

Abstract
Mitogen-activated protein kinase (MAPK) pathways are involved in the regulation of cellular responses, including cell proliferation, differentiation, cell growth, and apoptosis. Because these responses are tightly related to cellular energy level, AMP-activated protein kinase (AMPK), which plays an essential role in energy homeostasis, has emerged as another key regulator. In the present study, we demonstrate a novel signal network between AMPK and MAPK in HCT116 human colon carcinoma. Glucose deprivation activated AMPK and three MAPK subfamilies, extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 MAPK. Under these conditions, inhibition of endogenous AMPK by expressing a dominant-negative form significantly potentiated ERK activation, indicating that glucose deprivation-induced AMPK is specifically antagonizing ERK activity in HCT116 cells. Moreover, we provide novel evidence that AMPK activity is critical for p53-dependent expression of dual-specificity phosphatase (DUSP) 1 & 2, which are negative regulators of ERK. Notably, ERK exhibits pro-apoptotic effects in HCT116 cells under glucose deprivation. Collectively, our data suggest that AMPK protects HCT116 cancer cells from glucose deprivation, in part, via inducing DUSPs, which suppresses pro-apoptotic ERK, further implying that a signal network between AMPK and ERK is a critical regulatory point in coupling the energy status of the cell to the regulation of cell survival.
Keywords: AMP-activated Kinase (AMPK), Apoptosis, Carcinogenesis, Dual Specificity Phosphoprotein Phosphatase, ERK
Introduction
AMP-activated protein kinase (AMPK)2 is a heterotrimeric serine/threonine kinase that consists of an α catalytic subunit and regulatory β and γ subunits, and plays a central role in cellular adaptation to ATP-depleting stresses such as glucose deprivation. Increases in the cellular AMP:ATP ratio promotes AMPK activation through allosteric binding of AMP, which changes AMPK into a better substrate for phosphoactivation via an upstream kinase. Once activated, AMPK inhibits the ATP consuming pathway, while activating ATP-generating pathways, to optimize total cellular ATP levels (1). In fact, AMPK protects cells from ATP-depleting stresses (1). However, a pro-apoptotic role of AMPK was also reported, and such a role is mediated, in part, by tumor suppressor proteins associated with the AMPK signaling network, such as LKB1 (2), tuberous sclerosis complex (TSC2) (3), and p53 (4). Thus, an extremely sophisticated regulatory system involving AMPK exists for monitoring the level of cellular energy under stress conditions and then driving cells to either survival or apoptosis.
Mitogen-activated protein kinase (MAPK) pathways are involved in the regulation of cellular responses, including cell proliferation, differentiation, cell growth, and apoptosis. Because these cellular responses are tightly related to the cellular energy level, a signal network between AMPK and MAPKs has emerged as a key regulatory point of significance. The three major subfamilies of MAPK are extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 MAPK. In general, ERK has been implicated in the regulation of growth factor-induced cell proliferation, whereas JNK and p38 are known to contribute to stress-induced cell apoptosis (5–7). However, their role can vary depending upon the cell type, the stimulus, and the duration of activation (8). These MAPKs are activated by dual-specific upstream kinases through reversible phosphorylation of both threonine and tyrosine resides of the TXY motif (9, 10). Conversely, the dephosphorylation of either residue is sufficient for kinase inactivation, and this is achieved largely by MAPK phosphatases, also known as dual-specificity protein phosphatases (DUSP). More than 11 different DUSPs have been identified that are highly specific for MAPKs but differ in MAPK substrate specificity (11–13). Cross-talk between ERK and AMPK has been tested by several researchers, and most of these studies were performed using the artificial AMPK activator, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR). In certain cases, pharmacological activation of AMPK resulted in ERK activation, which induced proliferation of cardiac fibroblasts (14), catecholamine secretion in PC12 cells (15), glucose transport in rat EDL muscle cells (16), potentiation of insulin signaling in rat heart (17), and suppression of protein synthesis in C2C12 myotubes (18). In contrast, ERK activity induced by IGF-1 (19), angiotensin II (20), or transaortic constriction (21), was down-regulated by AICAR. Consequently, the molecular mechanisms that link the two signal pathways remain unclear.
We therefore examined the role of AMPK on the regulation of ERK in HCT116 human colon carcinoma under physiologically relevant conditions that activate both AMPK and ERK. As solid tumors outgrow the existing vasculature, they are continuously exposed to nutrient-depleted microenvironments, such as glucose deprivation, and must adapt to such environments for survival. Here, we demonstrate that AMPK induces the expression of DUSP 1 & 2 via p53 activation under glucose deprivation, which leads to suppression of ERK activity. Notably, the glucose deprivation-induced ERK activity is pro-apoptotic. Collectively, our data suggest that AMPK protects HCT116 cancer cells from glucose deprivation, in part, via induction of DUSPs, which suppresses pro-apoptotic ERK.
EXPERIMENTAL PROCEDURES
Materials and DNA Constructs
RPMI medium 1640, Dulbecco's modified Eagle's medium, and fetal bovine serum were from Invitrogen (Carlsbad, CA). PD98059 and U0126 were obtained from Sigma. The antibodies for the phosphoactivated form of AMPKα-Thr172, ERK1&2 (Thr202/Tyr204), SAPK/JNK (Thr183/Tyr185), p38 (Thr180/Tyr182), ACC-Ser79, MEK (Ser217/221), p53 (Ser46), and p53 (Ser15) were from Cell Signaling Technology (Danvers, MA). Antibodies for DUSP2, c-Myc, ERK, JNK, p38, p53, and α-actinin, and siRNA for control, DUSP1, and DUSP2 were from Santa Cruz Biotechnology (Santa Cruz, CA). Plasmid p21-WT-luc, the promoter construct of wild-type p21, the p21dΔp53-luc (p53 binding site deletion form of p21-WT promoter), p53-WT, and the p53-DN (p53-R175D, dominant-negative mutant of wild-type p53) was generously provided by Dr. Xiao-Fan Wang (Duke University Medical Center, Durham, NC), Dr. H. D. Youn (Seoul National University, Korea) and Dr. Karen H. Vousden (ABL Basic Research Program, NCI-FCRDC, Frederick, Maryland 21702). c-Myc-tagged AMPKα2-WT and -DN (AMPK-K45R in which Lys45 was replaced with Arg) construct were generously provided by Dr. Morris J. Birnbaum (Howard Hughes Medical Institute and University of Pennsylvania School of Medicine, Philadelphia, PA). The regulatory and promoter region of the human DUSP2 gene was cloned by PCR from normal human genomic DNA using corresponding primers based on the human genome data base. The DUSP2 promoter was subcloned into the pGL3-basic reporter (Promega). The deletion of the core palindrome sequence (CCCCAC) in DUSP2 of the pDUSP2-luc, pDUSP2Δp53-luc, was generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol (22).
