

Abstract
InhA, the enoyl-ACP reductase in Mycobacterium tuberculosis is an attractive target for the development of novel drugs against tuberculosis, a disease that kills more than two million people each year. InhA is the target of the current first line drug isoniazid for the treatment of tuberculosis infections. Compounds that directly target InhA and do not require activation by the mycobacterial catalase-peroxidase KatG are promising candidates for treating infections caused by isoniazid-resistant strains. Previously we reported the synthesis of several diphenyl ethers with nanomolar affinity for InhA. However, these compounds are rapid reversible inhibitors of the enzyme, and based on the knowledge that long drug target residence times are an important factor for in vivo drug activity, we set out to generate a slow onset inhibitor of InhA using structure-based drug design. 2-(o-Tolyloxy)-5-hexylphenol (PT70) is a slow, tight binding inhibitor of InhA with a K1 value of 22 pm. PT70 binds preferentially to the InhA·NAD+ complex and has a residence time of 24 min on the target, which is 14,000 times longer than that of the rapid reversible inhibitor from which it is derived. The 1.8 Å crystal structure of the ternary complex between InhA, NAD+, and PT70 reveals the molecular details of enzyme-inhibitor recognition and supports the hypothesis that slow onset inhibition is coupled to ordering of an active site loop, which leads to the closure of the substrate-binding pocket.
Keywords: Crystal Structure, Drug Design, Enzyme Inhibitors, Enzyme Kinetics, Fatty Acid-binding Protein, Fatty Acid Synthase
Introduction
Mycobacterium tuberculosis is the causative agent of tuberculosis, an infectious disease that is spread all over the world. Two billion people, one-third of the world population, are infected with tuberculosis, and M. tuberculosis is responsible for 8.8 million new infections and 1.6 million deaths each year (1). The emergence of multidrug-resistant M. tuberculosis strains that are resistant against the current frontline drugs isoniazid and rifampicin, contribute to the spread and worsen the situation by lengthening the treatment considerably from 6 months to nearly 2 years and thereby increasing the cost for therapy 20-fold. Extensively drug-resistant strains, which are almost untreatable with current chemotherapeutics, threaten both developing and industrialized countries (2). Novel drugs with activity against drug-resistant strains are therefore urgently needed to restrain the disease that was once thought to be under control.
One of the most effective and widely used drugs for the treatment of tuberculosis is isoniazid (INH).4 INH is a prodrug that has to be activated by KatG, the mycobacterial catalase-peroxidase, to form together with NAD(H), an INH-NAD adduct (3). This adduct is a slow onset inhibitor of InhA, the enoyl-ACP reductase of the mycobacterial type II fatty acid biosynthesis pathway (4, 5). In this pathway, very long chain fatty acids are generated that act as precursors for mycolic acids, which in turn are essential building blocks of the waxy cell wall of mycobacteria (6). Inhibition of InhA blocks mycolic acid biosynthesis, thereby impairing the integrity of the cell wall and eventually leading to cell death (7). Because the predominant mechanism of resistance against isoniazid arises from mutations in KatG (3), new compounds that directly target InhA and circumvent the activation step are promising candidates for combating multidrug-resistant strains of M. tuberculosis.
Triclosan inhibits InhA directly (8). Although it is a relatively weak inhibitor of mycobacterial InhA (Ki = 0.2 μm (9)), significant progress has been made to improve its affinity toward InhA; first generation diphenyl ethers have been developed using structure-based drug design that are nanomolar inhibitors of InhA with minimum inhibitory concentrations of 1–2 μg/ml against both drug-sensitive and drug-resistant strains of M. tuberculosis (10). In addition, Freundlich et al. (11) recently reported the development of potent triclosan derivatives that demonstrated inhibition of InhA in the nanomolar range with minimum inhibitory concentrations of 5–10 μg/ml. However, all compounds in Ref. 10 are rapid reversible inhibitors of the enzyme. This is significant given the increasing importance attached to compounds that have long residence times on their targets (see Ref. 12 and references therein), as recently demonstrated by the correlation between residence time and in vivo activity for a series of Francisella tularensis FabI inhibitors (13). In addition, the highly successful INH-NAD adduct was shown to be a slow onset inhibitor of InhA (5). The crystal structures of InhA (Protein Data Bank code 2nv6 (14)) and Escherichia coli FabI (Protein Data Bank code 1qg6 (15)) support the hypothesis that slow onset inhibition is coupled to ordering of an active site loop (residues 195–210 in InhA), which leads to a closure of the substrate-binding pocket.
Toward the design of a slow onset diphenyl ether, we speculated that there must be an entropic penalty for loop ordering. Thus, reducing the conformational flexibility of the lead diphenyl ether might enable ordering of the active site loop, thus in turn resulting in slow onset enzyme inhibition. Introduction of a methyl group ortho to the diphenyl ether linkage resulted in a compound, PT70 (Fig. 1), that is a slow onset inhibitor of InhA with a K1 value of 22 pm. The crystal structure of PT70 bound to InhA corroborates that slow onset inhibition is coupled to ordering of the substrate-binding loop. Significantly the B-ring methyl group of PT70 forms critical van der Waal's interactions with the NAD+ cofactor and with key amino acids of the formerly disordered substrate-binding loop. These studies provide information on the mechanistic imperatives for slow onset inhibition of enoyl ACP reductases, and the novel inhibitor has the potential to be effective against both drug-sensitive and drug-resistant strains of M. tuberculosis.
