

Abstract
Rationale
Calcification of heart valve structures is the most common form of valvular disease and is characterized by the appearance of bone-like phenotypes within affected structures. Despite the clinical significance, the underlying etiology of disease onset and progression is largely unknown and valve replacement remains the most effective treatment. The SRY-related transcription factor Sox9 is expressed in developing and mature heart valves, and its function is required for expression of cartilage-associated proteins, similar to its role in chondrogenesis. In addition to cartilage-associated defects, mice with reduced sox9 function develop skeletal bone prematurely, however the ability of sox9 deficiency to promote ectopic osteogenic phenotypes in heart valves has not been examined.
Objective
This study aims to determine the role of Sox9 in maintaining connective tissue homeostasis in mature heart valves using in vivo and in vitro approaches.
Methods and Results
Using histological and molecular analyses we report that Sox9fl/+;Col2a1-cre mice develop calcific lesions in heart valve leaflets from 3 months of age associated with increased expression of bone-related genes and activation of inflammation and matrix remodeling processes. Consistently, ectopic calcification is also observed following direct knockdown of Sox9 in heart valves in vitro. Further, we show that retinoic acid treatment in mature heart valves is sufficient to promote calcific processes in vitro, which can be attenuated by Sox9 overexpression.
Conclusions
This study provides insights into the molecular mechanisms of heart valve calcification and identifies reduced Sox9 function as a potential genetic basis for calcific valvular disease.
Keywords: Heart valves, calcification, Sox9, extracellular matrix, mouse model
Introduction
Calcification of heart valve structures affects more than 27% of the US population over age 65 and is the major contributor of heart valve malfunction.1 Despite the clinical significance, little is known about the mechanisms that underlie this multifactorial disease. Treatment options for valve calcification are limited and no known therapies prevent disease progression.2 Normal heart valve function requires organization of differentiated cell types and specialized extracellular matrix (ECM) arranged according to blood flow within the valve leaflet.3 This defined tissue architecture provides the mechanical resilience and compressibility required to open and close the valve orifices effectively during the cardiac cycle.4 In diseased heart valves, loss of ECM organization is associated with changes in mechanical properties, ultimately leading to dysfunction.5,6 One of the most striking alterations in valve ECM homeostasis is ectopic bone-like matrix mineralization observed in calcific valve diseae.7,8 At the functional level, this histopathological alteration results in stiffened leaflets, narrowing of the valve opening, and impaired blood flow.9
The mechanisms that promote the onset and progression of heart valve calcification are not clear, but recent reports suggest a complex process involving molecular and cellular phenotypes shared with bone formation and chronic inflammation.8 The contribution of these phenotypes in the onset and progression of calcific valvular disease is not known, however studies have identified clinical risk factors including older age and hypercholesterolemia.2 Recent reports have also shown that regulatory mechanisms common to osteogenesis play a major role in valvular calcification.10 Expression of bone-associated genes including Runx2, a transcription factor required for the osteoblast gene program, and downstream target genes including Osteopontin and Osteonectin, is increased in calcified lesions of human heart valves.7,10–12 The mechanisms initiating ectopic osteogenic processes in heart valves are not known, but recent reports have implicated a genetic basis, and therefore developmental origins.9,13,15,16
The diversified cell types and ECM that form the mature valve architecture are derived during embryogenesis from undifferentiated precursor cells of endocardial cushions.17–19 Following endothelial to mesenchymal transformation and cushion formation, valve precursor cells differentiate and secrete specialized ECM.17,20 The molecular mechanisms that regulate this process are not well defined, but are likely essential for normal structure and function of mature heart valves. Identification of such signaling pathways during development may improve understanding of adult valve pathologies associated with altered connective tissue composition and organization. Recent studies have identified parallel regulatory mechanisms between heart valve development and formation of other connective tissue systems including cartilage and tendon.21,22 Although aberrations in these signaling pathways are known to affect skeletogenesis, pathological effects on heart valve structure and function are not well defined.
First identified for its essential role in chondrocyte differentiation, the transcription factor Sox9 is known to play parallel roles in promoting expression of cartilaginous matrix proteins in developing heart valve structures.21,23 In addition to cartilage defects, mice with reduced Sox9 function develop premature skeletal ossification, suggesting an opposing role in bone formation.24,25 The mechanisms of Sox9 deficiency during skeletogenesis are not clear, although SOX9 function is sufficient to inhibit RUNX2-mediated activation of osteogenic target genes.26 Upstream regulators of Sox9 in cartilage and bone formation include retinoic acid (RA).27–29 RA treatment of chondrogenic cells in vitro leads to decreased Sox9 activity, associated with reduced expression of cartilage genes29,30 and significant increases in Runx2 and osteogenic processes.31,32 These studies suggest that RA and Sox9 signaling play pivotal roles in promoting cartilage and bone phenotypes. However, the potential for RA-Sox9 signaling to promote osteogenic processes in heart valve connective tissue is not known.
Previous studies have shown that heart valves from mice with targeted homozygous loss of Sox9 in type II collagen- (Col2a1-) derived cells express diminished levels of cartilage-associated proteins 21, and increased calcium deposition is observed on valves from viable heterozygotes (Sox9fl/+;Col2a1-cre) .21 However molecular, cellular and functional analyses were lacking in this previous study.21 Here, we report that Sox9fl/+;Col2a1-cre mice develop calcific lesions within heart valve leaflets from 3 months of age with significant increases in bone-related genes and ECM remodeling and inflammatory processes. This osteogenic phenotype is recapitulated following direct Sox9 knockdown in heart valve explants. Further, calcification in chick valve explants is promoted by RA treatment, which can be attenuated by Sox9 overexpression. These data suggest that Sox9 plays an important role in preventing calcific processes in normal heart valves and identifies RA-Sox9 signaling as a suitable pathway for therapeutic targets in the prevention and treatment of calcific valvular disease.