Cell Culture and Glucose Deprivation
HCT116p53+/+, HCT116p53−/− (human colon carcinoma), HepG2 (human hepatoma), and AGS (human gastric carcinoma) cells were maintained in RPMI supplemented with 25 mm glucose and 10% heat-inactivated fetal bovine serum and antibiotics at 37 °C with 95% air and 5% CO2. AMPKα+/+ or AMPKα−/− mouse embryo fibroblasts (MEF) were a generous gift from Dr. Benoit Viollet (René Descartes University, Paris, France) and maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum and antibiotics at 37 °C with 95% air and 5% CO2. Cells were rinsed three times with phosphate-buffered saline and then exposed to glucose-free RPMI medium 1640 (Invitrogen) containing 10% fetal bovine serum.
Transient Transfection
Plasmids were transfected into cells using GenePORTER transfection reagent (Gene Therapy Systems, San Diego, CA) according to the manufacturer's instructions. pcDNA was used as a blank plasmid to balance the amount of DNA introduced in transient transfection. After 24 h of transfection, cells were exposed to the low glucose condition or other stimuli for the indicated time period.
RNA Isolation and Real-time PCR
Total RNA was extracted with TRIzol Reagent (Invitrogen, Gaithersburg, MD). Whole-cell RNA was then reverse-transcribed into cDNA using avian myeloblastosis virus reverse transcriptase (Takara, Otsu, Japan) with oligonucleotide random primers. Real-time polymerase chain reaction was performed to quantify messenger RNA expressions using SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA) and the ABI PRISM® 7300 real-time PCR system (Applied Biosystems), according to the manufacturer's instructions. Relative messenger RNA expression was quantified using the comparative Ct (ΔCt) method and expressed as 2−ΔΔCt, where ΔΔCt = ΔE − ΔC, ΔE = CtEtarget − CtEβ-actin and ΔC = Ctctarget − Ctcβ-actin (E is the experimental result and C is controls). Each assay was done in triplicate and expressed as the mean ± S.D. A series of dilutions were prepared from a stock solution of total RNA to generate a standard curve to check the efficiencies of each reaction. A slope of −3.3 indicated reaction linearity. The primers for the PCR analysis were as follows: for DUSP1 Forward 5′-TTTGAGGGTCACTACCAG-3′, DUSP1 Reverse 5′-GAGATGATGCTTCGCC-3′; DUSP2 Forward 5′-AGTCACTCGTCAGACC-3′, DUSP2 Reverse 5′-TGTTCTTCACCCAGTCAAT-3′; DUSP3 Forward 5′-ACGTCAACACCAAT-GC-3′, DUSP3 Reverse 5′-ATGAGGTAGGCGATAACT-3′; DUSP4 Forward 5′-CAAA-GGCGGCTATGAG-3′, DUSP4 Reverse 5′-GGTTATCTTCCACTGGG-3′; DUSP5 Forward 5′-CTGAGTGTTGCGTGGA-3′, DUSP5 Reverse 5′-AGTCTATTGCTTCTTG-AAAGT-3′; DUSP6 Forward 5′-CGAGACCCCAATAGTGC-3′, DUSP6 Reverse 5′-AAT-GGCCTCAGGGAAA-3′; DUSP7 Forward 5′-TCATTGACGAAGCCCG-3′, DUSP7 Reverse 5′-GCGTATTGAGTGGGAACA-3′; DUSP8 Forward 5′-GACGCAAAATGGA-ATAAGC-3′, DUSP8 Reverse 5′-CTTCACGAACCTGTAGGC-3′; DUSP9 Forward 5′-ATCCGCTACATCCTCAA-3′, DUSP9 Reverse 5′-AGGTCATAGGCATCGTT-3′; DUSP10 Forward 5′-CTGAACATCGGCTACG-3′, DUSP10 Reverse 5′-GGTGTAAGGATTCTC-GGT-3′; DUSP14 Forward 5′-CTGCTCACTTAGGACTTTCT-3′, DUSP14 Reverse 5′-C-CTTGGTAGCGTGCTG-3′; DUSP16 Forward 5′-AGAATGGGATTGGTTATGTG-3′, DUSP16 Reverse 5′-TGTAGGCGATAGCGATG-3′; GAPDH Forward 5′-AAGGTCGGAGTCAACGGATT-3′, GAPDH Reverse 5′-CTCCTGGAAGATGGTGATGG-3′.
Fluorescence-activated Cell Sorting Analysis
Cells were harvested by trypsinization, collected by centrifugation, washed with phosphate-buffered saline, fixed in 70% ethanol, and resuspended in phosphate-buffered saline containing 10 μg/ml PI. After sorting out viable cells, fluorescence intensity was measured by flow cytometry (Becton Dickinson, San Jose, CA) using excitation and emission wavelengths of 488 and 525 nm, respectively.
Reporter Gene Assay
Transfection with the DUSP2-luc reporter, p21WT-luc reporter, p21Δp53-luc (p53 binding site deletion form of p21-WT promoter) reporter, or p53 response element-luc reporter constructs were performed. HCT116 cells were seeded onto 24-well culture plates at 4 × 104 cells/well and incubated for 24 h in medium. Plasmids were transfected into cells using GenePORTER transfection reagent (Gene Therapy Systems) according to the manufacturer's instructions. For co-transfections, a 1:1 ratio between DUSP2-luc and pcDNA containing AMPK-WT or AMPK-DN was used. After 24 h of transfection, cells were exposed to glucose deprivation. Luciferase activity was determined by mixing 20 μg of cell extract with 100 μl of luciferase assay reagent (Promega) and subsequent measurement of relative light units for 10 s in a luminometer (TD-20/20 luminometer, Turner Designs). At least three independent transfections were performed in triplicate.