FIGURE 1.

Structures of the rapid reversible inhibitor 6PP and the slow onset inhibitor PT70.
EXPERIMENTAL PROCEDURES
Synthesis of PT70
PT70 was synthesized using the procedure described previously for the synthesis of alkyl-substituted diphenyl ethers (16). Briefly, 2-iodotoluene (7.35 mmol), 4-bromo-2-methoxyphenol (14.7 mmol), Cs2CO3 (32.3 mmol), (CuOTf)2·PhH (0.735 mmol, 5.0 mol % copper), ethyl acetate (0.125 mmol, 5.0 mol %), 1-naphthoic acid (32.3 mmol), molecular sieves 4 Å (1.8 g), and toluene (15 ml) were added to an oven-dried 50-ml two-necked round-bottomed flask sealed with a septum that was purged with N2 and heated to 110 °C. After 24 h the reaction mixture was cooled to room temperature, dichloromethane was added, and the organic phase was obtained by filtration. The organic phase was washed with 5% NaOH, and the aqueous layer was then extracted with dichloromethane. The combined organic layers were washed with brine, dried over MgSO4, and concentrated under vacuum to give the crude product (1-(4-bromo-2-methoxyphenoxy)-2-methylbenzene), which was subsequently purified by flash column chromatography on silica gel (ethyl acetate/hexane). ZnCl2 (0.5 m solution in tetrahydrofuran; 9.0 ml, 4.52 mmol) was added to a round-bottomed flask sealed with a rubber septum and purged with nitrogen. Hexyl magnesium chloride (1.0 m solution in tetrahydrofuran; 4.0 ml, 4.26 mmol) was then added dropwise, and the resulting solution was stirred at room temperature for 20 min. N-Methylpyrrolidinone (4.7 ml) was then added to the flask, followed by 21.7 mg (0.0426 mmol) of Pd(P(t-Bu)3)2 and 500 mg (1.71 mmol) of 1-(4-bromo-2-methoxyphenoxy)-2-methylbenzene after 5 min. The flask was fitted with a reflux condenser and heated for 48 h at 130 °C. After cooling gradually to room temperature, 20 ml of a 1.0 m aqueous HCl solution was added to the flask. The resulting mixture was extracted with diethyl ether, and the ether extract was washed with water, dried over anhydrous MgSO4, and concentrated under vacuum to give the crude product 1-(4-hexyl-2-methoxyphenoxy)-2-methylbenzene, which was purified by flash column chromatography on silica gel (ethyl acetate/hexane). A solution of boron tribromide in 1.8 ml of dichloromethane (1.0 m solution) was added to a solution of the protected diphenyl ether (1.4 mmol) in 3 ml of dry dichloromethane at −78 °C for 1 h and then at room temperature for 3 h. When thin layer chromatography showed that the reaction had reached completion, the reaction mixture was quenched with methanol at −78 °C and concentrated to give an oil. A suspension of this oil in dichloromethane was washed with 10% aqueous sodium bicarbonate solution, and the organic layer was drawn off, washed sequentially with water then brine, and dried over MgSO4. The organic layer was filtered and concentrated under vacuum to give the crude product, which was then purified by flash column chromatography (ethyl acetate/hexane). 1H NMR (300 MHz, CDCl3): δ 0.87–0.92 (m, 3H), 1.27–1.36 (m, 6H), 1.57–1.63 (m, 2H), 2.30 (s, 3H), 2.52–2.57 (t, J = 7.5 Hz, 2H), 5.56 (s, 1H), 6.61–7.26 (m, 7H); ESI-MS (m/z): 285.1 (M+1).
Substrate Preparation
Trans-2-dodecenoyl-CoA (DD-CoA) was synthesized from trans-2-dodecenoic acid by using the mixed anhydride method as described previously (17). Aliquots of the substrate were flash frozen in liquid N2 and stored at −80 °C.
Expression and Purification of InhA
InhA, the M. tuberculosis enoyl-ACP reductase, was expressed in E. coli strain BL21(DE3)pLysS. The cells were induced with 1 mm isopropyl-β-d-thiogalactopyranoside at an A600 of 0.8 and harvested by centrifugation after shaking for 12 h at 25 °C. The bacterial pellet was resuspended in 20 mm Tris-HCl, pH 7.9, containing 500 mm NaCl and 5 mm imidazole and lysed by sonication followed by centrifugation at 50,000 × g to remove cell debris. The supernatant was applied to a nickel affinity column (GE Healthcare), and InhA was eluted using a gradient of 60–500 mm imidazole. Fractions containing InhA were pooled and exchanged into 30 mm PIPES buffer, pH 6.8, containing 150 mm NaCl and 1 mm EDTA via desalting columns (GE Healthcare). For further purification, size exclusion chromatography was carried out using a Superdex 200 column (GE Healthcare) and the same buffer.
Steady State Kinetic Assay
Kinetic assays using DD-CoA and wild-type InhA were performed as described previously (9). The reactions were initiated by the addition of InhA to solutions containing substrate, inhibitor, and NADH in 30 mm PIPES and 150 mm NaCl, pH 6.8. The IC50 values were determined by varying the concentration of inhibitor in reactions containing 250 μm NADH, 25 μm DD-CoA, and 100 nm InhA. When the assays were performed at 10 nm InhA, the enzyme was stabilized by the addition of glycerol (8%, v/v) and bovine serum albumin (0.1 mg/ml). The experimental data were analyzed using Equation 1, where I is the inhibitor concentration, and y is the percentage of activity.