Materials and Methods
Sox9fl/fl female mice33 were bred with Col2a1-cre males (Jackson Laboratories)34 to generate heterozygous offspring at expected Mendelian ratios. Sox9fl/+;Col2a1-cre mice and Sox9fl/+ littermate controls were aged and subject to echocardiography as described22. Following functional analysis, hearts were removed for RNA extraction from atrioventricular regions (containing the mitral and tricuspid valves) or whole hearts fixed, cryo-embedded, and sectioned for histological staining, in situ hybridization, and immunofluorescence as described.21 Real time PCR-based TaqMan Low Density Array (TLDA) cards were used to quantitatively identify changes in mRNA transcript levels of target genes as described.35 For in vitro studies, neonate mouse or embryonic day 10 (E10) chick mitral valve explants were treated with DMSO, 1µmol/L RA, adenovirus- (Ad-) GFP, Ad-Sox9 or Ad-Cre. Expanded Materials and Methods are available in the online data supplement at http://circres.ahajournals.org.
Results
Generation of mice with targeted reduction of Sox9 function in heart valves
Our previous studies have shown that Sox9 is highly expressed during early stages of endocardial cushion development.21 Using immunofluorescence, we also detect Sox9 expression in the mitral (mv) (Figure 1A), tricuspid, pulmonic (data not shown), and aortic (Ao) valve (Figure 1B) leaflets during remodeling stages at E17.5. In order to determine the role of Sox9 in murine heart valves we employed a targeted approach using the Cre/loxP system. Breedings of Col2a1-cre with Rosa26R reporter mice reveal recombination by X-gal staining in a subset of cells along the edges of the mitral (mv) (Figure 1C), aortic (Ao) (Figure 1D), tricuspid and pulmonic valve leaflets (data not shown) from E15.5, consistent with Sox9 expression (Figures 1A, B).21 In heart valves from 3 month old Sox9fl/+;Col2a1-cre−/− (Sox9fl/+) mice, Sox9 is expressed in cells throughout the valve leaflet (Figure 1E). However, following recombination in Sox9fl/+;Col2a1-cre mice, Sox9 expression is moderately reduced along the edges of the valve leaflets (arrows, Figure 1F) associated with Col2a1-derived cells (arrows, Figure 1C). This is in comparison to adjacent non-recombined cells (arrowheads, Figure 1F) and cells along the edges of valves from cre-negative Sox9fl/+ littermate controls (Figure 1E). Pecam staining indicates the endothelial cell layer (Figure 1E, F). These data validate successful recombination of Sox9 in targeted cells.
Figure 1. Sox9 expression is detected in developing and mature heart valve leaflets.

Sox9 (red) is expressed in the mitral (mv) (A) and aortic valve (Ao) leaflets at E17.5 (arrows) (B). X-gal staining shows Col2a1-cre specific recombination (blue) in a subset of cells of the mitral (C) and aortic valves (D) in E18.5 Col2a1-cre × Rosa26R reporter mice. In Sox9fl/+;Col2a1-cre mice (F), Sox9 expression (red) is reduced in recombined cells along the edge of the valve leaflets (arrows) compared to adjacent valve interstitial cells (arrowheads) not expressing the Col2a1-cre transgene. In control leaflets (E), cells with strong Sox9 expression are observed both at the leaflet surface and in the interstitium. Sox9 signal appears reduced in cells at the leaflet surface in Sox9fl/+;Col2a1-cre mice (F). Pecam (green) indicates endothelial cells (E, F). Nuclei are stained with DAPI (A,B,E,F).
Increased calcium deposition in heart valve leaflets from adult Sox9fl/+;Col2a1-cre mice
As determined by von Kossa reactivity, calcium deposits are observed in aortic (Ao) (Figure 2A, B), mitral (mv) (Figure 2C, D), and tricuspid (data not shown) valve leaflets of Sox9fl/+;Col2a1-cre mice from 3 months of age (Figure 2B, D), but not in leaflets from control Sox9fl/+ mice (Figure 2A, C). In all cases, von Kossa staining reveals calcium deposition on the leaflet surface adjacent to blood flow (red arrows). Lack of von Kossa reactivity in neighboring tissue sections from Sox9fl/+;Col2a1-cre mice treated with counterstain only (inset, Figure 2D), or decalcified with EDTA (data not shown), eliminates misidentification of pigmented melanocytes present in the valves as calcification.36 Notably, calcified lesions were observed only in leaflets and not other valvular structures. For quantitative comparisons, the area of von Kossa reactivity relative to the valve leaflet area as defined by Alcian blue counterstain was determined in tissue sections from mutant and control mice at 3, 6, and 12 months of age (Figure 2E, F). Heart valves from Sox9fl/+;Col2a1-cre mice have significantly increased von Kossa reactivity compared to littermate Sox9fl/+ mice at 3 months of age. Control Sox9fl/+ mice consistently had little or no reactivity (0.22%±0.24). Due to the increased variability in von Kossa reactivity in 6 and 12 month old Sox9fl/+;Col2a1-cre mice (Figure 2F), the area of calcium deposition did not significantly change with age, and differences compared to control mice were not observed (Figures 2E, F). However there is a trend toward increased lesion size with older age in Sox9fl/+;Col2a1-cre mice (Figure 2E). Survival is comparable to controls at 12 months of age. These findings suggest that reduced Sox9 function in Col2a1-derived cells during development promotes calcium deposition in adult heart valve leaflets.
Figure 2. Von Kossa reactivity reveals increased calcium deposition in valve leaflets from Sox9fl/+;Col2a1-cre mice.