RNA Interference
To knock down the DUSP1 and DUSP2, HCT116p53+/+ cells were transiently transfected with 0.25 μl/ml of chemically synthesized siRNAs targeting DUSP1 and DUSP2 or with the nonsilencing control siRNA (Santa Cruz Biotechnology) using GenePORTER transfection reagent (Gene Therapy Systems) according to the manufacturer's instructions. Total RNA and whole cell lysates were prepared 24 h after transfection.
Statistical Analysis
Results are expressed as the means ± S.E. We used Student's t test. Differences were considered significant at a p value of < 0.05.
RESULTS
AMPK Antagonizes ERK Activity after Glucose Deprivation in a MEK-independent Way
To examine the potential signal network between AMPK and MAPKs, we first examined the effect of glucose deprivation on the activity of these kinases after exposing HCT116p53+/+ cells to glucose-free medium for 24 h (Fig. 1A). Under these conditions, AMPK was significantly activated as assessed by the phosphorylation level of acetyl-CoA carboxylase (ACC) Ser79, a well-characterized substrate of AMPK (23). ERK, JNK, and p38, were also activated as determined by the phosphoactivated levels. Next, to examine the relationship between AMPK and MAPKs, cells were transfected with a eukaryotic expression vector encoding c-Myc-tagged AMPK wild-type (pAMPK-WT) or dominant-negative form (pAMPK-DN) to compare MAPK activation. Inhibition of endogenous AMPK function by AMPK-DN further increased ERK activation, but did not affect JNK and p38 MAPK (Fig. 1A). This relationship between AMPK and ERK was confirmed at glucose concentrations lower than 1 mm (Fig. 1B), and ERK induction by AMPK-DN was prominent at 24 h exposure to low glucose concentrations (Fig. 1C). These data suggest that AMPK inhibits ERK activity in response to glucose deprivation. The level of the phosphoactivated MEK, a direct upstream kinase of ERK, was not affected by AMPK-DN (Fig. 1D), suggesting that the AMPK target is not upstream of ERK.
FIGURE 1.

Inhibition of AMPK potentiates glucose deprivation-induced ERK activation in a MEK-independent manner. HCT116p53+/+ cells were transfected with pcDNA or c-myc-tagged pAMPK-WT or pAMPK-DN expression vector for 24 h and exposed to glucose deprivation for 24 h (A), the indicated concentration of glucose for 24 h (B, D), or glucose-free medium for the indicated time period (C, D). Under these conditions, the phosphorylated form of the indicated protein (P-ACC, P-ERK, P-JNK, P-p38, P-MEK) and the total form (ACC, ERK, JNK, p38, c-myc) was examined via Western blot analyses using specific antibodies. Experiments were repeated four times with similar results, and a representative result is shown. Each band was analyzed and quantified by densitometer. The data are expressed as the means ± S.E. (*, p < 0.05; compared with the indicated groups).
AMPK Is Critical for Induction of DUSP1 and -2 Expression under Glucose Deprivation
The steady-state level of ERK phosphorylation is a result of the balance between action of upstream kinases and phosphatases. Because MEK does not appear to be involved in AMPK-mediated ERK regulation (Fig. 1D), we hypothesized that MAPK phosphatase, also known as dual-specificity phosphatase (DUSP), might mediate the inhibitory effect of AMPK on ERK. We further suspected that long term regulation such as transcriptional control, rather than post-translational modification, may be responsible for this regulation because the effect required at least 24 h exposure to low glucose (Fig. 1C). To test this hypothesis, we examined the mRNA levels of 12 different DUSPs after glucose deprivation for 24 h, and found up-regulation of DUSP1 and DUSP2 (Fig. 2A), two genes that are known to be inducible by stress stimuli (22, 24–26). AMPK-DN significantly abrogated the induction of DUSP1 & 2 mRNA (Fig. 2B) and DUSP2 protein (Fig. 2C), whereas AMPK-WT showed positive effects.
FIGURE 2.

AMPK activation is critical for DUSP induction in response to glucose deprivation. A, HCT116p53+/+ cells were incubated in glucose-depleted medium for 24 h. Then, the mRNA levels of 12 DUSP members were determined by real-time PCR analysis, and the fold induction is expressed as a ratio of each DUSP: GAPDH mRNA. B, HCT116p53+/+ cells were transfected with pcDNA, pAMPK-WT, and pAMPK-DN expression vectors for 24 h, and maintained under glucose deprivation for 24 h. The amount of DUSP1 and -2 and GAPDH mRNA was evaluated by real-time PCR analysis. Results are the means ± S.E. for six determinations. C, HCT116p53+/+ cells were transfected with pcDNA, pAMPK-WT, and pAMPK-DN expression vectors for 24 h, and then were exposed to medium containing the indicated concentrations of glucose for 24 h (left panel) or incubated in glucose-free medium for the indicated times (right panel). The amount of DUSP2 protein was determined by Western blot analysis, and its level was quantified by densitometer. The data are expressed as the means ± S.E. (*, p < 0.05; compared with the indicated groups.) glucose+, 25 mm glucose; glucose-, 0 mm glucose.
To better understand the molecular mechanism, we constructed a luciferase reporter plasmid which contained a 0.7-kb DUSP2 promoter region (pDUSP2-luc) (22) and co-transfected it into HCT116p53+/+ cells with pcDNA, pAMPK-WT, or pAMPK-DN. Glucose deprivation for 24 h increased the luciferase activity of pDUSP2-luc ∼3∼4 fold, and this induction was significantly blocked by AMPK-DN (Fig. 3A). To confirm these results, we transfected pDUSP2-luc into mouse embryonic fibroblasts (MEF) where the AMPKα subunit is either expressed (AMPKα+/+) or deleted (AMPKα−/−), and then treated these cells with glucose-free medium for 24 h. Glucose deprivation increased luciferase activity 3-fold in AMPKα+/+ MEFs, but not AMPKα−/− MEFs (Fig. 3B, upper panel). There was essentially no difference in transfection efficiency between two cells as indicated by similar transfection levels of a GFP plasmid (Fig. 3B, lower panel). Therefore, our data suggest that AMPK activity is critical for DUSP2 expression at the transcription level after glucose deprivation.