Data fitting was performed using Grafit 4.0 (Erithacus Software Ltd.).
Progress Curve Analysis
The slow onset inhibition of InhA by PT70 was monitored by adding the enzyme (5 nm) to assay mixtures containing glycerol (8%), bovine serum albumin (0.1 mg/ml), DMSO (2% v/v), DD-CoA (300 μm), NADH (250 μm), NAD+ (200 μm), and inhibitor (0–480 nm). The reactions were monitored until the progress curve became linear, indicating that the steady state had been reached. To ensure that substrate depletion would not significantly affect the reaction rate, low enzyme concentrations and high substrate concentrations were used. Progress curves were analyzed as described previously (13, 18). This involved fitting the data to Equation 2,
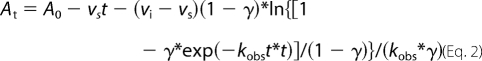 |
where γ = [E]*(1 − vs/vi)2/[I], vi and vs are the initial velocity and steady state velocity, and kobs is the observed rate constant for each progress curve. The values of kobs, vi, and vs obtained from Equation 2 were then fitted to Equations 3 and 4, which describe a two-step inhibition mechanism in which rapid binding of the inhibitor to the enzyme is followed by a second slow step that results in the final complex.
In these equations, K−1app and Kiapp are the apparent dissociation constants for the initial enzyme·inhibitor complex (E-I) and the final enzyme·inhibitor complex (E-I*), respectively.
In addition to monitoring the onset of enzyme inhibition, progress curves were also used to analyze the recovery of enzyme activity resulting from the slow dissociation of inhibitor from E-I*. InhA (0.5 μm) was preincubated with PT70 (0.3–0.9 μm) and NAD+ (200 μm) at room temperature. After 5 h, 5 μl of incubation mixture was diluted 100-fold into an assay mixture containing glycerol (8%), bovine serum albumin (0.1 mg/ml), DMSO (2% v/v), DD-CoA (300 μm), NADH (250 μm), and NAD+ (200 μm). Substrate consumption was monitored at 340 nm, and the resulting recovery progress curves were analyzed in a similar fashion to that described above (18, 19).
Preincubation Inhibition Assays for Slow Binding Inhibitors
Preincubation inhibition assays were performed to determine the preference of PT70 for the different cofactor-bound forms of InhA. These experiments were conducted as described previously (18). InhA (10 nm) was preincubated in the presence of a fixed concentration of DMSO (2%), NAD+ (10–200 μm), NADH (250 μm), and PT70 (0–1000 μm) for 5 h at 4 °C. The mixture was then warmed to room temperature, and the reaction was initiated by the addition of DD-CoA (30 μm). Equation 5 was used to estimate the apparent inhibition constant Ki′,
where v and v0 are the initial velocity in the presence and absence of inhibitor, respectively, and [I] is the inhibitor concentration. The Ki′ values obtained at different NAD+ concentrations were then fit to Equations 6–8, which describe the binding of the inhibitor to either E-NAD+ (Equation 6), E-NADH (Equation 7), or both E-NAD+ and E-NADH (Equation 8).
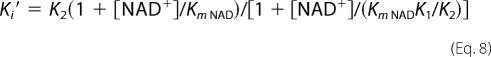 |
K1 and K2 are inhibition constants for inhibitor binding to the E-NAD+ and E-NADH forms.
Direct Determination of koff
Having shown that PT70 bound preferentially to the E-NAD+ product complex, the rate of dissociation of PT70 from InhA was monitored using 32P-NAD+ to provide a direct estimate for koff. These experiments followed a similar protocol to that described previously (5). InhA was incubated with PT70, NAD+, and 32P-NAD+ (800 Ci/mmol) for 5 h at room temperature to generate the ternary complex formed by InhA, NAD+, and PT70. After purification using a Sephadex G-75 spin column, 500 μl of the complex was diluted into 70 ml of buffer to initiate the dissociation of the inhibitor from the enzyme. Because NAD+ only has a weak affinity for the free enzyme (Kd > 2 mm), dissociation of PT70 from the ternary complex also leads to the release of NAD+ and 32P-NAD+ from the enzyme. Subsequently, 700 μl of the diluted complex solution was withdrawn at various time intervals, loaded into a microcon (Satorious 500, 10 kDa), and centrifuged in a microcentrifuge for 1 min at maximum speed. 450 μl of the filtrate was collected, and the amount of 32P-NAD+ was quantitated using a scintillation counter. A value for koff was obtained by fitting the data to Equation 9, where Nt is the number of radioactive counts (cpm) at time t, and N0 is the number of radioactive counts (cpm) following complete dissociation of the complex.
Crystallization
For the formation of the ternary InhA· NAD+·PT70 complex, the protein solution (10 mg/ml) was combined with NAD+ and PT70 in a molar ratio of 1:5:200. After 2 h of incubation on ice, the solution was centrifuged at 25,000 × g for 20 min, and the supernatant was used for crystallization by the hanging drop vapor diffusion technique. Equal volumes of protein solution and precipitant solution (12–16% (w/v) polyethylene glycol 4000, 1% DMSO, 100 mm N-(2-acetamido) iminodiacetic acid, pH 6.8, 100–250 mm ammonium acetate) were mixed and equilibrated against precipitant solution. Diffraction quality crystals of the complex grew within 4 days at 22 °C to a maximum size of 900 × 100 × 100 μm3. The crystals were transferred to precipitant solutions containing NAD+ and PT70 in excess and increasing amounts of DMSO in steps of 5% (v/v) to a final concentration of 25% and subsequently frozen in liquid nitrogen.