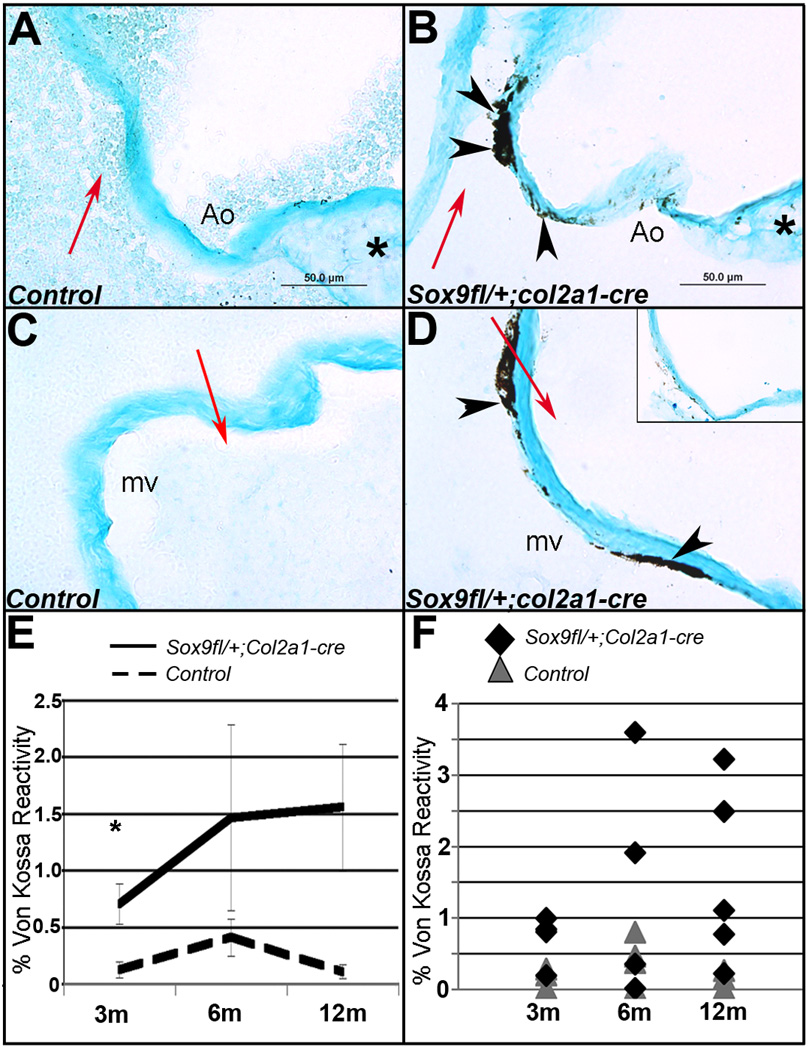
Von Kossa reactivity indicates calcium deposits (black, arrowheads) in aortic (Ao) (B) and mitral valve (mv) (D) leaflets adjacent to blood flow (red arrows) in tissue sections from Sox9fl/+;Col2a1-cre mice at 12 months of age. Von Kossa reactivity was not significantly detected in littermate Sox9fl/+ controls (A, C). Inset (D), no von Kossa treatment. Alcian blue counterstain defines the valve area (A–D), * indicates the base of the aortic valve, adjacent to the myocardium (A, B). Quantification of von Kossa reactivity as a percentage of valve area demonstrates significant increases in calcium deposits in Sox9fl/+;Col2a1-cre mitral valve leaflets with a trend toward increasing severity and variability with age (E, F). m, months. Low magnification images (A–D) are available in Online Figure I.
Heart valves from Sox9fl/+;Col2a1-cre mice show increased osteogenic signaling and inflammatory and ECM remodeling processes at 12 months of age
Recent studies have described human calcific valvular disease as an active process associated with activation of regulatory pathways common to bone development and increased inflammatory processes and ECM remodeling.37,7 To identify similar processes in Sox9fl/+;Col2a1-cre mice, high throughput quantitative real-time PCR was performed using custom TaqMan low-density array (TLDA) cards designed to identify changes in genes characteristic of advanced human heart valve calcification.10,38–44
In heart valves from 12 month old Sox9fl/+;Col2a1-cre mice transcript levels of several bone-related genes are significantly increased over controls. Similar to previous observations in human calcific valvular disease, these include increased expression of Runx2 (2-fold), Osteonectin (ON) (1.8-fold), Osteopontin (OP) (8.7-fold) Osteoprotegerin (OPG) (3.9-fold) and Smpd3 (1.9-fold) compared to Sox9fl/+ littermate controls (Figure 3A).12,45 Online table II shows results for all the genes examined in Sox9fl/+;Col2a1-cre mice at 3, 6, and 12 months of age. In situ hybridization confirms increased ON transcript in 12 month old Sox9fl/+;Col2a1-cre valve leaflets over Sox9fl/+ control (Figure 3B,C).
Figure 3. Transcript levels of bone-related genes are increased in 12 month old Sox9fl/+;Col2a1-cre mice.

(A) Normalized fold changes in transcripts levels of osteogenic-signaling genes Runx2, Osteonectin (ON), Osteopontin (OP), Osteoprotegrin (OPG) and Smpd3 in Sox9fl/+;Col2a1-cre versus Sox9fl/+ control mice at 3, 6, and 12 months as determined by TaqMan Low Density Array (TLDA). *p<0.05. Osteonectin, undetected by in situ hybridization in control mitral valve leaflets (B), is detected in a 12 month Sox9fl/+;Col2a1-cre mouse mitral valve leaflet (arrows, C). Low magnification images (B, C) are available in Online Figure II.
Consistent with previous observations in calcified human valves38,45,48–50, inflammatory responses appear active in heart valves from 12 month old Sox9fl/+;Col2a1-cre mice compared to controls. This is indicated by increased expression of Vascular adhesion molecule-1 (Vcam-1) (1.9-fold), a cell adhesion molecule involved in pro-inflammatory signal transduction51, Colony stimulating factor 1 receptor (Csf1r) (1.6-fold), indicative of macrophage infiltration, and Toll-like receptor 2 (Tlr2) (2.1-fold), important in cytokine release in immune response52,53 (Figure 4A). Spatially, increases in Vcam-1 expression by immunostaining were observed throughout the valve leaflet (data not shown). Further, increases in inflammation-related transcripts observed in Sox9fl/+;Col2a1-cre mice are significantly higher at 12 months compared to 3 months of age.
Figure 4. Inflammation and ECM remodeling-related genes are increased in 12 month old Sox9fl/+;Col2a1-cre mice.

(A) Inflammation-associated genes Vcam-1, Csfr1, and Tlr2 are increased inSox9fl/+;Col2a1-cre mice relative to controls at 3, 6, and 12 months (TLDA analysis). (B) Increases in ECM remodeling and fibrosis-related genes Col1a1, Col1a2, Timp1, and Tenascin-C (Ten-C) are also observed. *p<0.05 relative to age-matched controls, #p<0.05 relative to 3 month Sox9fl/+;Col2a1-cre mice. Elastin (green) is detected on the atrial surface of the mitral valve in control Sox9fl/+ mice (arrows, C). Elastin fibers are fragmented and disorganized in Sox9fl/+;Col2a1-cre mitral valve leaflets (arrows, D).