FIGURE 3.

The activation of DUSP2 promoter by glucose deprivation requires AMPK. A, HCT116p53+/+ cells were co-transfected with a luciferase reporter plasmid containing DUSP promoter (pDUSP2-luc) and pcDNA, pAMPK-WT, or pAMPK-DN at a 1:1 ratio. After 24 h, cells were exposed to glucose depleted-medium for 24 h, and then luciferase activity was measured. B, mouse embryonic fibroblasts with AMPKα-deleted (AMPKα−/−) or wild type (AMPKα+/+) were co-transfected with pDUSP2-luc and an expression vector for green fluorescence protein for 24 h, and then exposed to glucose-free medium for 24 h. Then cell lysates were subjected to the luciferase activity assay or the intensity of fluorescence was measured. The data represent means ± S.E. for six determinations. (*, p < 0.05; compared with the indicated groups.)
Next, to determine whether DUSP1 or DUSP2 is involved in the regulation of ERK phosphorylation, we knocked down DUSP expression by transfecting HCT116p53+/+ cells with control-, DUSP1-, and DUSP2-siRNA (Fig. 4). DUSP1 and DUSP2 siRNA specifically reduced target levels after glucose deprivation (Fig. 4A), and increased ERK phosphorylation (Fig. 4B). These data confirm the role of DUSP1 and DUSP2 in negative regulation of ERK activity after glucose deprivation. Collectively, our data suggest that AMPK activity is critical for the expression of a DUSP 1 and 2 expression, negative regulators of ERK activity, under glucose deprivation.
FIGURE 4.

Down-regulation of DUSP1 and DUSP2 enhances ERK activity. After transfection with siRNA for control, DUSP1, or DUSP2 for 24 h, HCT116p53+/+ cells were maintained in the absence of glucose for 24 h. Then, the mRNA level of DUSP1 and 2 was determined by RT-PCR (A), and phosphoactivated ERK (P-ERK), and its total form (ERK) were determined by Western blot analysis and compared via densitometer (B). Experiments were repeated four times with similar results, and a representative result is shown. (*, p < 0.05; compared with the indicated groups.)
AMPK Induces DUSP2 Expression in a p53-dependent Manner after Glucose Deprivation
It was recently reported that p53, a tumor suppressor and a regulator of cell cycle arrest and apoptosis, regulates transcription of several DUSPs, including DUSP1 and DUSP2. DUSP1 is a direct transcriptional target of p53 and negatively regulates G1 progression of the cell cycle response to growth factors (27), and DUSP2 is also regulated by p53 under specific stress conditions (22). Moreover, there are several reports showing highly complex interrelationship between AMPK and p53. For example, the gene encoding the β-subunit of AMPK is directly up-regulated by p53 (28), and p53 targets (Sestrin 1 and 2) activate AMPK (29). Conversely, p53 can be phosphorylated and stabilized in an AMPK-dependent manner (4, 30). Therefore, we tested a possibility that p53 mediates AMPK activity on DUSP regulation, and first measured p53 activity in HCT116p53+/+ cells by transfecting reporter plasmids that can be regulated by p53 (Fig. 5A). pG-13-luc, in which luciferase is under the control of 13 p53-resposive elements, was transfected into cells along with pAMPK-WT or pAMPK-DN. Glucose deprivation induced p53-dependent luciferase activity by 2∼3 fold, and this induction was almost completely blocked by AMPK-DN. Likewise, the promoter activity of p21cip1/waf1 (p21-luc), a target gene of p53, was also induced by glucose deprivation in an AMPK-dependent manner, whereas the promoter activity of p21cip1/waf1Δp53-luc, where the p53 binding site is deleted, did not respond to glucose deprivation, suggesting that glucose deprivation increases p53 activity through AMPK activation.
FIGURE 5.

AMPK increases DUSP2 promoter activity via p53 activation. A, HCT116p53+/+ cells were co-trasnfected with pcDNA, pAMPK-WT, or pAMPK-DN expression vectors, and a luciferase reporter containing a promoter with 13 p53 responsive elements (pG-13-luc), a wild-type p21cip1/waf1 promoter (p21-luc), p21 promoter with the p53 binding site deleted (p21cip1/waf1Δp53-luc), or pDUSP2-luc for 24 h. B, HCT116p53+/+ cells were transfected with a luciferase reporter with a wild-type DUSP2 promoter (pDUSP2-luc) or a DUSP2 promoter with the p53 binding site deleted (pDUSP2Δp53-luc) for 24 h. After exposure to glucose-depleted medium for 24 h, cell lysates were subjected to a luciferase activity assay. The data represent means ± S.E. for six determinations. C and D, HCT116p53+/+ and HCT116p53−/− cells were transiently co-transfected with pcDNA, pAMPK-WT, or pAMPK-DN expression vector, and pDUSP2-luc with a 1:1 ratio. After 24 h transfection, cells were exposed to glucose deprivation for 24 h, and then phosphorylation level of ACC and total ACC was measured by Western blot (C). Also, the luciferase activity assay (left panel) or mRNA level of DUSP2 was determined (right panel) (D). E, HCT116p53−/− cells were co-transfected with pDUSP2-luc and/or pcDNA, pAMPK-WT, pAMPK-DN, p53-WT, or p53-DN (p53-R175D) expression vectors at a 1:1:1 ratio. After 24 h transfection, cells were exposed to glucose deprivation for 24 h, and then luciferase activity (left panel) or mRNA level of DUSP2 (right panel) was measured. The data represent means ± S.E. for six determinations. (*, p < 0.05; compared with the indicated groups.)