Data Collection, Structure Determination, and Refinement
Two diffraction data sets were collected at a Rigaku MicroMaxTM-007HF x-ray generator with an Raxis HTC detector. The first crystal diffracted to 2.1 Å, and the second crystal diffracted to 1.8 Å. Data sets 1 and 2 were indexed and processed using Mosflm (20) and XDS (21), respectively. Although the crystals grew in the same drop, they belong to different space groups: C2221 for data set 1 and P21 for data set 2. The data collection statistics are listed in Table 2. The two structures were solved by molecular replacement using Phaser (22). As a search model for data set 1, a monomer was derived from the Protein Data Bank entry 2b37 (10), omitting the NAD+ cofactor and the inhibitor. Molecular replacement yielded two molecules in the crystallographic asymmetric unit that form half of a homotetramer. Model building and refinement were carried out using alternating rounds of Coot (23) for manual model building and Refmac (24) for maximum likelihood refinement. Five percent of all reflections were omitted throughout the refinement for the calculation of Rfree. In the initial refinement rounds, the density improved significantly and allowed the unambiguous modeling of the NAD+ molecule, the inhibitor PT70, and the missing residues of the substrate-binding loop into the electron density with Coot.
TABLE 2.
Crystallographic data and refinement statistics
| InhA NAD+ PT70 |
||
|---|---|---|
| Crystal 1 | Crystal 2 | |
| Data collection | ||
| Space group | C2221 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 89.80, 157.51, 91.23 | 88.48, 90.27, 89.56 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 118.76, 90.00 |
| Resolution (Å) | 36.16-2.10 (2.21-2.10) | 78.57-1.81 (1.9-1.81) |
| Rmerge | 0.080 (0.300) | 0.101 (0.438) |
| I/σ I | 16.0 (2.5) | 6.1 (1.7) |
| Completeness (%) | 98.1 (89.7) | 92.5 (77.8) |
| Redundancy | 4.4 (2.9) | 1.6 (1.4) |
| Refinement | ||
| Resolution (Å) | 2.10 | 1.81 |
| No. reflections | 38141 | 112861 |
| Rwork/Rfree (%) | 17.2/21.6 | 16.8/20.3 |
| No. atoms | ||
| Protein | 4063 | 8033 |
| PT70 | 42 | 84 |
| NAD+ | 88 | 176 |
| Water | 143 | 682 |
| B-factors | ||
| Protein | 26.2 | 10.4 |
| PT70 | 26.9 | 7.3 |
| NAD+ | 19.7 | 3.9 |
| Water | 29.9 | 16.0 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.013 | 0.015 |
| Bond angles (°) | 1.465 | 1.534 |
For data set 2, the coordinates of one monomer of the refined first structure, omitting only the inhibitor, were used as a search model. Molecular replacement yielded one homotetramer in the crystallographic asymmetric unit. Refinement and model building was carried out as described above. The refinement statistics for the two data sets are listed in Table 2. The Ramachandran statistics calculated by MolProbity (25) show that the structures are of good quality. In structure 1, 96.9% of all residues are in the favored regions, and 100% are in the allowed regions. 96.4% of all residues of structure 2 are in the favored regions, and 99.9% in the allowed regions. Residue Asp42, located in a surface exposed loop, is an outlier of the Ramachandran plot. All of the structural figures were prepared with Pymol (26).
RESULTS
Kinetic Characterization
The inhibition of InhA by PT70 was characterized by determining the IC50 values using steady state kinetic methods. The IC50 values obtained using 100 nm and 10 nm InhA were 50.3 ± 7.0 and 5.3 ± 0.4 nm, respectively, indicating that PT70 is a tight binding inhibitor. Progress curve analysis was used to determine whether PT70 is also a slow onset inhibitor of InhA. The reaction conditions were adjusted so that consumption of DD-CoA occurred at a linear rate for up to 30 min. Upon addition of the inhibitor, the turnover velocity decreased exponentially with time, from an initial velocity (vi) to a steady state velocity (vs). Additionally, with increasing concentrations of PT70, both vi and vs decreased, whereas kobs increased, and the time required to reach vs decreased (Fig. 2A). This behavior is a classic example of slow onset inhibition in which the rapid formation of the initial E-I complex is followed by a second slow step leading to the formation of the final E-I* complex (Fig. 3). Fitting the data to Equation 2 provided values for vi, vs, and kobs. The hyperbolic dependence of kobs on the concentration of PT70 was fitted to Equation 3, allowing the calculation of