Valvular disease is frequently associated with ECM disorganization and excess collagen deposition, leading to fibrosis.3 In Sox9fl/+;Col2a1-cre mice, transcript levels of genes associated with tissue fibrosis46,47 are increased, including Col1a1 (1.9-fold), Col1a2 (1.7-fold), and Tenascin-C (ten-C) (4.3-fold) (Figure 4B). In addition, the matrix metalloproteinase inhibitor Timp1 is increased 4-fold, indicative of valve leaflet remodeling. Disorganized elastic fibers have previously been reported with calcified heart valves.14 In valves from Sox9fl/+;Col2a1-cre mice, changes in Elastin transcript levels are not observed, however mature elastic fibers appear fragmented and ectopically distributed throughout the valve leaflet in Sox9fl/+;Col2a1-cre mice (Figure 4C), compared to parallel bundles localized along the atrial surface in controls (Figure 4D). Collectively these findings indicate that increased calcium deposition observed in Sox9fl/+;Col2a1-cre mice is associated with activation of osteogenic, inflammation, and ECM remodeling programs.
Knockdown of Sox9 in mature heart valves in vitro promotes calcific phenotypes
To support in vivo findings and determine a direct role for Sox9 in promoting heart valve calcification, atrioventricular valve explants from neonate Sox9fl/fl mice were infected with adenovirus (Ad) targeting Cre recombinase (Ad-Cre) (Figure 5C) or Ad-GFP (Figure 5B) as a negative control. In association with a >2-fold knockdown of Sox9, explants infected with Ad-Cre display 28% von Kossa reactivity (28%) (Figure 5B, C), compared to Ad-GFP controls (<5%) (Figure 5A, C), or explants from Sox9+/+ mice infected with Ad-Cre (data not shown). This increase in calcium deposition is associated with a significant increase in Osteopontin and a trend towards increased Runx2 expression. These data support a direct role for reduced Sox9 function in promoting calcific heart valve phenotypes.
Figure 5. Sox9 knockdown increases calcification phenotypes in mouse valve explants.
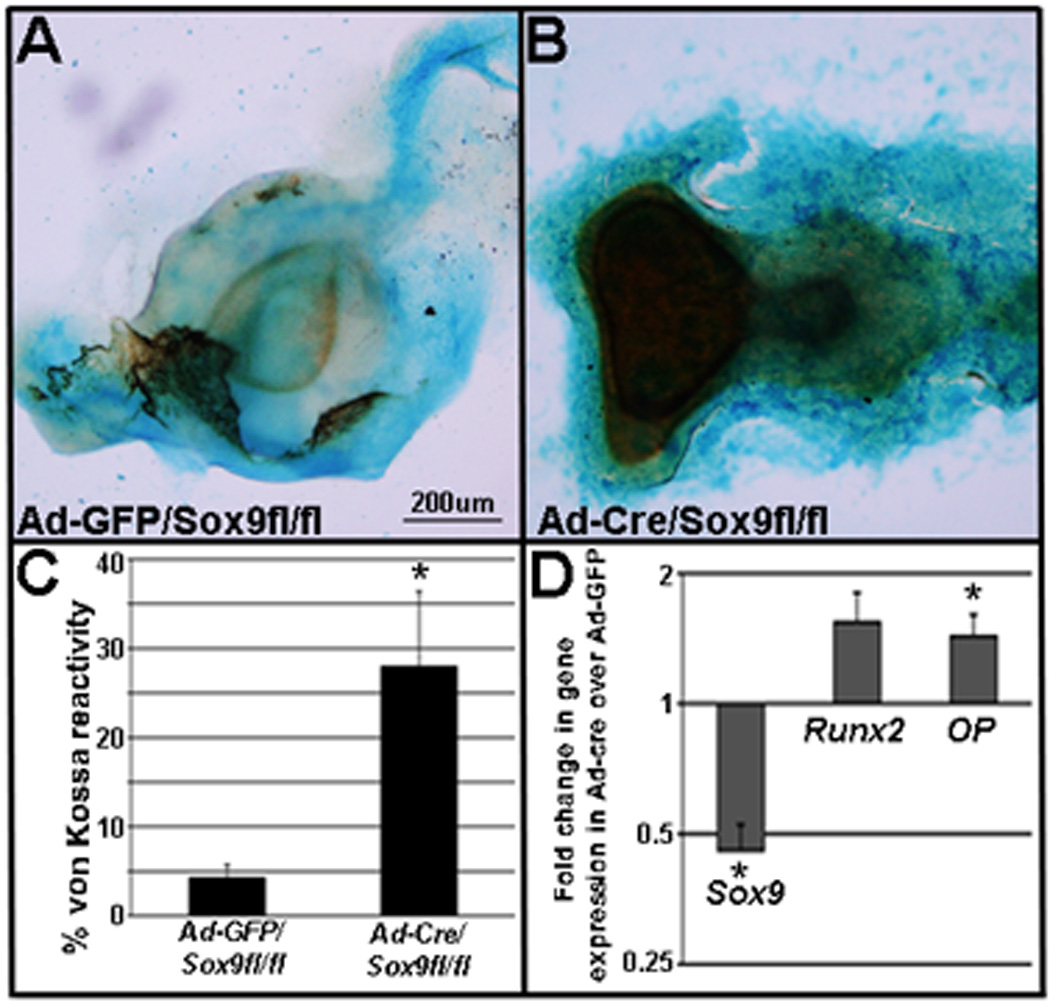
(A, B) von Kossa reactivity in Sox9fl/fl neonatal mouse valve explants infected with GFP-adenovirus (Ad-GFP) (A) or Cre-adenovirus (Ad-Cre) (B). (C) Quantification of von Kossa reactivity in Ad-GFP and Ad-Cre infected explants as a percentage of total area defined by Alcian blue. (D) Sox9 transcript levels are significantly decreased following infection with Ad-Cre, while Runx2 and Osteopontin (OP) are increased relative to GFP-treated controls. *p>0.05.
Retinoic acid treatment reduces Sox9 expression and promotes osteogenic-like processes in mature chick heart valves
Retinoic acid (RA) has previously been identified as an upstream regulator of Sox9 during cartilage and bone development.28,29 In order to determine similar roles in mature heart valves intact avian mitral valve explants were subject to RA treatment in vitro. At embryonic day 10 (E10), avian heart valves express high levels of Sox927 and exhibit highly organized ECM patterning indicative of maturation3. Following 1µmol/L RA treatment for 48 hours, von Kossa staining reveals significantly increased calcium deposition (21.7%) compared to DMSO controls (2.9%)(Figure 6A, B, G). Further, RA treatment is associated with significant decreases in Sox9 transcript levels (5.3-fold) (Figure 6H).