We next examined whether induction of DUSP2 promoter activity by glucose deprivation is mediated by p53. The DUSP2 promoter used in this study contains the p53 consensus binding site at a position from −202 to −191 (CCCCACGTGAGG). HCT116p53+/+ cells were transfected with wild-type pDUSP2-luc or pDUSP2Δp53-luc with the p53 binding site deleted (22). Contrast to wild-type pDUSP2-luc, glucose deprivation failed to induce the luciferase activity of pDUSP2Δp53-luc (Fig. 5B), indicating that p53 mediates the effect of glucose deprivation on DUSP2 promoter. Next, HCT116p53+/+ cells and p53-deficient HCT116p53−/− cells were co-transfected with pDUSP2-luc and pAMPK-WT or pAMPK-DN, and then promoter activity was measured after 24 h of glucose deprivation. In both cells, glucose deprivation activated AMPK, and AMPK-DN effectively blocked endogenous AMPK activity (Fig. 5C). DUSP2 promoter activity was induced in an AMPK-dependent manner in HCT116p53+/+ cells, but not in HCT116p53−/− cells, as was DUSP2 mRNA induction (Fig. 5D). To confirm these results, we performed reconstitution experiments by introducing p53 wild type (p53-WT) as well as dominant-negative mutant of p53 (p53-DN) into HCT116p53−/− cells (Fig. 5E). p53-DN is a mutant form where Arg175 is mutated to Asp to ablate both the apoptosis and cell cycle arrest functions of wild-type p53 (31). AMPK-dependent induction of the DUSP2 promoter after glucose deprivation was restored in HCT116p53−/− cells, where p53-WT, but not p53-DN, was reintroduced. Likewise, AMPK-dependent induction of DUSP2 mRNA was also restored in HCT116p53−/− cells transfected with p53-WT, but not with p53-DN. In addition, DUSP1 mRNA expression was also induced in an AMPK-dependent manner only in HCT116p53+/+ cells and not HCT116p53−/− cells (Fig. 6A). These results were further confirmed by identical reconstitution experiments as in Fig. 5E (Fig. 6B). Taken together, our results suggest that AMPK activity is critical for the promoter activity of DUSP1 and DUSP2 and their subsequent mRNA expression under glucose deprivation, and that p53 acts as a downstream mediator of AMPK in this signaling pathway.
FIGURE 6.

AMPK regulates DUSP1 mRNA expression via p53 activation in response to glucose deprivation. A, HCT116p53+/+ and HCT116p53−/− cells were transfected with pcDNA, pAMPK-WT, or pAMPK-DN expression vectors for 24 h, and then exposed to glucose-depleted medium for 24 h. Then, the mRNA level of DUSP1 was determined. B, under identical conditions as Fig. 5E (right panel), the mRNA level of DUSP1 was determined. The data represent means ± S.E. for six determinations. (*, p < 0.05; compared with the indicated groups.)
p53 is extensively phosphorylated in response to cell stress, including glucose deprivation, but the amino acid target is not clear (4, 30). Here, we observed that glucose deprivation induced phosphorylation at Ser46 and not Ser15, and this induction was blocked by AMPK-DN (Fig. 7). Total p53 also increased as Ser46 phosphorylation increased, suggesting that AMPK activates p53 through phosphorylation at Ser46.
FIGURE 7.
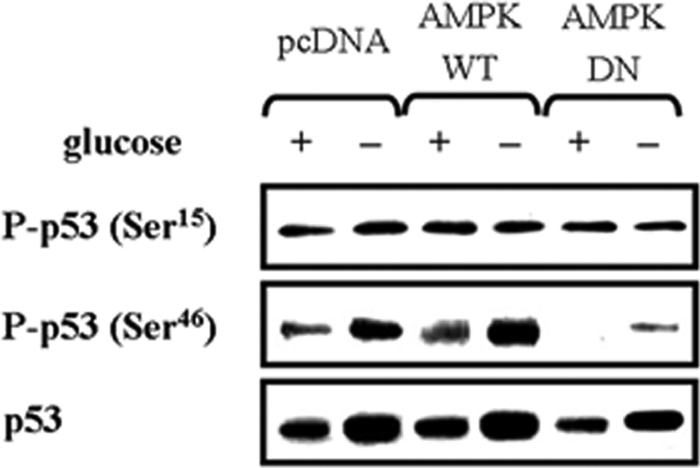
AMPK is required for p53 phosphorylation at Ser46, but not at Ser15, following glucose deprivation. HCT116p53+/+ cells were transfected with pcDNA, pAMPK-WT, or pAMPK-DN expression vectors for 24 h, and then incubated in glucose-depleted medium for 24 h. The phosphorylation level of p53 at Ser15 and Ser46 was analyzed by Western blot. Experiments were repeated three times with similar results, and a representative result is shown.
Negative Regulation of ERK by AMPK Requires p53
We next examined whether the negative regulation of ERK by AMPK is indeed dependent on p53. To this end, HCT116p53+/+ and HCT116p53−/− cells were transfected with pcDNA, pAMPK-WT or -DN, and then exposed to glucose deprivation for 24 h (Fig. 8A). AMPK-DN further induced ERK activation only in HCT116p53+/+ cells, but not HCT116p53−/− cells. JNK and p38 MAPK activity were not affected by AMPK. These results were further confirmed by a reconstitution experiment, where the effect of AMPK-DN was restored in HCT116p53−/− cells where p53-WT, but not p53-DN, was reintroduced (Fig. 8B). Taken together, our findings suggest that AMPK regulates DUSP1 and -2 expression via p53 in response to glucose deprivation, which in turn negatively regulates ERK.
FIGURE 8.

AMPK negatively regulates ERK phosphorylation in a p53-dependent manner. A, HCT116p53+/+ and HCT116p53−/− cells were transfected with pcDNA, pAMPK-WT, or pAMPK-DN expression vectors for 24 h, and then incubated in glucose depleted-medium for 24 h. B, HCT116p53−/− cells were co-transfected with pcDNA, pAMPK-WT, pAMPK-DN, and p53-WT, p53-DN (p53-R175D) expression vectors at a 1:1 ratio for 24 h, and then incubated in glucose-depleted medium for 24 h. Then, the phosphoactive form and total form of ERK, JNK, and p38 MAPK were analyzed by Western blot. Experiments were repeated three times with similar results, and a representative result is shown.
