (a) following 6-OHDA-induced lesions of the dorsal ascending noradrenergic bundle to produce profound cortical NE loss (see Robbins & Everitt 1995), (b) following lesions of NE terminals in the rat neocortex using antidopamine B-hydroxylase saporin (Milstein et al. 2007), and (c) following low-dose methylphenidate (Berridge et al. 2006).
Burgeoning evidence also indicates that the NE coeruleo-cortical projections are also implicated in cognitive flexibility as required in set shifting, in which attention must be shifted from one perceptual dimension to another (Birrell & Brown 2000, Dias et al. 1996). Converging new evidence suggests a selective effect of prefrontal NE manipulations on attentional set shifting. First, Lapiz & Morilak (2006) have shown that atipamezole, an α-2-adrenergic autoreceptor antagonist, improves set shifting when injected systemically and that its effects are blocked by mPFC infusions of a postsynaptic α-1 receptor antagonist, but not by mPFC infusions of a β-1 or β-2 receptor antagonist. Lapiz et al. (2007) described a similar improvement in set shifting following a regimen of subchronic treatment with the NE-reuptake blocker desipramine, leading to an upregulation of mPFC NE (as confirmed with microdialysis). Second, lesions ofthe dorsal noradrenergic bundle using 6-OHDA produce substantial depletions of cortical NE and selectively impair set shifting (Tait et al. 2007). Moreover, selective lesions of noradrenergic terminals within the PFC using antidopamine β-hydroxylase saporin produced a selective impairment of extradimensional set shifting (McGaughy et al. 2008), which was ameliorated by the selective norepinephrine reuptake inhibitor, atomoxetine (Newman et al. 2008). The deficits induced by this drug in sham-lesioned rats suggest once again the existence of an inverted U-shaped curve contributing to the control of attentional set shifting, consistent with the findings of Aston-Jones & Cohen (2005) and Arnsten and colleagues (Arnsten 2000) on working memory.
These effects of atomoxetine are intriguing in view of recent evidence that this drug enhances stop-signal performance in the rat, a paradigm that specifically addresses inhibitory response control and which depends on or-bitofrontal cortical substrates (see Eagle et al. 2008 for a review). Specifically, atomoxetine dose-dependently hastened the reaction time for inhibiting an initiated response without affecting the go reaction time (Robinson et al. 2008). Thus, doses of atomoxetine that impair attentional set shifting in normal rats appear to improve stop-signal performance (Newman et al. 2008). A similar effect of the nature of the task in fact permeates the literature on PFC function. These effects of atomoxetine, which have some clinical relevance, need to be shown definitively to depend on PFC NE, especially because the drug is known to enhance PFC DA as well as NE, according to findings obtained from in vivo microdialysis (Bymaster et al. 2002). However, the positive effects of atomoxetine on stop-signal reaction time are not seen with systemic administration of the DA-reuptake inhibitor GBR-12909 (Bari et al. 2009). Nevertheless, as in the case of working memory, we must eventually address the relative and, indeed, differential contributions of the NE and DA systems projecting to PFC in attentional performance.
Following findings from Granon et al. (2000), Chudasama & Robbins (2004) found that a D1-R agonist infused into the mPFC produced improvements in an attentional component of a combined sustained attention and memory paradigm that were at least as great as those produced during the delay period that constituted the working memory component of the task. Further insights have arisen from investigations of the effects of PFC DA depletion on the development of an attentional set (Crofts et al. 2001, Robbins & Roberts 2007). Specifically, PFC DA loss induced by local administration of 6-OHDA (also leading to lesser but nevertheless substantial cortical NE loss) produced impairments in serial intradimensional set shifting such that it produced no effect on learning of an initial visual compound discrimination task based on shapes with superimposed irrelevant lines, but the capacity of the marmosets to learn a dimensional rule over several subsequent discriminations (or intradimensional shifts) was impaired. Additionally, the marmosets with PFC DA loss were impaired when they had to resist distraction produced by introducing irrelevant cues and were sometimes even facilitated at learning discriminations on the basis of the previously irrelevant stimulus dimension (extradimensional shift). The authors suggested that the role of DA was congruent with one of stabilizing representations (e.g., rules) within the PFC, consistent also with a function in working memory in which representations must be maintained over time and resist distractions and other forms of interference.
Hampshire & Owen (2006) recently showed that the PFC mediation of intra- and extradimensional shifts depends on the ventrolateral and possibly dorsolateral regions of the human PFC on the basis of fMRI evidence. This finding is consistent with earlier evidence from the marmoset of a specific involvement of the lateral PFC in the mediation of extradimensional shifting and of the orbitofrontal cortex in reversal learning, another aspect of discrimination learning that requires cognitive flexibility (Dias et al. 1996). Further evidence supports this type of anatomical dissociation in both rats (McAlonan & Brown 2003) and mice (Bissonette et al. 2008), although the task used is based on odors and textures rather than on different visual dimensions, and the rodent extradimensional shift is impaired by lesions of the mPFC not the lateral region, presumably as a consequence of homologies among these areas between primates and rodents.
The possible role of DA in intradimensional set shifting in primates has been supported by a recent pharmacological study of the effects of the COMT inhibitor tolcapone in humans: This drug, which may enhance PFC DA function, improved intradimensional shift performance in volunteers (Apud et al. 2007). However, a possible discrepancy must be resolved with studies in rodents. First, tolcapone treatment in rats improved extradimensional rather than intradimensional set shifting (Tunbridge et al. 2004). Second, using other types of tasks that reflect cognitive flexibility, such as strategic shifts in response versus place learning, other investigators have found that both DA D1-R and DA D2-R antagonists infused into the mPFC impair cognitive flexibility (Floresco et al. 2006, Ragozzino 2007). Robbins & Roberts (2007) have suggested that these differences may arise from the fact that the primate test requires shifting between dimensions within a modality, whereas the rodent versions require shifting between sensory modalities, possibly resulting in less interference at set formation stages (i.e., intradimensional shifting). These considerations are pertinent to the considerable evidence of deficits in set formation and shifting performance in patient groups such as schizophrenia (e.g., Ceaser et al. 2008, Pantelis et al. 1999) and obsessive-compulsive disorder (Chamberlain et al. 2006a).
The attentional-set-shifting findings are also relevant to other evidence that PFC DA depletions can sometimes produce different effects compared with working memory functions (e.g., Collins et al. 1998). Recent evidence from Floresco et al. (2006) demonstrated no effect of intra-mPFC infusions ofa D1 receptor agonist on attentional set shifting at doses that affect working memory and continuous attentional performance in other studies (Chudasama & Robbins 2004). By contrast, agents working at the D4 receptor did modulate set-shifting performance. The DA D4 receptor antagonist L-7,45,870 improved and the DA D4 agonist PD-1,68,077 impaired performance. An inverted U-shaped function focusing on the D1 receptor does not seem readily able to explain these findings. However, if one considers the relative affinity of the different DA receptors at different levels of DA activity within the PFC, this intriguing difference could perhaps be linked to a modified Yerkes-Dodson principle, which links different degrees of monoaminergic modulation of the PFC optimally to the performance of distinct behavioral functions within the PFC (Seamans & Robbins 2009). Indeed, if D1 receptor stimulation shunts network inputs (Vijayraghavan et al. 2007), D1 receptor stimulation may be helpful only in contexts in which limiting or narrowing neuronal inputs is useful (Arnsten et al. 2009).