Figure 6. Retinoic acid treatment reduces Sox9 expression and promotes matrix mineralization.

Cultured E10 chick mitral valve explants were treated with DMSO (A, C, E) or RA (B, D, F) and subject to infection with Ad-GFP (C, D) or Ad-Sox9 (E, F), or cultured without virus (A, B). (G) Quantitation of von Kossa reactivity in treated explants, normalized to area (Alcian blue staining). *p<0.05 compared to relative DMSO control, #p<0.05 compared to RA and Ad-GFP infection. (H) Normalized fold change in endogenous chicken Sox9 (cSox9) and adenoviral mouse Sox9 (Ad mSox9). *p<0.05 compared to respective DMSO- or RA-treated controls. #p<0.05 compared to DMSO+Ad-GFP.
To determine the role of reduced Sox9 in RA-induced valve calcification, avian explants treated with DMSO or 1µmol/L RA were infected with adenovirus (Ad) expressing full length mouse Sox9 (Ad-Sox9) or GFP (Ad-GFP). Consistent with Figure 6A, negligible von Kossa reactivity is observed in DMSO-treated explants infected with Ad-GFP (Figure 6C) or Ad-Sox9 (Figure 6E). In DMSO treated explants, endogenous chicken Sox9 expression does not change, although mouse Sox9 expression is significantly increased in Ad-Sox9 infected explants (Figure 6H) confirming targeted overexpression. As expected, RA treatment in Ad-GFP infected explants significantly increases von Kossa reactivity (>30%) (Figure 6D) and decreases Sox9 expression (Figure 6H) compared to DMSO controls infected with Ad-GFP (Figure 6C, H). Notably, RA-treatment in Ad-Sox9 infected explants (Figure 6F) does not increase von Kossa reactivity to levels observed in RA-treated Ad-GFP infected (Figure (6D) explants. These findings suggest that increased Sox9 expression prevents RA-induced heart valve calcification in vitro, supporting a direct and causative role for reduced Sox9 function in promoting calcific valve phenotypes.
Discussion
Identification of signaling pathways that mediate disease onset or progression is critical for the development of new treatments for calcific valvular disease. Here, we present findings from a mouse model with targeted loss of Sox9 function during valve development that displays increased susceptibility to calcific valve phenotypes in adulthood. This is marked by the appearance of calcific lesions and increased expression of osteogenic genes including Runx2 in valve leaflets from adult Sox9fl/+;Col2a1-cre mice. Further, these calcification processes are associated with activation of genes characteristic of ECM remodeling and inflammation. Despite these pathological phenotypes, valve function in adult Sox9fl/+;Col2a1-cre mice is comparable to Sox9fl/+ littermate controls (Online Table I). The ability of Sox9 deficiency to directly promote valvular calcification is supported by increased von Kossa reactivity following Sox9 knockdown in murine valve explants in vitro. Further, similar calcification is observed following RA treatment in vitro, and overexpression of Sox9 is sufficient to attenuate RA-induced calcification. Collectively these findings suggest that Sox9 plays important roles in maintaining connective tissue homeostasis in mature heart valve structures and provide insights into a genetic basis for calcific valvular disease.
Sox9 is required to maintain heart valve connective tissue homeostasis
In mature heart valves, ECM composition and organization are essential for maintaining valve structure and function. 3,4,37. Previous studies have shown that mature valve leaflets are rich in proteoglycans and express markers characteristic of cartilage tissue.4 Further, our group has shown that cartilage-associated gene expression in developing heart valves requires Sox9, similar to findings in chondrogenic systems.21,23,54,55 These findings highlight an important role for Sox9 in establishing the desired connective tissue composition in normal heart valves. In this current study we show that reduced Sox9 function leads to ectopic formation of calcified matrix and therefore highlight an additional role for Sox9 in maintaining connective tissue homeostasis of mature valves. However, the mechanisms of Sox9 function in these processes are not clear, but our findings suggest that Sox9 plays pivotal roles in promoting cartilage-like phenotypes and preventing osteogenic processes in normal heart valve structures. Therefore misregulation of Sox9 function likely has profound effects in promoting proteoglycan or osteogenic-related valvular disease associated with ‘floppy’ or ‘stiffened’ valve function respectively.
Sox9 function and calcific valvular disease
Increased Sox9 expression has previously been observed in myxomatous valves associated with increased proteoglycan production.11 Conversely, Sox9fl/+;Col2a1-cre mice develop calcific lesions on the surface of heart valve leaflets adjacent to blood blow. These findings highlighting similarities between our mutant mouse model and human calcified valvular disease. 9 However it is appreciated that the gross changes in ECM organization observed in human calcified valves cannot be thoroughly analyzed in this model due to size limitations of rodent heart valves. 3 At the molecular level, we again observe similarities with clinical pathology including increased expression of bone-associated genes including Runx2 and downstream osteogenic target genes..7,53 The underlying etiology that promotes pathological bone signaling in heart valves are not understood, but recent studies have identified NOTCH1 mutations in patients with bicsupid aortic valve (BAV) and aortic valve calcification.15 Interestingly, both NOTCH1 and SOX9 have been shown to repress RUNX2 activity.15, 26,. Therefore one might predict that the osteogenic phenotypes observed in Sox9fl/+;Col2a1-cre mice are due to lost repression of Runx2. However Notch1 expression was not significantly different in heart valves from Sox9fl/+;Col2a1-cre mice and therefore interactions between Sox9 and Notch signaling on Runx2 activity cannot be discerned.