ERK Has Contrasting Effects Depending on the Stimulus, and AMPK Protects Cells from Glucose Deprivation-induced Apoptosis via Suppressing Pro-apoptotic ERK
To establish the physiological role of ERK, HCT116p53+/+ cells were exposed to various stimuli. ERK was activated in response to glucose deprivation, Fas (CD95/Apo-1), serum, and insulin following 12 h serum starvation of HCT116p53+/+ cells. Under these conditions, we blocked ERK activity with structurally two different MEK inhibitors, PD98059 and U0126, and then assessed the degree of apoptosis via fluorescence-activated cell scanning analysis of sub-G1 DNA content. Notably, glucose deprivation produced pro-apoptotic ERK activity, whereas ERK activation by Fas, serum, and insulin was anti-apoptotic (Fig. 9A). The degree of apoptosis gradually increased as cells were exposed to decreasing concentrations of glucose, and low glucose-induced apoptosis was attenuated by the addition of PD98059 (Fig. 9B). The expression of AMPK-DN further increased apoptosis induced by glucose deprivation, indicating that AMPK plays an anti-apoptotic role, and pretreatment with PD98059 significantly reduced apoptosis (Fig. 9C). These results show that AMPK protects cells from apoptosis after glucose deprivation, in part, via suppressing pro-apoptotic ERK. Next, we further examined the effect of AMPK activity on ERK phosphorylation and the role of ERK activity on apoptosis in several different human cancer cell lines, including HepG2 hepatoma and AGS gastric adenocarcinoma that contain the wild-type p53 (Fig. 10). HepG2 and AGS cells were transfected with pcDNA, pAMPK-WT, or -DN, and then exposed to glucose deprivation for 24 h. AMPK-DN further induced ERK activation in both cell lines as shown in HCT116p53+/+ cells (Fig. 10A). The expression of AMPK-DN further increased apoptosis induced by glucose deprivation, indicating that AMPK plays an anti-apoptotic role, and pretreatment with PD98059 significantly reduced apoptosis in both cell lines (Fig. 10B). Therefore, the suggested role of AMPK and ERK as well as their signal network may exist in a broad range of cancer types.
FIGURE 9.

ERK has contrasting effects depending on the stimulus, and AMPK protects cells from glucose deprivation-induced apoptosis via suppressing pro-apoptotic ERK. A, HCT116p53+/+ cells were pretreated with two different MEK inhibitors, PD98059 (25 μm) or U0126 (10 μm), for 30 min, and then cells were exposed for 24 h to glucose-deprived medium, Fas (CD95/Apo-1)-activating mouse anti-Fas monoclonal antibody (Fas), 10% serum, or insulin (100 nm) following 12 h of serum starvation. Cells were then subjected to fluorescence-activated cell scanning analysis for the percentage of nuclei containing subdiploid amounts of DNA (sub-G1 fraction) (upper panel) and phosphoactivated ERK (P-ERK) was measured (lower panel). B, HCT116p53+/+ cells were exposed to a medium containing the indicated concentrations of glucose for 24 h with or without 25 μm PD98059 and then analyzed for apoptosis via fluorescence-activated cell scanning analysis. C, HCT116p53+/+ cells were transfected with pcDNA, pAMPK-WT, or pAMPK-DN expression vectors for 24 h and maintained under glucose deprivation for 24 h with or without PD98059 (25 μm). The sub-G1 fraction of the cells was measured as a percentage. Experiments were repeated three times with similar results, and a representative result is shown. (*, p < 0.05; compared with the indicated groups.)
FIGURE 10.

Inhibition of AMPK activation by glucose deprivation promotes pro-apoptotic ERK in various human cancer cell lines. A, HepG2 and AGS cells were transfected with pcDNA, pAMPK-WT, or pAMPK-DN expression vectors for 24 h, and then incubated in glucose-depleted medium for 24 h. Then, the phosphoactive form (P-ERK) and total form of ERK (ERK) were analyzed by Western blot. Experiments were repeated three times with similar results and a representative result is shown. B, HepG2 and AGS cells were transfected with pcDNA, pAMPK-WT, or pAMPK-DN expression vectors for 24 h and maintained under glucose deprivation for 24 h with or without PD98059 (25 μm). Percentages of sub-G1 cells via FACS analysis are indicated. Experiments in triplicate were repeated three times. (*, p < 0.05; compared with the indicated groups.)
DISCUSSION
Recently, a number of attempts have been made to reveal a relationship between AMPK and ERK signal pathways, but there is no consensus on how two signal pathways interact (14–21). In most of these studies, an artificial AMPK activator, AICAR, has been used to determine its role in different cells and tissues. AICAR is converted to ZMP in a cell, thereby acting as an AMP analogue and activating AMPK. For this reason, AICAR has been widely used to demonstrate the role of AMPK, but it exerts adverse effects. Here, we demonstrated that glucose deprivation activated both AMPK and ERK in HCT116 colon cancer cells. Inhibition of AMPK by AMPK-DN significantly potentiated ERK activation, indicating that AMPK normally suppresses ERK activity. Moreover, we provide novel evidence that AMPK activity is critical for inducing DUSPs expression, which are negative regulators of ERK. In contrast to a general perception that ERK mediates anti-apoptotic effects, our results with two different MEK inhibitors revealed that ERK exerts pro-apoptotic effects in HCT116 cells after glucose deprivation (Fig. 9). Indeed, ERK acts as a survival factor under Fas (CD95/Apo1)-, serum-, and insulin-treated conditions in the same cells (Fig. 9). Because inhibition of AMPK potentiated glucose deprivation-induced apoptosis and increased pro-apoptotic ERK, we concluded that AMPK protects HCT116 cells from glucose deprivation via suppressing ERK. Indeed, ERK can function in a pro-apoptotic manner under different stimuli in different tissues, and ERK-mediated cell death was also reported in many animal models (32, 33). As a downstream effector of cell death-promoting ERK, cellular components involved in intrinsic as well as extrinsic apoptosis pathway have been suggested. The suppression of survival pathways such as phosphatidylinositol 3-kinase/Akt could mediate ERK-induced apoptosis (33). Furthermore, the kinetics and duration of ERK activation, its subcellular localization, and model system, affect cell fate (32, 34, 35). Nevertheless, the mechanism by which ERK executes anti- or pro-apoptotic function is far from clear.