the constants for the conversion of E-I to E-I* (k2 and k−2 in Fig. 3) and also providing a value for K−1app, the dissociation constant of E-I (Fig. 2B). The dependence of vs/v0 on the concentration of PT70 was analyzed using Equation 4 to give Kiapp, the dissociation constant of the final E-I* complex. The thermodynamic and kinetic constants describing the inhibition of InhA by PT70 are summarized in Table 1. PT70 initially binds to InhA with a K−1app value of 62 nm, whereas the dissociation constant of the final E-I* complex Kiapp is ∼10 times smaller than K−1app (7.8 nm). Data analysis using Equations 2 and 3 also provided a value for k−2, the rate constant for conversion of E-I* to E-I. Assuming that k−1 ≫ k2 and k−2, then k−2 will be equal to koff, the dissociation rate constant for the formation of active enzyme from E-I*. The value of k−2 obtained from progress curve analysis of 0.041 min−1 is the same within experimental error to the values obtained when the recovery of enzyme activity was monitored (Fig. 2C; 0.044 min−1) and when koff was determined directly by following the dissociation of 32P-NAD+ from the enzyme·inhibitor ternary complex (0.043 min−1). Thus, the residence time of PT70 on the enzyme (1/koff) is 24 min, which is ∼3-fold smaller than that of the INH-NAD adduct (Table 1). Most diphenyl ethers preferentially bind to the E-NAD+ product complex of InhA and other FabI enzymes and only occasionally prefer the E-NADH form of the enzyme (9, 10, 13, 27–29). Preincubation studies were used to examine which form of the enzyme PT70 prefers. InhA and PT70 were preincubated in the presence of saturating NADH (250 μm) and at different fixed concentrations of NAD+ (10, 30, 50, 100, and 200 μm). Assays were initiated by adding the substrate DD-CoA to obtain the apparent inhibition constant Ki′ at each concentration of NAD+. The dependence of Ki′ on the concentration of NAD+ was fit to Equations 6–8 using a Km NAD value of 4 mm and a Km NADH value of 48 μm. The dependence of Ki′ on the concentration of NAD+ was best described by Equation 8 (Fig. 2D), indicating that PT70 binds to both the E-NAD+ and E-NADH forms of the enzyme, albeit with a strong (4000-fold) preference for the enzyme-oxidized cofactor product complex (K1 = 0.022 nm, K2 = 90.6 nm; Table 1). The value of K1 is significantly smaller than that obtained for Kiapp (7.8 nm); however, the latter was determined at a fixed concentration of NAD+.
FIGURE 2.

Progress curve analysis for the inhibition of InhA by PT70 and effect of NAD+ on the apparent inhibition constant of PT70. A, progress curves were obtained for inhibitor concentrations ranging from 0 to 480 nm. The solid curves are the best fits of the data to Equation 2. B, kobs plotted as a hyperbolic function of [PT70] using Equation 3. C, progress curves of InhA activity recovery were obtained for inhibitor concentrations ranging from 0 to 9 nm. The solid curves are the best fits of the data to Equation 2 to obtain kobs, which was then plotted against [PT70] using Equation 3 to obtain k−2. D, effect of NAD+ on the apparent inhibition constant of PT70. The fitted curves are shown for Equation 6 (dashed line; K1 = 0.0146 ± 0.0009 nm), Equation 7 (dotted line; K2 = 13.6 ± 5.5 nm), and Equation 8 (solid line; K1 = 0.022 ± 0.001 nm, K2 = 90.6 ± 9.7 nm).
FIGURE 3.

Kinetic scheme for the slow onset inhibition of InhA by PT70.
TABLE 1.
Inhibition of InhA by PT70 and the INH-NAD adduct
| Inhibitor | k2 | k−2 | t½a | Residence timea | K−1app | Kiapp | koffb | K1c |
|---|---|---|---|---|---|---|---|---|
| min−1 | min−1 | min | min | nm | nm | min−1 | pm | |
| PT70 | 0.46 ± 0.003 | 0.041 ± 0.003e | 17 ± 1 | 24 ± 2 | 62 ± 2 | 7.8 ± 0.4 | 0.043 ± 0.006 | 22 ± 1 |
| PT70 | 0.46 ± 0.003 | 0.044 ± 0.008f | 17 ± 1 | 24 ± 2 | 62 ± 2 | 7.8 ± 0.4 | 0.043 ± 0.006 | 22 ± 1 |
| INH-NADd | 0.13 ± 0.01 | 0.016 ± 0.007 | 43 ± 12 | 63 ± 27 | 100 ± 75 | 5.0 ± 0.5 | 0.017 ± 0.001 |
a t½ = 0.693/koff and residence time = 1/koff, where koff = k−1k−2/(k−1 + k2 + k−2). Assuming that k−1 ≪ k2 and k−2, then koff ≈ k−2.
b koff was determined by monitoring the rate of release of 32P-NAD+ from the ternary enzyme·inhibitor complex.
c K1 was determined from preincubation experiments.
d The data were taken from Ref. 5.
e k−2 was determined by progress curve of enzyme·inhibitor complex formation.
f k−2 was determined by progress curve of enzyme activity recovery.