Unlike calcified human valves where pathological calcification is associated with functional defects including stenosis, heart valve function in Sox9fl/+;Col2a1-cre mice is normal and using conventional echocardiography valve leaflet fusion was not observed. This suggests that the calcific lesion size (<4%) within the valve leaflet is not sufficient to affect mechanical properties of the valve at 12 months of age. Studies in mouse models susceptible to valve calcification have effectively aggravated phenotypes with additional exposure to known clinical risk factors including hypercholesterolemia and renal dysfunction, highlighting the multifactorial nature of this disease, and therefore exposing Sox9fl/+;Col2a1-cre mice to such factors would be a plausible approach for future studies.14,56,57
The RA-Sox9 pathway is a potential therapeutic target for treatment or prevention of calcific valvular disease
Previous approaches in the treatment of calcific valvular disease have focused on treating underlying risk factors. However clinical trials using lipid-lowering therapies have been inconclusive and patient outcomes are not improved58–60. Therefore alternative strategies are needed to improve disease prognosis and insights will likely be gained by targeting signaling pathways active during the onset and progression of calcific disease. Our findings suggest that RA signaling regulates Sox9 function to promote osteogenic processes in mature heart valves, highlighting RA-Sox9 signaling as a potential target for alternative therapeutic approaches. Manipulation of the RA signaling pathway has already proven pharmacologically effective in the treatment of several bone-associated pathologies including the use of small-molecule retinoic acid inhibitors as anti-osteogenic agents.61–63 Therefore future work investigating the benefits of retinoid pathway antagonism in the prevention or treatment of calcific valvular disease is warranted. Collectively our studies identify a previously unappreciated role for Sox9 function in maintaining connective tissue homeostasis in mature heart valve structures. In addition, there is evidence to suggest that reduced Sox9 function during embryonic development later leads to calcific valvular disease manifested in the adult.
Novelty and Significance.
What is known?
Heart valve calcification is the most frequently acquired valvular disease, but the etiology is not clear.
Sox9 is expressed in developing and mature heart valves and is required for expression of cartilage-related proteins.
Mice deficient for Sox9 fail to form skeletal cartilage and develop bone prematurely.
What new information does this article contribute?
Reduced Sox9 function promotes calcific valve phenotypes in vitro and in vivo.
Heart valve calcification in mice with reduced Sox9 function is associated with increased osteogenic signaling and activation of inflammatory and extracellular matrix processes.
Retinoic acid treatment promotes calcific valve phenotypes that can be rescued by Sox9 overexpression.
Calcification of heart valve structures is the most common form of valvular disease and most often results in surgical replacement. Despite the significance the mechanisms that promote disease onset and progression are largely unknown. In this current study, we have identified that reduced Sox9 function in a subset of type II collagen-derived valve cells during embryonic development promotes calcific valve phenotypes in vivo. This pathologic state is associated with increased signaling of genes active during bone development, and activation of inflammatory and matrix remodeling process in calcified valves from Sox9 mutant mice. Further, we have identified retinoic acid as an upstream repressor of Sox9 function in promoting calcification in vitro. This study has generated a mouse model of human pathology, and identified a novel genetic-based mechanism for calcific valve disease. Findings from this study provide insights into the molecular mechanisms that promote the onset of heart valve calcification that will undoubtedly contribute to the development of alternative therapeutic strategies.
Supplementary Material
Acknowledgments
We thank Drs. Ralf Kist and Gerd Scherer for their collegiate sharing of Sox9 floxed mice, Dr. Katherine Yutzey for providing in situ hybridization probes, Dr. Micheal Wegner for the Sox9 antibody and Harriet Hammond for editorial assistance in preparing this manuscript. In addition we thank Dr. Kyle Padgett for scientific and technical assistance.
Sources of Funding
This work was supported by the Florida Heart Research Institute (JL), Florida Biomedical Research Program (07KN-07, JL), NIH Training Program in Cardiovascular Signaling (5T32HL007188-28) and American Heart Association Pre-doctoral fellowship (09PRE2050088, JDP).
Non-standard Abbreviations and Acronyms
- Ao
aortic valve
- Ad
adenovirus
- DMSO
dimethyl sulfoxide
- E
embryonic day
- EC
endocardial cushions
- ECM
extracellular matrix
- ON
osteonectin
- OP
osteopontin
- OPG
osteoprotegrin
- m
months
- mv
mitral valve
- tv
tricuspid valve
- RA
retinoic acid
- TLDA
TaqMan Low Density Array
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Subject codes: [19] Valvular heart disease, [130] Animal models of human disease, [139] Developmental biology, [142] Gene expression, [143] Gene regulation
Disclosures
None.
References
- 1.Stewart B, Siscovick D, Lind B, Gardin J, Gottdiener J, Smith V, Kitzman D, Otto C. Clinical factors associated with calcific aortic valvular disease. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 2.Otto CM. Calcific aortic stenosis - time to look more closely at the valve. N Engl J Med. 2008;359:1395–1398. doi: 10.1056/NEJMe0807001. [DOI] [PubMed] [Google Scholar]
- 3.Hinton RB, Jr, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- 4.Lincoln J, Lange AW, Yutzey KE. Hearts and bones: Shared regulatory mechanisms in heart valve, cartilage, tendon, and bone development. Developmental Biology. 2006;294:292–302. doi: 10.1016/j.ydbio.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 5.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 6.Schoen FJ. Cardiac valves and valvular pathology: Update on function, disease, repair, and replacement. Cardiovascular Pathology. 2005;14:189–194. doi: 10.1016/j.carpath.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Brien KD. Pathogenesis of calcific aortic valvular disease: A disease process comes of age (and a good deal more) Arterioscler Thromb Vasc Biol. 2006;26:1721–1728. doi: 10.1161/01.ATV.0000227513.13697.ac. [DOI] [PubMed] [Google Scholar]