Certain tasks requiring PFC involvement do not appear to require DA-ergic modulation at all. This was first manifest in a self-ordered working memory task that is dramatically impaired by excitotoxic lesions of the marmoset PFC, but not by PFC DA depletion using 6-OHDA (Collins et al. 1998). An even more graphic example is that of visual reversal learning. Reversal learning depends on the integrity of the OFC in both monkeys (e.g., Dias et al. 1996) and rodents (e.g., Bissonette et al. 2008, Chudasama & Robbins 2003). Although attentional set formation and shifting are affected by PFC DA loss, this is not true for reversal learning, including serial reversal learning (Clarke et al. 2005, 2007; Roberts et al. 1994), even when the DA loss is confined to OFC regions. This finding is theoretically significant because 5-HT depletion in the OFC produces robust and substantial deficits in reversal learning (Clarke et al. 2004, 2005, 2007). One cannot argue that DA depletion from the OFC is insufficient for functional effects because a recent study has shown that depletion results in a major resistance to extinction in an instrumental paradigm (Walker et al. 2008). Moreover, other evidence in the rat indicates that DA in the OFC (or in the ventromedial PFC (Hitchcott et al. 2007)] is implicated in decisional processes concerned with reward (Floresco & Magyar 2006, Kheramin et al. 2004, Winstanley et al. 2006). Thus it appears likely that DA modulation subserves different functions (although perhaps through common mechanisms) in different sectors of the PFC and that these differ from those of 5-HT and probably from NE and other neuromodulators. Figure 2 summarizes some of the comparative evidence regarding the effects of monoamine manipulation on different PFC tasks and is based largely on evidence from nonhuman primates.
Figure 2.

a: Schematic summary of differential impact of ascending monoamine systems on different tasks mediated by different sectors of the PFC. b: Speculative interpretation of underlying PFC processes regionally differentially affected by monoamines. DR, dorsal raphé: 5-HT system; LC, coeruleo-cortical NE system; SSRT, stop-signal reaction time task; VTA, mesocortical DA system.
ROLE OF MONOAMINES IN HUMAN FRONTO-EXECUTIVE PROCESSING
It has always been difficult to attribute effects ofmonoaminergic drugs or manipulations on executive functioning to actions within the human PFC because such agents may act at other sites, especially the striatum for agents affecting DA or 5-HT. This has been mitigated to some extent by the use of neuroimaging techniques, which combine pharmacological treatments with task-dependent activations of the blood-oxygen-level-dependent (BOLD) response, and also by the discovery of genetic polymorphisms such as COMT, which have a degree of specificity for PFC.
A notable recent study (Cools et al. 2007) has compared the effects of the D2 receptor agent bromocriptine on different aspects of attention and working memory in the striatum as well as in the PFC by observing the BOLD response in these regions during task performance in normal volunteers. Bromocriptine improved the flexible up-dating (switching) of information in subjects previously screened to exhibit high levels of impulsivity using the Barrett Impulsivity Scale, but it impaired that of low-impulsive subjects. Bromocriptine modulated the striatum during switching but not during distracting conditions of the working memory task. In contrast, the lateral frontal cortex was affected during distraction but not during switching. These data are consistent with another recent fMRI study (Dodds et al. 2008) on the effects of the indirect DA and NE agonist methylphenidate. Using a probabilistic reversal learning procedure, the study demonstrated that methylphenidate reduced the BOLD response in the ventral striatum during response switching after negative feedback but modulated activity in the anterior cingulate and ventrolateral PFC when subjects maintained their present response set, consistent with the study by Crofts et al. (2001) in nonhuman primates. The effects of the catecholamine manipulation contrasted with those produced by the tryptophan depletion technique, which leads to transient reductions in central 5-HT on the same task. Thus, Evers et al. (2005) reported that volunteers subjected to tryptophan depletion exhibited increased BOLD activation of a dorsomedial region of the PFC specifically following negative feedback. This result is consistent with the recent findings that the same treatment leads to a potentiation of punishment prediction (Cools et al. 2008a) and that the dorsomedial PFC is especially sensitive to effects of low tryptophan modulation (Evers et al. 2006, Talbot & Cooper 2006). However, we still need careful interpretation about the possible role of 5-HT mechanisms in punishment prediction (Cools et al. 2008b). Moreover, Rubia et al. (2005) found a significant reduction of BOLD signal with tryptophan depletion in the right inferior and orbitofrontal cortex during a go/no-go task.
Overall, these findings obviously need further investigation, but they are nonetheless consistent with the hypothesis that the monoamines modulate different aspects of cognitive function, often represented in distinct PFC regions, consistent with findings in the animal literature. They also highlight the role of the monoaminergic innervations of the PFC in sometimes contrasting cognitive processes: for example, working memory and attentional switching. These processes can also be reconstrued in terms of catecholaminergic activity in the PFC helping to provide stabilization and switching between different representations, for example of rules or objects, that guide behavior (Durstewictz et al. 2000).
This conceptualization may be substantiated by considering the discovery of genetic polymorphisms that appear to modulate catecholamine activity, especially in the PFC. Thus, the val108/58met COMT polymorphism has been linked to respectively low (val-val) or high (met-met) PFC DA (Egan et al. 2001), although catecholamines in other regions are also likely to be directly or indirectly affected. The finding of impaired working memory function in val-val individuals is consistent with the classic inverted-U-shaped dogma (Egan et al. 2001) but also with the view that the val-val phenotype may be associated with enhanced distractibility and switching (Bilder et al. 2004). These authors also hypothesize that the met-met alleles promote, in addition to elevated PFC DA, stable and persistent representations, a hypothesis supported by the finding of enhanced learning but impaired reversal, relative to val-val individuals (Nolan et al. 2004). This formulation is also consistent with the findings of Crofts et al. (2001) in nonhuman primates and possibly with the findings of Dodds et al. (2008), described above. However, Frank et al. (2007) found on a trial-to-trial basis during discrimination learning that val/val homozygotes were less likely than met/met carriers to switch responses after negative feedback, which appears inconsistent with the hypothesis of Bilder et al. Clearly, further investigations are warranted to establish the significance of the COMT polymorphism in terms of PFC function.
So far, relatively few studies have employed fMRI to examine PFC functions modulated by NE. However, a recent investigation of the stop-signal performance task after atomoxetine in humans has found that this selective noradrenergic reuptake inhibitor, consistent with its effect selectively to speed stop signal reaction time in healthy volunteers (Chamberlain et al. 2006b), enhanced the activation in the right inferior frontal gyrus associated with stopping. Furthermore, the activation associated with successful stopping was correlated with individual plasma atomoxetine levels (Chamberlain et al. 2009). This result is consistent with previous work in the rat (Blondeau & Dellu-Hagedorn 2007, Robinson et al. 2008) and in nonhuman primates in go/no-go performance (Ma et al. 2003), thus presenting good evidence of translatability across species. The key question remains whether the drug is exerting its beneficial effect on SSRT via cortical NE and/or DA mechanisms (Bymaster et al. 2002).
IMPLICATIONS FOR TREATMENT OF EXECUTIVE DEFICITS IN NEUROPSYCHIATRIC DISORDERS SUCH AS ADHD
The powerful beneficial effects of catecholamines on PFC function are highly relevant to the etiology and treatment of attention deficit hyperactivity disorder (ADHD). ADHD is often associated with genetic alterations in catecholamine signaling (Castellanos & Tannock 2002). For example, a polymorphism in the enzyme that synthesizes NE-dopamine β hydroxylase (DBH) leads to reduced NE synthesis, weaker sustained attention (Bellgrove et al. 2006), impaired executive function (Kieling et al. 2007), and poor impulse control (Hess et al. 2008). Most effective treatments for ADHD, including the stimulants amphetamine and methylphenidate, as well as the atypical agent atomoxetine, enhance catecholamine transmission in PFC. Therapeutic doses of stimulants such as methylphenidate increase NE (and to a lesser extent, DA) in the PFC, while producing lesser effects in sub-cortical regions (Berridge et al. 2006). Atomoxetine increases both NE and DA actions in the PFC (Bymaster et al. 2002) but has less effect on striatal DA. Therefore, it is of interest to compare the effects of stimulants such as methylphenidate and nonstimulants such as atomoxetine. Although it is clear that methylphenidate exerts therapeutic effects in both juvenile and adult ADHD (see Chamberlain & Sahakian 2007), atomoxetine has been less studied to date (Faraone et al. 2005). However, some recent evidence supports a therapeutic effect on impulsivity using objective tests in adult ADHD (Chamberlain et al. 2007).