Structure of the Ternary InhA·PT70·NAD+ Complex
The binding of PT70 to InhA and the basis for the slow binding step was further characterized by structural studies. Two structures of the ternary InhA·NAD+·PT70 complex were solved. The first structure was solved at a resolution of 2.1 Å resolution and belongs to space group C2221, the same space group as in the ternary InhA complex with 5-octyl-2-phenoxyphenol and NAD+ (8PP; Protein Data Bank code 2b37 (10)). The second structure was obtained at 1.8 Å resolution and belongs to space group P21, which has not been reported for InhA so far (structural data are summarized in Table 2) (Fig. 4). The slow onset inhibitor PT70 binds to the substrate-binding site with the two rings of the inhibitor oriented almost 90° to each other, comparable with the rings of the diphenyl ethers 5-pentyl-2-phenoxyphenol and 8PP described previously (10). Hydrogen bonds are formed between the inhibitor hydroxyl group and Tyr158, and a hydrogen bonding network is formed between the 2′-hydroxyl group of NAD+ and Lys165 (Fig. 5, red dotted lines). A π-π stacking interaction between the B-ring of PT70 and the nicotinamide ring of NAD+ further stabilizes the conformation of the inhibitor. The alkyl chain of PT70 extends into the hydrophobic environment of the substrate binding cavity and forms hydrophobic interactions with residues Phe149 and Tyr158. Most importantly, however, is the substrate-binding loop (residues 195–210, helix α6) of the structures reported here, which is ordered and forms a helix that covers the entrance to the active site (Fig. 4). Hydrophobic interactions are formed between the phenyl rings of the inhibitor and amino acids Ala198, Ile202, Val203, and Met199 of the substrate-binding loop (Fig. 5). Ala198 forms hydrophobic interactions with the B-ring methyl group at a distance of 3.4 Å, Ile202 interacts with the B-ring at a distance of 3.7 Å, and Met199 interacts with the A-ring at a distance of 3.8 Å. Val203 forms hydrophobic interactions with both phenyl rings at a distance of 4 Å and with the acyl chain of PT70 at 3.7 Å.
FIGURE 4.

Loop ordering upon slow binding inhibition. A, one monomer of the ternary InhA·NAD+·8PP complex (Protein Data Bank code 2b37) is shown in cartoon representation with the NAD+ molecule in cyan and the 8PP molecule in black and all-bonds representation. The substrate-binding loop is disordered in the 8PP structure, and the loop ends are depicted in red. B, monomer of the ternary InhA·NAD+·PT70 complex using the same colors and orientation as in A. The substrate-binding loop is ordered in this structure and covers the binding pocket (red cartoon). Secondary structure elements for both molecules were assigned with STRIDE (34).
FIGURE 5.

Close-up of the binding pocket of InhA with bound NAD+ and PT70. Hydrogen bonds between PT70 (black) and Tyr158 (yellow) as well as the NAD+ molecule (cyan) are indicated as red dotted lines. The important hydrophobic residues Ala198, Met199, Ile202, and Val203 of the substrate-binding loop are shown in all bonds representation (red). An overlay of different triclosan inhibitors (PT70, black; triclosan, gray; 8PP, blue; JPL, green) in the binding pocket of InhA displays the differences in the orientation of the B-ring.
Additionally, the PT70 methyl group forms van der Waal's contacts to the phosphate groups of the NAD+ cofactor, resulting in a 1 Å shift of the B-ring upward relative to its position in 8PP. In contrast, when triclosan is bound to InhA the B-ring is tilted by ∼25° toward Ile202 of the substrate-binding loop and thereby interferes sterically with the loop residues, pushing the loop away from the substrate cavity instead of keeping it in place (Fig. 5).
DISCUSSION
We have used structure-based design to develop a slow onset inhibitor that directly targets InhA, the enoyl-ACP reductase of M. tuberculosis. Previous work resulted in the development of a series of alkyl-diphenyl ethers that are nanomolar inhibitors of InhA (10). The best inhibitor of this series, 8PP, is active against drug-sensitive and drug-resistant strains of MTB. Although these inhibitors have high affinity for InhA, they are still rapid reversible inhibitors. However, based on these encouraging results, we rationally modified the alkyl-diphenyl ethers to promote interactions between the inhibitor and the loop that becomes ordered during slow onset inhibition.
The resulting inhibitor, PT70, shows improved affinity for InhA compared with the first generation compounds reported previously (10, 11). Like the parent compound 5-hexyl-2-phenoxyphenol that lacks the methyl group (6PP; Fig. 1), PT70 binds preferentially to the E-NAD+ product. However, the K1 value of 22 pm for PT70 is ∼430-fold smaller than the Ki′ value of 9.4 nm for 6PP (10), and thus introduction of the methyl group has dramatically increased the affinity of the inhibitor for the enzyme complex. More importantly, this inhibitor now also displays slow onset inhibition, which is expected to be crucial for its in vivo antibacterial activity. Assuming that kon for 6PP is limited by the rate of encounter of enzyme and inhibitor (109 m−1 s−1), then the Ki′ value of 9.4 nm allows koff for 6PP to be estimated at 9.4 s−1, giving a residence time of 0.1 s for 6PP on the enzyme. This value is in stark contrast to the residence time of 24 min for PT70, a 14,000-fold increase compared with 6PP.
The crystal structures of the ternary InhA·NAD+·PT70 complex reveal how the inhibitor is bound to the active site. The observed hydrophobic interactions and the hydrogen bonding network of PT70 are similar to the observed interactions of previously described triclosan derivatives (10). In addition, however, PT70 generates hydrophobic interactions to amino acids Ala198, Met199, Ile202, and Val203, which are part of the substrate-binding loop (residues 195–210 in InhA). This substrate-binding loop is disordered in InhA structures in the presence of triclosan as well as in the structures with the improved 5-pentyl-2-phenoxyphenol and 8PP triclosan derivatives that, despite their improved IC50 values, remain to be rapid reversible inhibitors (10). In contrast, triclosan is a slow onset inhibitor of E. coli FabI, and the corresponding crystal structure shows that the substrate-binding loop is ordered (15). The substrate-binding loop is also ordered in the crystal structure of InhA inhibited by the slow onset INH-NAD adduct (14). These observations prompted us to speculate that compounds with the ability to cause loop ordering will be slow, tight binding inhibitors of InhA (10). Although loop ordering with respect to INH-NAD binding remains elusive, the triclosan bound E. coli FabI structure clearly reveals that the inhibitor forms hydrophobic interactions with the loop amino acids Ala196 and Ala197, which correspond to Ala198 and Met199 in InhA. Additionally, Rozwarski et al. (30) identified in the crystal structure of InhA with a C16 fatty acyl substrate that hydrophobic amino acids of the loop are important for proper substrate binding into the cavity. Interestingly, the last few carbon atoms of the fatty acid interact with the hydrophobic amino acids Ala198, Met199, and Ile202. A fatty acid shorter than 16 carbons might not be accommodated correctly by the enzyme because of missing interactions with Ala198, Met199, and Ile202.