- 9.Mohler ER., III Mechanisms of aortic valve calcification. The American Journal of Cardiology. 2004;94:1396–1402. doi: 10.1016/j.amjcard.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Rajamannan NM, Nealis TB, Subramaniam M, Pandya S, Stock SR, Ignatiev CI, Sebo TJ, Rosengart TK, Edwards WD, McCarthy PM, Bonow RO, Spelsberg TC. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation. 2005;111:3296–3301. doi: 10.1161/CIRCULATIONAHA.104.473165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caira F, Stock S, Gleason T, McGee E, Huang J, Bonow R, Spelsberg T, McCarthy P, Rahimtoola S, Rajamannan N. Human degenerative valvular disease is associated with up-regulation of low-density lipoprotein receptor-related protein 5 receptor-mediated bone formation. Journal of the American College of Cardiology. 2006;47:1707–1712. doi: 10.1016/j.jacc.2006.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 13.Yoshioka M, Yuasa S, Matsumura K, Kimura K, Shiomi T, Kimura N, Shukunami C, Okada Y, Mukai M, Shin H, Yozu R, Sata M, Ogawa S, Hiraki Y, Fukuda K. Chondromodulin-I maintains cardiac valvular function by preventing angiogenesis. Nat Med. 2006;12:1151–1159. doi: 10.1038/nm1476. [DOI] [PubMed] [Google Scholar]
- 14.Aikawa E, Aikawa M, Libby P, Figueiredo J, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi G, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valvular disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 16.Garg V. Insights into the genetic basis of congenital heart disease. Cellular and Molecular Life Sciences. 2006;63:1141–1148. doi: 10.1007/s00018-005-5532-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Person AD, Klewer SE, Runyan RB. Cell biology of cardiac cushion development. In: Kwang W. Jeon., editor. International Review of Cytology. Vol Volume 243. Academic Press; 2005. pp. 287–335. [DOI] [PubMed] [Google Scholar]
- 18.de Lange FJ, Moorman AFM, Anderson RH, Manner J, Soufan AT, Vries CdG, Schneider MD, Webb S, van den Hoff MJB, Christoffels VM. Lineage and morphogenetic analysis of the cardiac valves. Circ Res. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong EJ, Bischoff J. Heart valve development: Endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Developmental Dynamics. 2004;230:239–250. doi: 10.1002/dvdy.20051. [DOI] [PubMed] [Google Scholar]
- 21.Lincoln J, Kist R, Scherer G, Yutzey KE. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Developmental Biology. 2007;305:120–132. doi: 10.1016/j.ydbio.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levay AK, Peacock JD, Lu Y, Koch M, Hinton RB, Jr, Kadler KE, Lincoln J. Scleraxis is required for cell lineage differentiation and extracellular matrix remodeling during murine heart valve formation in vivo. Circ Res. 2008;103:948–956. doi: 10.1161/CIRCRESAHA.108.177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation; 10.1038/8792. Nat Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 24.Bi W, Huang W, Whitworth DJ, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci USA. 2001;98:6698–6703. doi: 10.1073/pnas.111092198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci USA. 2003;100:9360–9365. doi: 10.1073/pnas.1631288100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci USA. 2006;103:19004–19009. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lincoln J, Alfieri C, Yutzey K. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Developmental Biology. 2006;292:290–302. doi: 10.1016/j.ydbio.2005.12.042. [DOI] [PubMed] [Google Scholar]
- 28.Yoon BS, Lyons KM. Multiple functions of BMPs in chondrogenesis. J Cell Biochem. 2004;93:93–103. doi: 10.1002/jcb.20211. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman LM, Weston AD, Underhill TM. Molecular mechanisms regulating chondroblast differentiation. J Bone Joint Surg Am. 2003;85:124–132. doi: 10.2106/00004623-200300002-00017. [DOI] [PubMed] [Google Scholar]
- 30.Sekiya I, Koopman P, Tsuji K, Mertin S, Harley V, Yamada Y, Shinomiya K, Niguji A, Noda M. Transcriptional suppression of Sox9 expression in chondrocytes by retinoic acid. J Cell Biochem. 2001;81:71–78. doi: 10.1002/jcb.1077. [DOI] [PubMed] [Google Scholar]
- 31.Iwamoto M, Shapiro IM, Yagami K, Boskey AL, Leboy PS, Adams SL, Pacifici M. Retinoic acid induces rapid mineralization and expression of mineralization-related genes in chondrocytes. Exp Cell Res. 1993;207:413–420. doi: 10.1006/excr.1993.1209. [DOI] [PubMed] [Google Scholar]
- 32.Jimenez MJG, Balbin M, Alvarez J, Komori T, Bianco P, Holmbeck K, Birkedal-Hansen H, Lopez JM, Lopez-Otin C. A regulatory cascade involving retinoic acid, Cbfa1, and matrix metalloproteinases is coupled to the development of a process of perichondrial invasion and osteogenic differentiation during bone formation. J Cell Biol. 2001;155:1333–1344. doi: 10.1083/jcb.200106147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kist R, Schrewe H, Balling R, Scherer G. Conditional inactivation of Sox9: A mouse model for campomelic dysplasia. Genesis. 2002;32:121–123. doi: 10.1002/gene.10050. [DOI] [PubMed] [Google Scholar]
- 34.Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146. [PubMed] [Google Scholar]
- 35.Peacock JD, Lu Y, Koch M, Kadler KE, Lincoln J. Temporal and spatial expression of collagens during murine atrioventricular heart valve development and maintenance. Developmental Dynamics. 2008;237:3051–3058. doi: 10.1002/dvdy.21719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- 37.Schoen FJ. Evolving concepts of cardiac valve dynamics: The continuum of development, functional structure, pathobiology, and tissue engineering. Circulation. 2008;118:1864–1880. doi: 10.1161/CIRCULATIONAHA.108.805911. [DOI] [PubMed] [Google Scholar]