RELATIONSHIPS OF CATECHOLAMINE SIGNALING PATHWAYS TO THE GENETICS OF MENTAL ILLNESS
Recent genetic studies of mental illness have revealed alterations in genes encoding molecules that contribute to glutamate signaling, cortical development, and notably for this article, molecules that serve as brakes on the intracellular stress pathways activated by high levels of monoamines (Owen et al. 2005). These include DISC1 (disrupted in schizophrenia 1), RGS4 (regulator of G-protein signaling 4), and DGK (DAG kinase) (Figure 3). Loss of function of these inhibitory molecules may explain why patients with mental illness are so vulnerable to stress e.g., (Hammen et al. 1992) and why pharmacological treatments that inhibit these pathways are helpful in restoring function.
Figure 3.
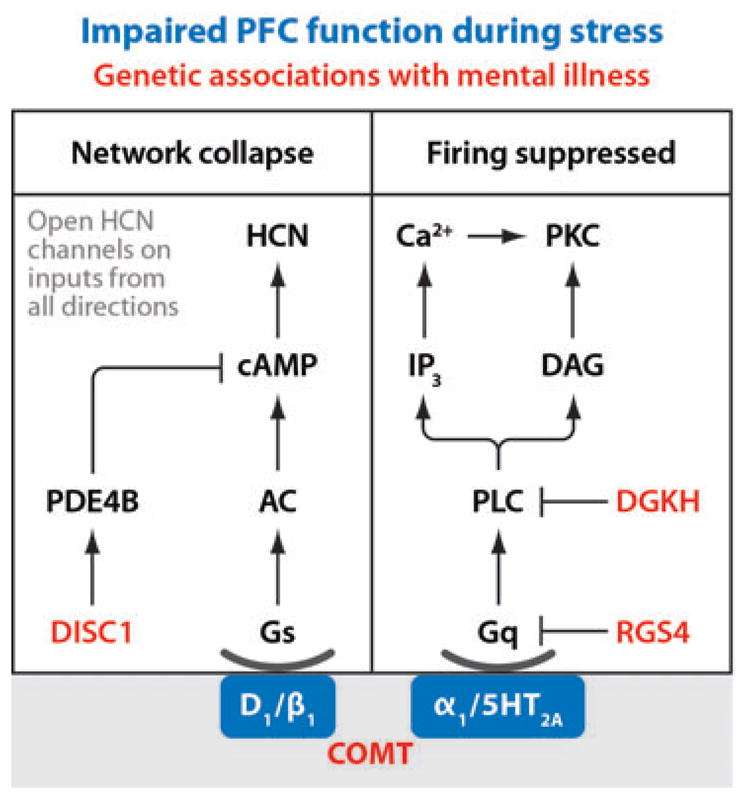
Schematic diagram illustrating some of the important intracellular signaling cascades activated by high levels of catecholamine release in the PFC during stress exposure. Activation of these pathways reduces PFC cell firing and impairs spatial working memory performance. Molecules that are often genetically altered in mental illness are denoted in red; these substances normally inhibit the stress pathways but are weakened by genetic alterations. Arrows, activate; T, inhibit.
DISC1
Several independent genetic association and linkage studies indicate that disc1 is a major susceptibility gene for major mental illnesses, including schizophrenia, bipolar disorder, and major depressive disorder (e.g., Blackwood & Muir 2004, Mackie et al. 2007). Translocations within the gene lead to a loss of DISC1 function. DISC1 normally interacts with phosphodiesterase 4B (PDE4B) to increase the hydrolysis of cAMP under conditions of high cAMP concentrations (Millar et al. 2005, 2007). Thus, impaired DISC1 function would likely lead to overactivation of cAMP-HCN signaling and weakening of PFC network connections (Wang et al. 2007). In this regard, alterations in DISC1 are associated with impairments in cognitive function in both schizophrenia patients and healthy individuals (Blackwood & Muir 2004, Cannon et al. 2005). Furthermore, DISC1 alterations are also implicated in structural changes to the PFC. DISC1 regulates cortical development in animals (Hikida et al. 2007, Ishizuka et al. 2006) and likely in patients with schizophrenia (Cannon et al. 2005).
RGS4
RGS4 belongs to a family of regulatory molecules that act as GTPase activating proteins (GAPs) that drive Gα subunits into the inactive GDP-bound form, thereby decreasing their activity. RGS4 inhibits Gq signaling (i.e., phosphotidyl inositol signaling), and sometimes Gi signaling as well. In a nonbiased gene array study, RGS4 was the most consistently altered transcript in the PFC of schizophrenia patients (Mirnics et al. 2001). A postmortem study found significantly lower RGS4 mRNA and protein levels in the dlPFC of schizophrenia patients compared with healthy controls (Erdely et al. 2006), and RGS4 SNPs predicted poorer performance on clinical ratings scales in a recent study performed in a population of Han-Chinese schizophrenia patients (Lane et al. 2008). Reduced RGS4 in the dlPFC would lead to excessive protein kinase C signaling and impaired PFC cognitive function. These patients also responded best to risperidone (Lane et al. 2008), which reduces PI signaling by blocking extracellular receptors coupled to Gq (see below).
DGKH
DGKH encodes the eta isoform of DGK. A genome-wide association study, which examined more than550,000 genetic polymorphisms in independent samples, found that mutations in DGKH are most robustly linked to bipolar disorder (Baum et al. 2008), and this finding has now been replicated in a separate, large sample (Wellcome 2007). DGKs are a family of lipid kinases that catalyze the conversion of DAG to phosphatidic acid, thereby leading to reductions in DAG. DAG is a necessary co-factor for the activation of many isoforms of protein kinase C. Thus, loss of DGKH activity would lead to increases in protein kinase C signaling. These genetic findings are consistent with numerous lines of evidence linking dysregulation of the protein kinase C signaling cascade to bipolar disorder, and particularly mania (Arnsten & Manji 2008, Friedman et al. 1993, Manji et al. 1999, Manji & Lenox 1999). Reduced DGK activity through mutations in DGKH provides a potential mechanism for PKC dysregulation in bipolar disorder.
Manji and colleagues were the first to recognize that protein kinase C pathways were overactive in bipolar disorder and that treatments for this disorder (lithium, valproate) inhibit protein kinase C signaling (Manji et al. 1999, Manji & Lenox 1999). This hypothesis has been confirmed by a proof-of-concept study in which high doses of tamoxifen, which inhibit protein kinase C, were administered to patients with bipolar disorder (Yildiz et al. 2008, Zarate et al. 2007). Tamoxifen successfully reduced manic symptoms, encouraging the development of other protein kinase C inhibitors. Atypical antipsychotics also target this pathway, blocking the receptors that activate Gq-PI signaling: α-1-AR and 5HT2A receptors (Deutch et al. 1991). Reducing activity of this signaling pathway may ameliorate genetic and/or environmental insults to intracellular stress pathways in the PFC.
In summary, the monoamine neuromodulators have marked effects on PFC function. These influences likely contribute to the fluctuations in cognition and self-control in normal subjects, and more dramatically in those with mental illness. The monoamines represent promising means of modulating PFC dysfunction to ameliorate cognitive deficits in disorders such as ADHD, bipolar disorder, and schizophrenia. Recent advances in understanding the molecular concomitants of catecholamine receptor stimulation are leading to a new era of psychopharmacology based in part on the use of agents affecting the downstream molecular cascades associated with these receptors in a way that may be consistent with recent studies of the genetic underpinning of these neuropsychiatric disorders.