These observations strongly suggest that the interactions with the three amino acids (Ala198, Met199, and Ile202) are important determinants for loop ordering. An inhibitor, like PT70, that is able to directly interact with these residues leads to a defined loop structure. The ordered substrate-binding loop covers the entrance to the binding pocket and thereby locks the inhibitor into the cavity and increases its residence time. It is conceivable that the conformational change of the loop poses the slow step observed in the binding studies.
It is important to note that Freundlich et al. (11) recently reported InhA structures inhibited by triclosan derivatives that have an ordered substrate-binding loop in two of the four structures (Protein Data Bank codes 3fng and 3fnh). However, it was not reported, whether the compounds are slow binding inhibitors. The possibility that loop ordering is due to intermolecular contacts in space group I4122, which has not been reported so far for InhA structures, cannot be excluded. In this space group residues 197–205 of the substrate-binding loop are closely packed with atoms of symmetry-related molecules so that interactions caused by crystallographic packing could be the reason for the ordered substrate-binding loop. This contention is also supported by the fact that the cocrystal structures of InhA with the other inhibitors from this series that crystallized in space group C2 (Protein Data Bank codes 3fne and 3fnf) remain disordered in the loop segment. In the case of our PT70 structures that crystallized in space groups C2221 and P21, crystal contacts can be ruled out to be responsible for loop ordering, and instead the hydrophobic interactions of the residues with the inhibitor itself lead to the formation of the stable helix of the substrate-binding loop. Close inspection of the solvent areas between the molecules in the crystal packing demonstrate that the closest contact between two of the loops within the tetramer and the symmetry-related molecule is at least ∼5 Å, and no salt bridges or hydrogen bonds exist between the molecules that directly stabilize the loop.
The substrate-binding loop in the InhA·NAD+·PT70 structure adopts a different conformation compared with that observed in other InhA complexes in which the substrate-binding loop is also ordered. The loop in the PT70-bound structures is close to the substrate-binding pocket because of hydrophobic interactions with the inhibitor, whereas the loops in the other structures are located farther from the pocket. Although the fatty acid in the substrate-bound structure (Protein Data Bank code 1bvr (30)) does interact with residues Ala198, Ile202, and Met199, movement of the loop in this case is thought to be necessary to widen the binding pocket to accommodate the long chain fatty acid. However, in the InhA complexes with the triclosan derivative JPL (Protein Data Bank code 3fng (11)) and the pyrrolidine carboxamide inhibited form (Protein Data Bank code 2h7m (31)), movement of the loop away from the substrate-binding pocket could be due to the reduced number of contacts to Ala198, Met199, and Ile202. Although a number of contacts are present between enzyme and inhibitor in the PT70 structure, a detailed analysis of the structural data suggests that interactions with the side chain of Ile202, which is turned toward the PT70 B-ring, is particularly important for orientation of the substrate-binding loop close to the active site.
Although measurements of inhibitor residence time are not normally incorporated into drug discovery programs, there is growing evidence that residence time is a critical factor for in vivo drug activity (12, 13, 32, 33). In particular, slow onset inhibitors will spend longer times bound to their targets compared with rapid reversible inhibitors and will remain bound even when free drug concentrations are low. Recent studies with the FabI enzyme from F. tularensis highlight the importance of this concept, where it was seen that residence time was a better predictor of in vivo activity than the thermodynamic affinity of the inhibitor for the enzyme (13).
The new characteristics of PT70 facilitate the hydrophobic interactions to the important loop residues of InhA, resulting in slow tight binding inhibition. Our work supports the prediction that slow onset inhibition of enoyl-ACP reductases is indeed coupled to loop ordering. Slow binding and improved residence time are expected to result in significant improvements in in vivo antibacterial activity.
This work was supported, in whole or in part, by National Institutes of Health Grant AI44639 (to P. J. T.). This work was also supported by Deutsche Forschungsgemeinschaft Grant SFB 630 and Forschungszentrum Grant FZ82 (to C. K.).