Similar to ERK, AMPK has a pleiotropic role in cell growth and apoptosis. AMPK plays a central role for energy conservation in response to metabolic stresses, revealing anti-apoptotic properties (36, 37). Recently, a pro-apoptotic function of AMPK was also reported; AMPK can block cell growth via tumor suppressors. Identified as one of the upstream activating AMPK kinases, LKB1 is a tumor suppressor kinase, and its gene mutations cause a dominantly inherited a cancer pre-disposition disease, Peutz-Jeghers syndrome (38–40). The TSC2 tumor suppressor is a direct substrate of AMPK, providing a link between AMPK activation and the suppression of protein synthesis (3, 41, 42). Moreover, there are several recent reports showing a positive interplay between AMPK and tumor suppressor p53 (4, 30, 43). In relation to dual roles of AMPK, our current data raises an intriguing possibility that AMPK can potentially exert either anti- or pro-apoptotic role through regulating ERK. For example, we demonstrated here that AMPK suppresses pro-apoptotic ERK in HCT116 cells after glucose deprivation, but we and others previously reported that AMPK can suppress ERK to block cell proliferation; e.g. pharmacological activation of AMPK by AICAR inhibits IGF-1- or angiotensin-induced ERK activation (19, 20). More than 150 ERK substrates have been identified, including protein kinases, protein phosphatases, transcription factors, cytoskeleton components, apoptosis-related proteins, and other types of proteins. Therefore, it would be highly intriguing to examine whether AMPK activation under different conditions can affect selection of ERK downstream substrates and determine the outcome of ERK activation on cell death.
Conversely, the RAF-MEK-ERK kinase cascade also negatively regulates AMPK signal pathway; oncogenic B-RAF suppresses LKB1 and AMPK function in melanoma cells (44). Because both LKB1 and AMPK suppress cell growth, suppression of LKB1-AMPK by oncogenic RAF could allow tumor cells to gain growth advantage by turning off signaling for stopping cell growth. Similarly, serum inhibited AMPK via ERK activation in neonatal rat cardiac fibroblasts (45). Thus, a highly complex and sophisticated inhibitory signaling network may exist to balance AMPK and ERK activity. ERK signaling is involved in the regulation of cell proliferation, differentiation, tumorigenesis, cell survival, and death; constant changes in intracellular ATP and AMP levels occur in these cellular processes. Thus, our results suggest that a signal network that connects AMPK and ERK plays a central role in linking intracellular energy levels to the regulation of a broad spectrum of cellular processes.
In this study, we have shown that AMPK induces DUPSs in a p53-dependent manner. The effect of AMPK inhibition on the DUSP mRNA levels, its promoter activity, and ERK phosphorylation was observed only in HCT116p53+/+ cells, but not HCT116p53−/− cells. In reconstitution experiments, the introduction of p53 into HCT116p53−/− cells restored the effect of AMPK inhibition, indicating that p53 acts as a downstream mediator of AMPK. However, the exact mechanism linking p53 and AMPK remains elusive. It was recently reported that p53 phosphorylation at Ser15 was essential for mediating the effect of AMPK on p53-dependent cell cycle arrest of mouse embryonic fibroblasts, but p53 is phosphorylated at Ser46 in human osteosarcoma-derived U2OS cells after glucose deprivation (4, 30). Although we found that Ser46 is more important for regulation by AMPK, more elaborate studies are required for understanding the regulatory mechanisms between AMPK and p53. Indeed, AMPK may suppress p53 stability under other conditions (46). Moreover, p53 can regulate the phosphorylation of AMPKα or gene expression of AMPK β, indicating a complicated interaction (28, 29).
Abnormalities in MAPK signaling pathways occur in a wide range of human malignancies, and significant overexpression of DUSP1 was observed in the early phases of prostate, colon, and bladder cancer, falling progressively in tumors of higher histological grade and in metastasis. Expression of DUSP2 occurs in serous carcinomas of the ovary and is associated with a poor outcome in terms of overall survival (13). Therefore, further studies on the novel signaling network of AMPK, DUSPs, and MAPKs, will enhance our understanding of the initiation, genesis, and progression of cancer.
This work was supported by Korea Science and Engineering Foundation (KOSEF) grants funded by the Korean government (MEST) (20090080996 and 20090091345).
- AMPK
- AMP-activated protein kinase
- ACC
- acetyl-CoA carboxylase
- FACS
- fluorescence-activated cell sorting
- WT
- wild type
- DN
- dominant negative
- ERK
- extracellular signal-regulated kinase
- JNK
- c-Jun NH2-terminal kinase
- DUSP
- dual-specificity phosphatase
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
REFERENCES
- 1.Hardie D. G., Carling D., Carlson M. (1998) Annu. Rev. Biochem. 67, 821–855 [DOI] [PubMed] [Google Scholar]
- 2.Shackelford D. B., Shaw R. J. (2009) Nat. Rev. Cancer 9, 563–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inoki K., Ouyang H., Zhu T., Lindvall C., Wang Y., Zhang X., Yang Q., Bennett C., Harada Y., Stankunas K., Wang C. Y., He X., MacDougald O. A., You M., Williams B. O., Guan K. L. (2006) Cell 126, 955–968 [DOI] [PubMed] [Google Scholar]
- 4.Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M. J., Thompson C. B. (2005) Mol. Cell 18, 283–293 [DOI] [PubMed] [Google Scholar]
- 5.Xia Z., Dickens M., Raingeaud J., Davis R. J., Greenberg M. E. (1995) Science 270, 1326–1331 [DOI] [PubMed] [Google Scholar]