Acknowledgments
We offer thanks to our work colleagues and to Nicola Richmond for her work preparing the manuscript. The Behavioural and Clinical Neuroscience Institute is supported by a joint award from the Medical Research Council and the Wellcome Trust. The Arnsten lab is supported by PHS grants MERIT Award AG06036, P50MH068789, PO1AG030004, and RL1AA017536 as part of U54RR024350, as well as the Kavli Neuroscience Institute at Yale, and a NARSAD Distinguished Investigator Award to AFTA.
- Executive function
the set of processes that help to optimize performance in complex conditions requiring several components of cognitive function
- PFC
prefrontal cortex
- Working memory
a form of short-term memory enabling the construction of representations of the world and the guidance of action
- DA
dopamine
- NE
norepinephrine
- 5-HT
serotonin
- Set
a predisposition to respond in a particular way or to exhibit a form of bias toward a particular type of stimulus
- COMT
catecholamine-o-methyl-transferase
- fMRI
functional magnetic resonance imaging
- mPFC
medial PFC
- Reversal learning
when the stimuli stay the same but which are rewarded and punished are swapped
- Set-shifting
an alteration of choice against the prevailing set
- Temporal discounting of reward (sometimes called intertemporal choice)
the temporal gradient of choice between a small immediate reward (or punishment) and a large, delayed one
- OFC
orbital frontal cortex
- AR
adrenergic receptor
- dlPFC
dorsolateral prefrontal cortex
- HCN channels
hyperpolarization-activated cyclic nucleotide gated cation channels
- DAG
diacyl glycerol
- Response inhibition (inhibitory response control)
a hypothetical process that is recruited when ongoing responding must be suppressed
- go/no-go (discrimination)
when it is correct on go trials to respond and on no-go trials to refrain from responding
- 6-OHDA
6-hydroxydopamine
- Extradimensional shift
when the shifted response is from one perceptual dimension or sensory modality to another
- Stop-signal reaction time
measured in a task enabling the time spent in stopping or canceling an initiated motor response to be estimated
- Intradimensional shift
when the shifted response is from one set of exemplars to another, in the same perceptual dimension or sensory modality
- Set-formation
a product of learning that leads to a predisposition or set
- BOLD
blood-oxygen-level-dependent
- ADHD
attention deficit hyperactivity disorder
- DISC1
disrupted in schizophrenia 1
- RGS4
regulator of G-protein signaling 4
- DGK
DAG kinase
Footnotes
DISCLOSURE STATEMENT
AFTA and Yale University have license agreements with Shire Pharmaceuticals for the development of guanfacine for the treatment of ADHD and related disorders, and with Marinus Pharmaceuticals for the development of chelerythrine for the treatment of bipolar disorder and related conditions. T.W.R. discloses consultancy for Cambridge Cognition, E. Lilly, Pfizer, Wyeth, and Roche.
Contributor Information
T.W. Robbins, Email: twr2@cam.ac.uk.
A.F.T. Arnsten, Email: amy.arnsten@yale.edu.
LITERATURE CITED
- Amat J, Baratta MV, Paul E, Bland ST, Watkins LR, Maier SF. Medial prefrontal cortex determines how stressor controllability affects behavior and dorsal raphe nucleus. Nat Neurosci. 2005;8:365–71. doi: 10.1038/nn1399. [DOI] [PubMed] [Google Scholar]
- Apud JA, Mattay V, Chen J, Kolachana BS, Callicott JH, et al. Tolcapone improves cognition and cortical information processing in normal human subjects. Neuropsychopharmacology. 2007;32:1011–20. doi: 10.1038/sj.npp.1301227. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT. Through the looking glass: differential noradrenergic modulation of prefrontal cortical function. Neural Plasticity. 2000;7:133–46. doi: 10.1155/NP.2000.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AFT, Goldman-Rakic PS. Selective prefrontal cortical projections to the region of the locus coeruleus and raphe nuclei in the rhesus monkey. Brain Res. 1984;306:9–18. doi: 10.1016/0006-8993(84)90351-2. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT, Manji HK. Mania: a rational neurobiology. Fut Neurol. 2008;3 In press. [Google Scholar]
- Arnsten AFT, Vijayraghavan S, Wang M, Gamo NJ, Paspalas CD. Dopamine’s influence on prefrontal cortical cognition: actions and circuits in behaving primates. In: Bjorklund A, Dunnett S, Iversen L, Iversen S, editors. Dopamine Handbook. Oxford, UK: Oxford Univ. Press; 2009. In press. [Google Scholar]
- Aron AR, Robbins TW, Poldrack RA. Inhibition and the right inferior frontal cortex. Trends Cogn Sci. 2004;8:170–77. doi: 10.1016/j.tics.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD. An integrative theory of locus coeruleus-norepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–50. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Rajkowski J, Cohen J. Locus coeruleus and regulation of behavioral flexibility and attention. Prog Brain Res. 2000;126:165–82. doi: 10.1016/S0079-6123(00)26013-5. [DOI] [PubMed] [Google Scholar]
- Baddeley AD. Working memory. Oxford, UK: Oxford Univ. Press; 1986. [Google Scholar]
- Bari A, Eagle DM, Mar AC, Robinson ES, Robbins TW. Dissociable effects of noradrenaline dopamine and serotonin uptake blockers on stop task performance. Psychopharmacology (Berl) 2009 doi: 10.1007/s00213-009-1537-0. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum AE, Akula N, Cabanero M, Cardona I, Corona W, et al. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2008;13:197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellgrove MA, Hawi Z, Gill M, Robertson IH. The cognitive genetics of attention deficit hyperactivity disorder (ADHD): sustained attention as a candidate phenotype. Cortex. 2006;42:838–45. doi: 10.1016/s0010-9452(08)70426-x. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Devilbiss DM, Andrzejewski ME, Arnsten AF, Kelley AE, et al. Methylphenidate preferentially increases catecholamine neurotransmission within the prefrontal cortex at low doses that enhance cognitive function. Biol Psychiatry. 2006;60:1111–20. doi: 10.1016/j.biopsych.2006.04.022. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Bilder RM, Volavka J, Lachman HM, Grace AA. The catechol-O-methyltransferase polymorphism: relations to the tonic-phasic dopamine hypothesis and neuropsychiatric phenotypes. Neuropsychopharmacology. 2004;29:1943–61. doi: 10.1038/sj.npp.1300542. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Yuan PX, Wang M, Vijayraghavan S, Bloom AK, et al. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science. 2004;306:882–84. doi: 10.1126/science.1100021. [DOI] [PubMed] [Google Scholar]
- Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–24. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonette GB, Martins GJ, Franz TM, Harper ES, Schoenbaum G, Powell EM. Double dissociation of the effects of medial and orbital prefrontal cortical lesions on attentional and affective shifts in mice. J Neurosci. 2008;28:11124–30. doi: 10.1523/JNEUROSCI.2820-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood DH, Muir WJ. Clinical phenotypes associated with DISCI, a candidate gene for schizophrenia. Neurotox Res. 2004;6:35–41. doi: 10.1007/BF03033294. [DOI] [PubMed] [Google Scholar]