The atomic coordinates and structure factors (codes 2x22 and 2x23) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- INH
- isoniazid
- 6PP
- 5-hexyl-2-phenoxyphenol
- 8PP
- 5-octyl-2-phenoxyphenol
- ACP
- acyl carrier protein
- DD-CoA
- trans 2-dodecenyl-CoA
- PT70
- 2-(o-tolyloxy)-5-hexylphenol
- PIPES
- 1,4-piperazinediethanesulfonic acid
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1.WHO (2007) WHO Report, WHO, Switzerland [Google Scholar]
- 2.Jain A., Mondal R. (2008) FEMS Immunol. Med. Microbiol. 53, 145–150 [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y., Heym B., Allen B., Young D., Cole S. (1992) Nature 358, 591–593 [DOI] [PubMed] [Google Scholar]
- 4.Banerjee A., Dubnau E., Quemard A., Balasubramanian V., Um K. S., Wilson T., Collins D., de Lisle G., Jacobs W. R., Jr. (1994) Science 263, 227–230 [DOI] [PubMed] [Google Scholar]
- 5.Rawat R., Whitty A., Tonge P. J. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 13881–13886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barry C. E., 3rd, Lee R. E., Mdluli K., Sampson A. E., Schroeder B. G., Slayden R. A., Yuan Y. (1998) Prog. Lipid Res. 37, 143–179 [DOI] [PubMed] [Google Scholar]
- 7.Vilchèze C., Morbidoni H. R., Weisbrod T. R., Iwamoto H., Kuo M., Sacchettini J. C., Jacobs W. R., Jr. (2000) J. Bacteriol. 182, 4059–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McMurry L. M., Oethinger M., Levy S. B. (1998) Nature 394, 531–532 [DOI] [PubMed] [Google Scholar]
- 9.Parikh S. L., Xiao G., Tonge P. J. (2000) Biochemistry 39, 7645–7650 [DOI] [PubMed] [Google Scholar]
- 10.Sullivan T. J., Truglio J. J., Boyne M. E., Novichenok P., Zhang X., Stratton C. F., Li H. J., Kaur T., Amin A., Johnson F., Slayden R. A., Kisker C., Tonge P. J. (2006) ACS Chem. Biol. 1, 43–53 [DOI] [PubMed] [Google Scholar]
- 11.Freundlich J. S., Wang F., Vilchèze C., Gulten G., Langley R., Schiehser G. A., Jacobus D. P., Jacobs W. R., Jr., Sacchettini J. C. (2009) Chem. Med. Chem. 4, 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Copeland R. A., Pompliano D. L., Meek T. D. (2006) Nat. Rev. 5, 730–739 [DOI] [PubMed] [Google Scholar]
- 13.Lu H., England K., am Ende C., Truglio J. J., Luckner S., Reddy B. G., Marlenee N. L., Knudson S. E., Knudson D. L., Bowen R. A., Kisker C., Slayden R. A., Tonge P. J. (2009) ACS Chem. Biol. 4, 221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vilchèze C., Wang F., Arai M., Hazbón M. H., Colangeli R., Kremer L., Weisbrod T. R., Alland D., Sacchettini J. C., Jacobs W. R., Jr. (2006) Nat. Med. 12, 1027–1029 [DOI] [PubMed] [Google Scholar]
- 15.Ward W. H., Holdgate G. A., Rowsell S., McLean E. G., Pauptit R. A., Clayton E., Nichols W. W., Colls J. G., Minshull C. A., Jude D. A., Mistry A., Timms D., Camble R., Hales N. J., Britton C. J., Taylor I. W. (1999) Biochemistry 38, 12514–12525 [DOI] [PubMed] [Google Scholar]
- 16.am Ende C. W., Knudson S. E., Liu N., Childs J., Sullivan T. J., Boyne M., Xu H., Gegina Y., Knudson D. L., Johnson F., Peloquin C. A., Slayden R. A., Tonge P. J. (2008) Bioorg. Med. Chem. Lett. 18, 3029–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parikh S., Moynihan D. P., Xiao G., Tonge P. J. (1999) Biochemistry 38, 13623–13634 [DOI] [PubMed] [Google Scholar]
- 18.Copeland R. A. (2005) Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, pp. 141–154, Wiley, Hoboken, NJ: [PubMed] [Google Scholar]
- 19.Williams J. W., Morrison J. F., Duggleby R. G. (1979) Biochemistry 18, 2567–2573 [DOI] [PubMed] [Google Scholar]
- 20.Leslie A. G. (1992) in Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography, No. 26 [Google Scholar]
- 21.Kabsch W. (1988) J. Appl. Crystallogr. 21, 67–71 [Google Scholar]
- 22.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emsley P., Cowtan K. (2004) Acta Crystallogr. Sect. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 24.Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. Sect. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 25.Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, Palo Alto, CA [Google Scholar]
- 27.Sivaraman S., Sullivan T. J., Johnson F., Novichenok P., Cui G., Simmerling C., Tonge P. J. (2004) J. Med. Chem. 47, 509–518 [DOI] [PubMed] [Google Scholar]
- 28.Rafi S. B., Cui G., Song K., Cheng X., Tonge P. J., Simmerling C. (2006) J. Med. Chem. 49, 4574–4580 [DOI] [PubMed] [Google Scholar]
- 29.Lu H., Tonge P. J. (2008) Acc. Chem. Res. 41, 11–20 [DOI] [PubMed] [Google Scholar]
- 30.Rozwarski D. A., Vilchèze C., Sugantino M., Bittman R., Sacchettini J. C. (1999) J. Biol. Chem. 274, 15582–15589 [DOI] [PubMed] [Google Scholar]
- 31.He X., Alian A., Stroud R., Ortiz de Montellano P. R. (2006) J. Med. Chem. 49, 6308–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewandowicz A., Tyler P. C., Evans G. B., Furneaux R. H., Schramm V. L. (2003) J. Biol. Chem. 278, 31465–31468 [DOI] [PubMed] [Google Scholar]
- 33.Tummino P. J., Copeland R. A. (2008) Biochemistry 47, 5481–5492 [DOI] [PubMed] [Google Scholar]
- 34.Heinig M., Frishman D. (2004) Nucleic Acids Res. 32, W500–W502 [DOI] [PMC free article] [PubMed] [Google Scholar]