- 6.Paraskevas S., Aikin R., Maysinger D., Lakey J. R., Cavanagh T. J., Hering B., Wang R., Rosenberg L. (1999) FEBS Lett. 455, 203–208 [DOI] [PubMed] [Google Scholar]
- 7.Sasaki K., Chiba K. (2004) Mol. Biol. Cell 15, 1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janes K. A., Albeck J. G., Gaudet S., Sorger P. K., Lauffenburger D. A., Yaffe M. B. (2005) Science 310, 1646–1653 [DOI] [PubMed] [Google Scholar]
- 9.Waskiewicz A. J., Cooper J. A. (1995) Curr. Opin. Cell Biol. 7, 798–805 [DOI] [PubMed] [Google Scholar]
- 10.Su B., Karin M. (1996) Curr. Opin. Immunol. 8, 402–411 [DOI] [PubMed] [Google Scholar]
- 11.Camps M., Nichols A., Arkinstall S. (2000) FASEB J. 14, 6–16 [PubMed] [Google Scholar]
- 12.Patterson K. I., Brummer T., O'Brien P. M., Daly R. J. (2009) Biochem. J. 418, 475–489 [DOI] [PubMed] [Google Scholar]
- 13.Keyse S. M. (2008) Cancer Metastasis Rev. 27, 253–261 [DOI] [PubMed] [Google Scholar]
- 14.Hattori Y., Akimoto K., Nishikimi T., Matsuoka H., Kasai K. (2006) Hypertension 47, 265–270 [DOI] [PubMed] [Google Scholar]
- 15.Fukuda T., Ishii K., Nanmoku T., Isobe K., Kawakami Y., Takekoshi K. (2007) J. Neuroendocrinol. 19, 621–631 [DOI] [PubMed] [Google Scholar]
- 16.Chen H. C., Bandyopadhyay G., Sajan M. P., Kanoh Y., Standaert M., Farese R. V., Jr., Farese R. V. (2002) J. Biol. Chem. 277, 23554–23562 [DOI] [PubMed] [Google Scholar]
- 17.Longnus S. L., Ségalen C., Giudicelli J., Sajan M. P., Farese R. V., Van Obberghen E. (2005) Diabetologia 48, 2591–2601 [DOI] [PubMed] [Google Scholar]
- 18.Williamson D. L., Bolster D. R., Kimball S. R., Jefferson L. S. (2006) Am. J. Physiol. Endocrinol. Metab. 291, E80–E89 [DOI] [PubMed] [Google Scholar]
- 19.Kim J., Yoon M. Y., Choi S. L., Kang I., Kim S. S., Kim Y. S., Choi Y. K., Ha J. (2001) J. Biol. Chem. 276, 19102–19110 [DOI] [PubMed] [Google Scholar]
- 20.Nagata D., Takeda R., Sata M., Satonaka H., Suzuki E., Nagano T., Hirata Y. (2004) Circulation 110, 444–451 [DOI] [PubMed] [Google Scholar]
- 21.Li H. L., Yin R., Chen D., Liu D., Wang D., Yang Q., Dong Y. G. (2007) J. Cell. Biochem. 100, 1086–1099 [DOI] [PubMed] [Google Scholar]
- 22.Yin Y., Liu Y. X., Jin Y. J., Hall E. J., Barrett J. C. (2003) Nature 422, 527–531 [DOI] [PubMed] [Google Scholar]
- 23.Ha J., Daniel S., Broyles S. S., Kim K. H. (1994) J. Biol. Chem. 269, 22162–22168 [PubMed] [Google Scholar]
- 24.Keyse S. M., Emslie E. A. (1992) Nature 359, 644–647 [DOI] [PubMed] [Google Scholar]
- 25.Li J., Gorospe M., Hutter D., Barnes J., Keyse S. M., Liu Y. (2001) Mol. Cell. Biol. 21, 8213–8224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y., Gorospe M., Yang C., Holbrook N. J. (1995) J. Biol. Chem. 270, 8377–8380 [DOI] [PubMed] [Google Scholar]
- 27.Li M., Zhou J. Y., Ge Y., Matherly L. H., Wu G. S. (2003) J. Biol. Chem. 278, 41059–41068 [DOI] [PubMed] [Google Scholar]
- 28.Feng Z., Hu W., de Stanchina E., Teresky A. K., Jin S., Lowe S., Levine A. J. (2007) Cancer Res. 67, 3043–3053 [DOI] [PubMed] [Google Scholar]
- 29.Budanov A. V., Karin M. (2008) Cell 134, 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okoshi R., Ozaki T., Yamamoto H., Ando K., Koida N., Ono S., Koda T., Kamijo T., Nakagawara A., Kizaki H. (2008) J. Biol. Chem. 283, 3979–3987 [DOI] [PubMed] [Google Scholar]
- 31.Ryan K. M., Vousden K. H. (1998) Mol. Cell. Biol. 18, 3692–3698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Z., Xu S. (2006) IUBMB Life 58, 621–631 [DOI] [PubMed] [Google Scholar]
- 33.Zhuang S., Schnellmann R. G. (2006) J Pharmacol. Exp. Ther. 319, 991–997 [DOI] [PubMed] [Google Scholar]
- 34.Fan M., Chambers T. C. (2001) Drug Resist. Updat. 4, 253–267 [DOI] [PubMed] [Google Scholar]
- 35.Shaul Y. D., Seger R. (2007) Biochim. Biophys. Acta 1773, 1213–1226 [DOI] [PubMed] [Google Scholar]
- 36.Shibata R., Sato K., Pimentel D. R., Takemura Y., Kihara S., Ohashi K., Funahashi T., Ouchi N., Walsh K. (2005) Nat. Med. 11, 1096–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Föller M., Sopjani M., Koka S., Gu S., Mahmud H., Wang K., Floride E., Schleicher E., Schulz E., Münzel T., Lang F. (2009) FASEB J. 23, 1072–1080 [DOI] [PubMed] [Google Scholar]
- 38.Hawley S. A., Boudeau J., Reid J. L., Mustard K. J., Udd L., Mäkelä T. P., Alessi D. R., Hardie D. G. (2003) J. Biol. 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong S. P., Leiper F. C., Woods A., Carling D., Carlson M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8839–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G., Neumann D., Schlattner U., Wallimann T., Carlson M., Carling D. (2003) Curr. Biol. 13, 2004–2008 [DOI] [PubMed] [Google Scholar]
- 41.Inoki K., Zhu T., Guan K. L. (2003) Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 42.Corradetti M. N., Inoki K., Bardeesy N., DePinho R. A., Guan K. L. (2004) Genes Dev. 18, 1533–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cao C., Lu S., Kivlin R., Wallin B., Card E., Bagdasarian A., Tamakloe T., Chu W. M., Guan K. L., Wan Y. (2008) J. Biol. Chem. 283, 28897–28908 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Zheng B., Jeong J. H., Asara J. M., Yuan Y. Y., Granter S. R., Chin L., Cantley L. C. (2009) Mol. Cell 33, 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du J., Guan T., Zhang H., Xia Y., Liu F., Zhang Y. (2008) Biochem. Biophys. Res. Commun. 368, 402–407 [DOI] [PubMed] [Google Scholar]
- 46.Galbán S., Martindale J. L., Mazan-Mamczarz K., Lopez de Silanes I., Fan J., Wang W., Decker J., Gorospe M. (2003) Mol. Cell. Biol. 23, 7083–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]