- Blondeau C, Dellu-Hagedorn F. Dimensional analysis of ADHD subtypes in rats. Biol Psychiatry. 2007;61:1340–50. doi: 10.1016/j.biopsych.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Brennan AR, Dolinsky B, Vu MA, Stanley M, Yeckel MF, Arnsten AF. Blockade of IP3-mediated SK channel signaling in the rat medial prefrontal cortex improves spatial working memory. Learn Mem. 2008;15:93–96. doi: 10.1101/lm.767408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozoski T, Brown RM, Rosvold HE, Goldman PS. Cognitive deficit caused by regional depletion of dopamine in prefrontal cortex of rhesus monkey. Science. 1979;205:929–31. doi: 10.1126/science.112679. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27:699–711. doi: 10.1016/S0893-133X(02)00346-9. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Hennah W, van Erp TG, Thompson PM, Lonnqvist J, et al. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry. 2005;62:1205–13. doi: 10.1001/archpsyc.62.11.1205. [DOI] [PubMed] [Google Scholar]
- Castellanos FX, Tannock R. Neuroscience of attention-deficit/hyperactivity disorder: the search for endophenotypes. Nat Rev Neurosci. 2002;3:617–28. doi: 10.1038/nrn896. [DOI] [PubMed] [Google Scholar]
- Ceaser AE, Goldberg TE, Egan MF, McMahon RP, Weinberger DR, Gold JM. Set-shifting ability and schizophrenia: a marker of clinical illness or an intermediate phenotype? Biol Psychiatry. 2008;64:782–88. doi: 10.1016/j.biopsych.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SR, Del Campo N, Dowson J, Muller U, Clark L, et al. Atomoxetine improved response inhibition in adults with attention deficit/hyperactivity disorder. Biol Psychiatry. 2007;62:977–84. doi: 10.1016/j.biopsych.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Chamberlain SR, Fineberg NA, Blackwell AD, Robbins TW, Sahakian BJ. Motor inhibition and cognitive flexibility in obsessive-compulsive disorder and trichotillomania. Am J Psychiatry. 2006a;163:1282–84. doi: 10.1176/ajp.2006.163.7.1282. [DOI] [PubMed] [Google Scholar]
- Chamberlain SR, Hampshire A, Muller U, Rubia K, Campo ND, et al. Atomoxetine modulates right inferior frontal activation during inhibitory control: a pharmacological functional magnetic resonance imaging study. Biol Psychiatry. 2009 doi: 10.1016/j.biopsych.2008.10.014. In press. [DOI] [PubMed] [Google Scholar]
- Chamberlain SR, Muller U, Blackwell AD, Clark L, Robbins TW, Sahakian BJ. Neurochemical modulation of response inhibition and probabilistic learning in humans. Science. 2006b;311:861–63. doi: 10.1126/science.1121218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SR, Sahakian BJ. The neuropsychiatry of impulsivity. Curr Opin Psychiatry. 2007;20:255–61. doi: 10.1097/YCO.0b013e3280ba4989. [DOI] [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW. Dissociable contributions of the orbitofrontal and infralimbic cortex to Pavlovian autoshaping and discrimination reversal learning: further evidence for the functional heterogeneity of the rodent frontal cortex. J Neurosci. 2003;23:8771–80. doi: 10.1523/JNEUROSCI.23-25-08771.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW. Dopaminergic modulation of visual attention and working memory in the rodent prefrontal cortex. Neuropsychopharmacology. 2004;29:1628–36. doi: 10.1038/sj.npp.1300490. [DOI] [PubMed] [Google Scholar]
- Clarke HF, Dalley JW, Crofts HS, Robbins TW, Roberts AC. Cognitive inflexibility after prefrontal serotonin depletion. Science. 2004;304:878–80. doi: 10.1126/science.1094987. [DOI] [PubMed] [Google Scholar]
- Clarke HF, Walker SC, Crofts HS, Dalley JW, Robbins TW, Roberts AC. Prefrontal serotonin depletion affects reversal learning but not attentional set shifting. J Neurosci. 2005;25:532–38. doi: 10.1523/JNEUROSCI.3690-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke HF, Walker SC, Dalley JW, Robbins TW, Roberts AC. Cognitive inflexibility after prefrontal serotonin depletion is behaviorally and neurochemically specific. Cereb Cortex. 2007;17:18–27. doi: 10.1093/cercor/bhj120. [DOI] [PubMed] [Google Scholar]
- Collins P, Roberts AC, Dias R, Everitt BJ, Robbins TW. Perseveration and strategy in a novel spatial self-ordered sequencing task for nonhuman primates: effects of excitotoxic lesions and dopamine depletions of the prefrontal cortex. J Cogn Neurosci. 1998;10:332–54. doi: 10.1162/089892998562771. [DOI] [PubMed] [Google Scholar]
- Cools R, Roberts AC, Robbins TW. Serotoninergic regulation of emotional and behavioural control processes. Trends Cogn Sci. 2008a;12:31–40. doi: 10.1016/j.tics.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Cools R, Robinson OJ, Sahakian B. Acute tryptophan depletion in healthy volunteers enhances punishment prediction but does not affect reward prediction. Neuropsychopharmacology. 2008b;33:2291–99. doi: 10.1038/sj.npp.1301598. [DOI] [PubMed] [Google Scholar]
- Cools R, Sheridan M, Jacobs E, D’Esposito M. Impulsive personality predicts dopamine-dependent changes in frontostriatal activity during component processes of working memory. J Neurosci. 2007;27:5506–14. doi: 10.1523/JNEUROSCI.0601-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofts HS, Dalley JW, Collins P, Van Denderen JC, Everitt BJ, et al. Differential effects of 6-OHDA lesions of the frontal cortex and caudate nucleus on the ability to acquire an attentional set. Cereb Cortex. 2001;11:1015–26. doi: 10.1093/cercor/11.11.1015. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Theobald DE, Eagle DM, Passetti F, Robbins TW. Deficits in impulse control associated with tonically-elevated serotonergic function in rat prefrontal cortex. Neuropsychopharmacology. 2002;26:716–28. doi: 10.1016/S0893-133X(01)00412-2. [DOI] [PubMed] [Google Scholar]
- Dazzi L, Serra M, Spiga F, Pisu MG, Jentsch JD, Biggio G. Prevention of the stress-induced increase in frontal cortical dopamine efflux of freely moving rats by long-term treatment with antidepressant drugs. Eur Neuropsychopharmacol. 2001;11:343–49. doi: 10.1016/s0924-977x(01)00105-5. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Moghaddam B, Innis RB, Krystal JH, Aghajanian GK, et al. Mechanisms of action of atypical antipsychotic drugs. Implications for novel therapeutic strategies for schizophrenia. Schizophr Res. 1991;4:121–56. doi: 10.1016/0920-9964(91)90030-u. [DOI] [PubMed] [Google Scholar]
- Dias R, Robbins TW, Roberts AC. Dissociation in prefrontal cortex of affective and attentional shifts. Nature. 1996;380:69–72. doi: 10.1038/380069a0. [DOI] [PubMed] [Google Scholar]
- Dodds CM, Muller U, Clark L, van Loon A, Cools R, Robbins TW. Methylphenidate has differential effects on blood oxygenation level-dependent signal related to cognitive subprocesses of reversal learning. J Neurosci. 2008;28:5976–82. doi: 10.1523/JNEUROSCI.1153-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan J, Seitz RJ, Kolodny J, Bor D, Herzog H, et al. A neural basis for general intelligence. Science. 2000;289:457–60. doi: 10.1126/science.289.5478.457. [DOI] [PubMed] [Google Scholar]
- Durstewictz D, Seamans JK, Sejnowski T. Dopamine-mediated stabilization of delay-period activity in a network model of prefrontal cortex. J Neurophysiology. 2000;19:2807–22. doi: 10.1152/jn.2000.83.3.1733. [DOI] [PubMed] [Google Scholar]