- 38.Yang X, Fullerton DA, Su X, Ao L, Cleveland JC, Jr, Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 39.Pohjolainen V, Taskinen P, Soini Y, Rysä J, Ilves M, Juvonen T, Ruskoaho H, Leskinen H, Satta J. Noncollagenous bone matrix proteins as a part of calcific aortic valvular disease regulation. Human Pathology. 2008;39:1695–1701. doi: 10.1016/j.humpath.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 40.Hadian M, Corcoran BM, Han RI, Grossmann JG, Bradshaw JP. Collagen organization in canine myxomatous mitral valvular disease: An X-ray diffraction study. Biophys J. 2007;93:2472–2476. doi: 10.1529/biophysj.107.107847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yeghiazaryan K, Skowasch D, Bauriedel G, Schild H, Golubnitschaja O. Could activated tissue remodeling be considered as early marker for progressive valve degeneration? comparative analysis of checkpoint and ECM remodeling gene expression in native degenerating aortic valves and after bioprosthetic replacement. Amino Acids. 2007;32:109–114. doi: 10.1007/s00726-006-0376-0. [DOI] [PubMed] [Google Scholar]
- 42.Kaden JJ, Bickelhaupt S, Grobholz R, Haase KK, Sarιkoç A, Kιlιç R, Brueckmann M, Lang S, Zahn I, Vahl C, Hagl S, Dempfle C, Borggrefe M. Receptor activator of nuclear factor κB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol. 2004;36:57–66. doi: 10.1016/j.yjmcc.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 43.Alexopoulos A, Bravou V, Peroukides S, Kaklamanis L, Varakis J, Alexopoulos D, Papadaki H. Bone regulatory factors NFATc1 and osterix in human calcific aortic valves. International Journal of Cardiology. doi: 10.1016/j.ijcard.2008.10.014. In Press, Corrected Proof. [DOI] [PubMed] [Google Scholar]
- 44.Mohler ER, 3rd, Chawla MK, Chang AW, Vyavahare N, Levy RJ, Graham L, Gannon FH. Identification and characterization of calcifying valve cells from human and canine aortic valves. J Heart Valve Dis. 8:254–260. [PubMed] [Google Scholar]
- 45.Schroeder TM, Jensen ED, Westendorf JJ. Runx2: A master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Research Part C: Embryo Today: Reviews. 2005;75:213–225. doi: 10.1002/bdrc.20043. [DOI] [PubMed] [Google Scholar]
- 46.Kuhn C, Mason RJ. Immunolocalization of SPARC, tenascin, and thrombospondin in pulmonary fibrosis. Am J Pathol. 1995;147:1759–1769. [PMC free article] [PubMed] [Google Scholar]
- 47.Herpel E, Pritsch M, Koch A, Dengler TJ, Schirmacher P, Schnabel PA. Interstitial fibrosis in the heart: Differences in extracellular matrix proteins and matrix metalloproteinases in end-stage dilated, ischaemic and valvular cardiomyopathy. Histopathology. 2006;48:736–747. doi: 10.1111/j.1365-2559.2006.02398.x. [DOI] [PubMed] [Google Scholar]
- 48.Mohler ER, III, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103:1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 49.Akat K, Borggrefe M, Kaden JJ. Aortic valve calcification - basic science to clinical practice. Heart. 2008;95:616–623. doi: 10.1136/hrt.2007.134783. [DOI] [PubMed] [Google Scholar]
- 50.Ghaisas NK, Foley JB, O’Briain DS, Crean P, Kelleher D, Walsh M. Adhesion molecules in nonrheumatic aortic valvular disease: Endothelial expression, serum levels and effects of valve replacement. J Am Coll Cardiol. 2000;36:2257–2262. doi: 10.1016/s0735-1097(00)00998-0. [DOI] [PubMed] [Google Scholar]
- 51.Lobb R, Chi-Rosso G, Leone D, Rosa M, Newman B, Luhowskyj S, Osborn L, Schiffer S, Benjamin c, Dougas I, Hession C, Chow P. Expression and functional characterization of a soluble form of vascular cell adhesion molecule 1. Biochem Biophys Res Commun. 1991;178:1498–1504. doi: 10.1016/0006-291x(91)91063-i. [DOI] [PubMed] [Google Scholar]
- 52.Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N. Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J Endocrinol. 2007;193:323–330. doi: 10.1677/JOE-07-0067. [DOI] [PubMed] [Google Scholar]
- 53.Meng X, Ao L, Song Y, Babu A, Yang X, Wang M, Weyant MJ, Dinarello CA, Cleveland JC, Jr, Fullerton DA. Expression of functional toll-like receptors 2 and 4 in human aortic valve interstitial cells: Potential roles in aortic valve inflammation and stenosis. Am J Physiol Cell Physiol. 2008;294:C29–C35. doi: 10.1152/ajpcell.00137.2007. [DOI] [PubMed] [Google Scholar]
- 54.Bridgewater LC, Walker MD, Miller GC, Ellison TA, Holsinger LD, Potter JL, Jackson TL, Chen RK, Winkel VL, Zhang Z, McKinney S, de Crombrugghe B. Adjacent DNA sequences modulate Sox9 transcriptional activation at paired sox sites in three chondrocyte-specific enhancer elements. Nucl Acids Res. 2003;31:1541–1553. doi: 10.1093/nar/gkg230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akiyama H, Chaboissier M, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002;16:2813–2828. doi: 10.1101/gad.1017802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weiss RM, Ohashi M, Miller JD, Young SG, Heistad DD. Calcific aortic valve stenosis in old hypercholesterolemic mice. Circulation. 2006;114:2065–2069. doi: 10.1161/CIRCULATIONAHA.106.634139. [DOI] [PubMed] [Google Scholar]
- 57.Drolet M, Roussel E, Deshaies Y, Couet J, Arsenault M. A high Fat/High carbohydrate diet induces aortic valvular disease in C57BL/6J mice. J Am Coll Cardiol. 2006;47:850–855. doi: 10.1016/j.jacc.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 58.Cawley PJ, Otto CM. Prevention of calcific aortic valve stenosis—fact or fiction? Ann Med. 2009;41:100. doi: 10.1080/07853890802331394. [DOI] [PubMed] [Google Scholar]
- 59.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA the Scottish Aortic Stenosis and Lipid Lowering Trial,Impact on Regression (SALTIRE) Investigators. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 60.Rossebø AB, Pedersen TR, Allen C, Boman K, Chambers J, Egstrup K, Gerdts E, Gohlke-Bärwolf C, Holme I, Kesäniemi VAY, Malbecq W, Nienaber C, Ray S, Skjærpe T, Wachtell K, Willenheimer R. Design and baseline characteristics of the simvastatin and ezetimibe in aortic stenosis (SEAS) study. Am J Cardiol. 2007;99:970–973. doi: 10.1016/j.amjcard.2006.10.064. [DOI] [PubMed] [Google Scholar]
- 61.Kafienah W, Mistry S, Perry MJ, Politopoulou G, Hollander AP. Pharmacological regulation of adult stem cells: Chondrogenesis can be induced using a synthetic inhibitor of the retinoic acid receptor. Stem Cells. 2007;25:2460–2468. doi: 10.1634/stemcells.2007-0059. [DOI] [PubMed] [Google Scholar]
- 62.Lin C, Chen C, Lin T, Tung JC, Wang S. Anti-inflammation activity of fruit essential oil from cinnamomum insularimontanum hayata. Bioresour Technol. 2008;99:8783–8787. doi: 10.1016/j.biortech.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 63.Nagpal S, Lu J, Boehm MF. Vitamin D analogs: Mechanism of action and therapeutic applications. Curr Med Chem. 2001;8:1661. doi: 10.2174/0929867013371950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.