- Eagle DM, Bari A, Robbins TW. The neuropsychopharmacology of action inhibition: cross-species translation of the stop-signal and go/no-go tasks. Psychopharmacology (Berl) 2008;199:439–56. doi: 10.1007/s00213-008-1127-6. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Kolachana BS, Callicott JH, Mazzanti CM, et al. Effect of COMT val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci USA. 2001;98:6917–22. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdely HA, Tamminga CA, Roberts RC, Vogel MW. Regional alterations in RGS4 protein in schizophrenia. Synapse. 2006;59:472–79. doi: 10.1002/syn.20265. [DOI] [PubMed] [Google Scholar]
- Evers EA, Cools R, Clark L, Van Der Veen FM, Jolles J, et al. Serotonergic modulation of prefrontal cortex during negative feedback in probabilistic reversal learning. Neuropsychopharmacology. 2005;30:1138–47. doi: 10.1038/sj.npp.1300663. [DOI] [PubMed] [Google Scholar]
- Evers EA, Van Der Veen FM, van Deursen JA, Schmitt JA, Deutz NE, Jolles J. The effect of acute tryptophan depletion on the BOLD response during performance monitoring and response inhibition in healthy male volunteers. Psychopharmacology (Berl) 2006;187:200–8. doi: 10.1007/s00213-006-0411-6. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Spencer T, Michelson D, Adler L, et al. Atomoxetine and stroop task performance in adult attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2005;15:664–70. doi: 10.1089/cap.2005.15.664. [DOI] [PubMed] [Google Scholar]
- Feenstra MG, Vogel M, Botterblom MH, Joosten RN, de Bruin JP. Dopamine and noradrenaline efflux in the rat prefrontal cortex after classical aversive conditioning to an auditory cue. Eur J Neurosci. 2001;13:1051–54. doi: 10.1046/j.0953-816x.2001.01471.x. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Magyar O. Mesocortical dopamine modulation of executive functions: beyond working memory. Psychopharmacology (Berl) 2006;188:567–85. doi: 10.1007/s00213-006-0404-5. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Magyar O, Ghods-Sharifi S, Vexelman C, Tse MT. Multiple dopamine receptor subtypes in the medial prefrontal cortex of the rat regulate set-shifting. Neuropsychopharmacology. 2006;31:297–309. doi: 10.1038/sj.npp.1300825. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Phillips AG. Delay-dependent modulation of memory retrieval by infusion of a dopamine D1 agonist into the rat medial prefrontal cortex. Behav Neurosci. 2001;115:934–39. [PubMed] [Google Scholar]
- Frank MJ, Moustafa AA, Haughey HM, Curran T, Hutchison KE. Genetic triple dissociation reveals multiple roles for dopamine in reinforcement learning. Proc Natl Acad Sci USA. 2007;104:16311–16. doi: 10.1073/pnas.0706111104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman E, Hoau YW, Levinson D, Connell TA, Singh H. Altered platelet protein kinase C activity in bipolar affective disorder, manic episode. Biol Psychiatry. 1993;33:520–25. doi: 10.1016/0006-3223(93)90006-y. [DOI] [PubMed] [Google Scholar]
- Gibbs SE, D’Esposito M. A functional magnetic resonance imaging study of the effects of pergolide, a dopamine receptor agonist, on component processes of working memory. Neuroscience. 2006;139:359–71. doi: 10.1016/j.neuroscience.2005.11.055. [DOI] [PubMed] [Google Scholar]
- Gilbert SJ, Burgess PW. Executive function. Curr Biol. 2008;18:R110–14. doi: 10.1016/j.cub.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Giorgi O, Lecca D, Piras G, Driscoll P, Corda MG. Dissociation between mesocortical dopamine release and fear-related behaviours in two psychogenetically selected lines of rats that differ in coping strategies to aversive conditions. Eur J Neurosci. 2003;17:2716–26. doi: 10.1046/j.1460-9568.2003.02689.x. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Circuitry of primate prefrontal cortex and the regulation of behavior by representational memory. In: Plum F, Mountcastle V, editors. Handbook of Physiology. Bethesda, MD: Am. Physiol. Soc; 1987. pp. 373–417. [Google Scholar]
- Goldstein LE, Rasmusson AM, Bunney SB, Roth RH. Role of the amygdala in the coordination of behavioral, neuroendocrine and prefrontal cortical monoamine responses to psychological stress in the rat. J Neurosci. 1996;16:4787–98. doi: 10.1523/JNEUROSCI.16-15-04787.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granon S, Passetti F, Thomas KL, Dalley JW, Everitt BJ, Robbins TW. Enhanced and impaired attentional performance after infusion of D1 dopaminergic receptor agents into rat prefrontal cortex. J Neurosci. 2000;20:1208–15. doi: 10.1523/JNEUROSCI.20-03-01208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston AM, Fitzpatrick JS, Yeckel MF. MGluR-mediated calcium waves that invade the soma regulate firing in layer V medial prefrontal cortical pyramidal neurons. Cereb Cortex. 2008;18:407–23. doi: 10.1093/cercor/bhm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammen C, Davila J, Brown G, Ellicott A, Gitlin M. Psychiatric history and stress: predictors of severity of unipolar depression. J Abnorm Psychol. 1992;101:45–52. doi: 10.1037//0021-843x.101.1.45. [DOI] [PubMed] [Google Scholar]
- Hampshire A, Owen AM. Fractionating attentional control using event-related fMRI. Cereb Cortex. 2006;16:1679–89. doi: 10.1093/cercor/bhj116. [DOI] [PubMed] [Google Scholar]
- Hess C, Reif A, Strobel A, Boreatti-Hümmer A, Heine M, et al. A functional dopamine-beta-hydroxylase gene promoter polymorphism is associated with impulsive personality styles, but not with affective disorders. J Neural Transm. 2008 doi: 10.1007/s00702-008-0138-0. In press. [DOI] [PubMed] [Google Scholar]
- Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci USA. 2007;104:14501–6. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcott PK, Quinn JJ, Taylor JR. Bidirectional modulation of goal-directed actions by prefrontal cortical dopamine. Cereb Cortex. 2007;17:2820–27. doi: 10.1093/cercor/bhm010. [DOI] [PubMed] [Google Scholar]
- Horvath TL, Peyron C, Diano S, Ivanov A, Aston-Jones G, et al. Hypocretin (orexin) activation and synaptic innervation of the locus coeruleus noradrenergic system. J Comparative Neurol. 1999;415:145–59. [PubMed] [Google Scholar]
- Ishizuka K, Paek M, Kamiya A, Sawa A. A review of disrupted-in-schizophrenia-1 (DISC1): neurodevelopment, cognition, and mental conditions. Biol Psychiatry. 2006;59:1189–97. doi: 10.1016/j.biopsych.2006.03.065. [DOI] [PubMed] [Google Scholar]
- Jodo E, Chiang C, Aston-Jones G. Potent excitatory influence of prefrontal cortex activity on noradrenergic locus coeruleus neurons. Neuroscience. 1998;83:63–79. doi: 10.1016/s0306-4522(97)00372-2. [DOI] [PubMed] [Google Scholar]
- Kheramin S, Body S, Ho MY, Velazquez-Martinez DN, Bradshaw CM, et al. Effects of orbital prefrontal cortex dopamine depletion on inter-temporal choice: a quantitative analysis. Psychopharmacology (Berl) 2004;175:206–14. doi: 10.1007/s00213-004-1813-y. [DOI] [PubMed] [Google Scholar]
- Kieling C, Genro JP, Hutz MH, Rohde LA. The -1021 C/T DBH polymorphism is associated with neuropsychological performance among children and adolescents with ADHD. Am J Med Genet B Neuropsychiatr Genet. 2007 doi: 10.1002/ajmg.b.30636. In press. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Olausson P, Horst NK, Taylor JR, Aghajanian GK. Hypocretin and nicotine excite the same thalamocortical synapses in prefrontal cortex: correlation with improved attention in rat. J Neurosci. 2005;25:5225–29. doi: 10.1523/JNEUROSCI.0719-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane HY, Liu YC, Huang CL, Chang YC, Wu PL, et al. RGS4 polymorphisms predict clinical manifestations and responses to risperidone treatment in patients with schizophrenia. J Clin Psychopharmacol. 2008;28:64–68. doi: 10.1097/jcp.0b013e3181603f5a. [DOI] [PubMed] [Google Scholar]
- Lapiz MD, Bondi CO, Morilak DA. Chronic treatment with desipramine improves cognitive performance of rats in an attentional set-shifting test. Neuropsychopharmacology. 2007;32:1000–10. doi: 10.1038/sj.npp.1301235. [DOI] [PubMed] [Google Scholar]
- Lapiz MD, Morilak DA. Noradrenergic modulation of cognitive function in rat medial prefrontal cortex as measured by attentional set shifting capability. Neuroscience. 2006;137:1039–49. doi: 10.1016/j.neuroscience.2005.09.031. [DOI] [PubMed] [Google Scholar]
- Li B-M, Mao Z-M, Wang M, Mei Z-T. Alpha-2 adrenergic modulation of prefrontal cortical neuronal activity related to spatial working memory in monkeys. Neuropsychopharmacology. 1999;21:601–10. doi: 10.1016/S0893-133X(99)00070-6. [DOI] [PubMed] [Google Scholar]
- Li B-M, Mei Z-T. Delayed response deficit induced by local injection of the alpha-2 adrenergic antagonist yohimbine into the dorsolateral prefrontal cortex in young adult monkeys. Behav Neural Biol. 1994;62:134–39. doi: 10.1016/s0163-1047(05)80034-2. [DOI] [PubMed] [Google Scholar]
- Ma CL, Qi XL, Peng JY, Li BM. Selective deficit in no-go performance induced by blockade of prefrontal cortical alpha 2-adrenoceptors in monkeys. Neuroreport. 2003;14:1013–16. doi: 10.1097/01.wnr.0000070831.57864.7b. [DOI] [PubMed] [Google Scholar]
- Mackie S, Millar JK, Porteous DJ. Role of DISC1 in neural development and schizophrenia. Curr Opin Neurobiol. 2007;17:95–102. doi: 10.1016/j.conb.2007.01.007. [DOI] [PubMed] [Google Scholar]
- Manji HK, Bebchuk JM, Moore GJ, Glitz D, Hasanat KA, Chen G. Modulation of CNS signal transduction pathways and gene expression by mood-stabilizing agents: therapeutic implications. J Clin Psychiatry. 1999;60:27–39. discussion 40–41, 113–16. [PubMed] [Google Scholar]
- Manji HK, Lenox RH. Protein kinase C signaling in the brain: molecular transduction of mood stabilization in the treatment of manic-depressive illness. Biol Psychiatry. 1999;46:1328–51. doi: 10.1016/s0006-3223(99)00235-8. [DOI] [PubMed] [Google Scholar]
- McAlonan K, Brown VJ. Orbital prefrontal cortex mediates reversal learning and not attentional set shifting in the rat. Behav Brain Res. 2003;146:97–103. doi: 10.1016/j.bbr.2003.09.019. [DOI] [PubMed] [Google Scholar]
- McGaughy J, Ross RS, Eichenbaum H. Noradrenergic, but not cholinergic, deafferentation of prefrontal cortex impairs attentional set-shifting. Neuroscience. 2008;153:63–71. doi: 10.1016/j.neuroscience.2008.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, Mackie S, Clapcote SJ, Murdoch H, Pickard BS, et al. Disrupted in schizophrenia 1 and phosphodiesterase 4B: towards an understanding of psychiatric illness. J Physiol. 2007;584:401–5. doi: 10.1113/jphysiol.2007.140210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, Pickard BS, Mackie S, James RS, Christie S, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–91. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- Milstein JA, Lehmann O, Theobald DE, Dalley JW, Robbins TW. Selective depletion of cortical noradrenaline by anti-dopamine beta-hydroxylase-saporin impairs attentional function and enhances the effects of guanfacine in the rat. Psychopharmacology (Berl) 2007;190:51–63. doi: 10.1007/s00213-006-0594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P. Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry. 2001;6:293–301. doi: 10.1038/sj.mp.4000866. [DOI] [PubMed] [Google Scholar]
- Moore RY, Abrahamson EA, Van Den Pol A. The hypocretin neuron system: an arousal system in the human brain. Arch Ital Biol. 2001;139:195–205. [PubMed] [Google Scholar]
- Newman LA, Darling J, McGaughy J. Atomoxetine reverses attentional deficits produced by noradrenergic deafferentation of medial prefrontal cortex. Psychopharmacology (Berl) 2008;200:39–50. doi: 10.1007/s00213-008-1097-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan KA, Bilder RM, Lachman HM, Volavka J. Catechol O-methyltransferase val158Met polymorphism in schizophrenia: differential effects of val and met alleles on cognitive stability and flexibility. Am J Psychiatry. 2004;161:359–61. doi: 10.1176/appi.ajp.161.2.359. [DOI] [PubMed] [Google Scholar]
- Owen MJ, Craddock N, O’Donovan MC. Schizophrenia: genes at last? Trends Genet. 2005;21:518–25. doi: 10.1016/j.tig.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Pantelis C, Barber FZ, Barnes TR, Nelson HE, Owen AM, Robbins TW. Comparison of set-shifting ability in patients with chronic schizophrenia and frontal lobe damage. Schizophr Res. 1999;37:251–70. doi: 10.1016/s0920-9964(98)00156-x. [DOI] [PubMed] [Google Scholar]
- Phillips AG, Ahn S, Floresco SB. Magnitude of dopamine release in medial prefrontal cortex predicts accuracy of memory on a delayed response task. J Neurosci. 2004;24:547–53. doi: 10.1523/JNEUROSCI.4653-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragozzino ME. The contribution of the medial prefrontal cortex, orbitofrontal cortex, and dorsomedial striatum to behavioral flexibility. Ann N Y Acad Sci. 2007;1121:355–75. doi: 10.1196/annals.1401.013. [DOI] [PubMed] [Google Scholar]
- Ramos B, Colgan L, Nou E, Ovadia S, Wilson SR, Arnsten AFT. The beta-1 adrenergic antagonist, betaxolol, improves working memory performance in rats and monkeys. Biol Psychiatry. 2005;58:894–900. doi: 10.1016/j.biopsych.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS. Isodirectional tuning of adjacent interneurons and pyramidal cells during working memory: evidence for microcolumnar organization in PFC. J Neurophysiol. 1999;81:1903–16. doi: 10.1152/jn.1999.81.4.1903. [DOI] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA(A) blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–94. doi: 10.1523/JNEUROSCI.20-01-00485.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW. Dissociating executive functions of the prefrontal cortex. Philos Trans R Soc London B Biol Sci. 1996;351:1463–70. doi: 10.1098/rstb.1996.0131. discussion 70–71. [DOI] [PubMed] [Google Scholar]
- Robbins TW. Controlling stress: how the brain protects itself from depression. Nat Neurosci. 2005;8:261–62. doi: 10.1038/nn0305-261. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ. Central norepinephrine neurons and behavior. In: Bloom FE, Kupfer D, editors. Psychopharmacology—4th Generation of Progress. New York: Raven; 1995. pp. 363–72. [Google Scholar]
- Robbins TW, Roberts AC. Differential regulation of fronto-executive function by the monoamines and acetylcholine. Cereb Cortex. 2007;17(Suppl 1):i151–60. doi: 10.1093/cercor/bhm066. [DOI] [PubMed] [Google Scholar]
- Roberts AC, De Salvia MA, Wilkinson LS, Collins P, Muir JL, et al. 6-hydroxydopamine lesions of the prefrontal cortex in monkeys enhance performance on an analog of the Wisconsin Card Sort Test: possible interactions with subcortical dopamine. J Neurosci. 1994;14:2531–44. doi: 10.1523/JNEUROSCI.14-05-02531.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson ES, Dalley JW, Theobald DE, Glennon JC, Pezze MA, et al. Opposing roles for 5-HT2A and 5-HT2C receptors in the nucleus accumbens on inhibitory response control in the 5-choice serial reaction time task. Neuropsychopharmacology. 2008;33:2398–406. doi: 10.1038/sj.npp.1301636. [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Carboni S. Noradrenaline and dopamine elevations in the rat prefrontal cortex in spatial working memory. J Neurosci. 2005;25:2322–29. doi: 10.1523/JNEUROSCI.3038-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubia K, Lee F, Cleare AJ, Tunstall N, Fu CH, et al. Tryptophan depletion reduces right inferior prefrontal activation during response inhibition in fast, event-related fMRI. Psychopharmacology (Berl) 2005;179:791–803. doi: 10.1007/s00213-004-2116-z. [DOI] [PubMed] [Google Scholar]
- Runyan JD, Dash PK. Distinct prefrontal molecular mechanisms for information storage lasting seconds versus minutes. Learn Mem. 2005;12:232–38. doi: 10.1101/lm.92405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runyan JD, Moore AN, Dash PK. A role for prefrontal calcium-sensitive protein phosphatase and kinase activities in working memory. Learn Mem. 2005;12:103–10. doi: 10.1101/lm.89405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24:726–31. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- Sara SJ, Herve-Minvielle A. Inhibitory influence of frontal cortex on locus coeruleus. Proc Natl Acad Sci USA. 1995;92:6032–36. doi: 10.1073/pnas.92.13.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Hasselmo ME, Bruno JP, Givens B. Unraveling the attentional functions of cortical cholinergic inputs: interactions between signal-driven and cognitive modulation of signal detection. Brain Res Brain Res Rev. 2005;48:98–111. doi: 10.1016/j.brainresrev.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Goldman-Rakic PS. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science. 1991;251:947–50. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- Sawaguchi T, Goldman-Rakic PS. The role of D1-dopamine receptors in working memory: local injections of dopamine antagonists into the prefrontal cortex of rhesus monkeys performing an oculomotor delayed response task. J Neurophysiol. 1994;71:515–28. doi: 10.1152/jn.1994.71.2.515. [DOI] [PubMed] [Google Scholar]
- Schultz W. The phasic reward signal of primate dopamine neurons. Adv Pharmacol. 1998;42:686–90. doi: 10.1016/s1054-3589(08)60841-8. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci USA. 2001;98:301–6. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Robbins TW. Dopamine modulation of the prefrontal cortex and cognitive function. In: Neve K, editor. Dopamine Receptors. Totowa, NJ: Humana; 2009. In press. [Google Scholar]
- Stefani MR, Moghaddam B. Rule learning and reward contingency are associated with dissociable patterns of dopamine activation in the rat prefrontal cortex, nucleus accumbens, and dorsal striatum. J Neurosci. 2006;26:8810–18. doi: 10.1523/JNEUROSCI.1656-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait DS, Brown VJ, Farovik A, Theobald DE, Dalley JW, Robbins TW. Lesions of the dorsal noradrenergic bundle impair attentional set-shifting in the rat. Eur J Neurosci. 2007;25:3719–24. doi: 10.1111/j.1460-9568.2007.05612.x. [DOI] [PubMed] [Google Scholar]
- Talbot PS, Cooper SJ. Anterior cingulate and subgenual prefrontal blood flow changes following tryptophan depletion in healthy males. Neuropsychopharmacology. 2006;31:1757–67. doi: 10.1038/sj.npp.1301022. [DOI] [PubMed] [Google Scholar]
- Thierry AM, Tassin JP, Blanc G, Glowinski J. Selective activation of mesocortical DA system by stress. Nature. 1976;263:242–44. doi: 10.1038/263242a0. [DOI] [PubMed] [Google Scholar]
- Tunbridge EM, Bannerman DM, Sharp T, Harrison PJ. Catechol-o-methyltransferase inhibition improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J Neurosci. 2004;24:5331–35. doi: 10.1523/JNEUROSCI.1124-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DC, Robbins TW, Clark L, Aron AR, Dowson J, Sahakian BJ. Cognitive enhancing effects of modafinil in healthy volunteers. Psychopharmacology. 2003;165:260–69. doi: 10.1007/s00213-002-1250-8. [DOI] [PubMed] [Google Scholar]
- Van Der Meulen JA, Joosten RN, de Bruin JP, Feenstra MG. Dopamine and noradrenaline efflux in the medial prefrontal cortex during serial reversals and extinction of instrumental goal-directed behavior. Cereb Cortex. 2007;17:1444–53. doi: 10.1093/cercor/bhl057. [DOI] [PubMed] [Google Scholar]
- Vijayraghavan S, Wang M, Birnbaum SG, Bruce CJ, Williams GV, Arnsten AFT. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10:376–84. doi: 10.1038/nn1846. [DOI] [PubMed] [Google Scholar]
- Vittoz NM, Schmeichel B, Berridge CW. Hypocretin/orexin preferentially activates caudomedial ventral tegmental area dopamine neurons. Eur J Neurosci. 2008;28:1629–40. doi: 10.1111/j.1460-9568.2008.06453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SC, Robbins TW, Roberts AC. Differential contributions of dopamine and serotonin to or-bitofrontal cortex function in the marmoset. Cereb Cortex. 2009 doi: 10.1093/cercor/bhn136. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Ramos B, Paspalas C, Shu Y, Simen A, et al. Alpha2A-adrenoceptor stimulation strengthens working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Kodama T, Hikosaka K. Increase of extracellular dopamine in primate prefrontal cortex during a working memory task. J Neurophysiol. 1997;78:2795–98. doi: 10.1152/jn.1997.78.5.2795. [DOI] [PubMed] [Google Scholar]
- Wellcome T. Genome-wide association study of 14000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS. Blockade of dopamine D1 receptors enhances memory fields of prefrontal neurons in primate cerebral cortex. Nature. 1995;376:572–75. doi: 10.1038/376572a0. [DOI] [PubMed] [Google Scholar]
- Williams-Gray CH, Hampshire A, Robbins TW, Owen AM, Barker RA. Catechol O-methyltransferase val158met genotype influences frontoparietal activity during planning in patients with Parkinson’s disease. J Neurosci. 2007;27:4832–38. doi: 10.1523/JNEUROSCI.0774-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley CA, Theobald DE, Dalley JW, Cardinal RN, Robbins TW. Double dissociation between serotonergic and dopaminergic modulation of medial prefrontal and orbitofrontal cortex during a test of impulsive choice. Cereb Cortex. 2006;16:106–14. doi: 10.1093/cercor/bhi088. [DOI] [PubMed] [Google Scholar]
- Yildiz A, Guleryuz S, Ankerst DP, Ongur D, Renshaw PF. Protein kinase C inhibition in the treatment of mania: a double-blind, placebo-controlled trial of tamoxifen. Arch Gen Psychiatry. 2008;65:255–63. doi: 10.1001/archgenpsychiatry.2007.43. [DOI] [PubMed] [Google Scholar]
- Young CE, Yang CR. Dopamine D1/D5 receptor modulates state-dependent switching of somadendritic Ca2+ potentials via differential protein kinase A and C activation in rat prefrontal cortical neurons. J Neurosci. 2004;24:8–23. doi: 10.1523/JNEUROSCI.1650-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahrt J, Taylor JR, Mathew RG, Arnsten AF. Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci. 1997;17:8528–35. doi: 10.1523/JNEUROSCI.17-21-08528.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Quiroz J, Jolkovsky L, et al. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 2007;9:561–70. doi: 10.1111/j.1399-5618.2007.00530.x. [DOI] [PubMed] [Google Scholar]