

Abstract
The Na-K-2Cl cotransporter (NKCC1) participates in epithelial transport and in cell volume maintenance by mediating the movement of ions and water across plasma membranes. Functional studies have previously demonstrated that NKCC1 activity is stimulated by protein phosphatase 1 (PP1) inhibitors. In this study, we utilized both in vivo (heterologous cRNA expression in Xenopus laevis oocytes) and in vitro (32P-phosphorylation assays with glutathione S-transferase fusion proteins) experiments to determine whether PP1 exerts its inhibitory effect directly on the cotransporter, or indirectly by affecting the activating kinase. We found that PP1 reduced NKCC1 activity in oocytes under both isotonic and hypertonic conditions to the same level as in water-injected controls. Interestingly, mutation of key residues in the PP1 binding motif located in the N-terminal tail of NKCC1 significantly reduced the inhibitory effect of PP1. In vitro experiments performed with recombinant PP1, SPAK (Ste20-related proline/alanine-rich kinase, which activates NKCC1), and the N terminus of NKCC1 fused to glutathione S-transferase demonstrated that PP1 dephosphorylated both the kinase and the cotransporter in a time-dependent manner. More importantly, PP1 dephosphorylation of SPAK was significantly greater when protein-protein interaction between the kinase and the N-terminal tail of NKCC1 was present in the reaction, indicating the necessity of scaffolding the phosphatase and kinase in proximity to one another. Taken together, our data are consistent with PP1 inhibiting NKCC1 activity directly by dephosphorylating the cotransporter and indirectly by dephosphorylating SPAK.
Keywords: Chloride Transport, Potassium Transport, Protein Phosphatase, Protein Phosphorylation, Sodium Transport, Xenopus
Introduction
The Na-K-2Cl cotransporter (NKCC1)2 is expressed in the basolateral membranes of a variety of chloride-secreting epithelia and participates in cell volume maintenance and regulation by mediating the movement of ions and water across the plasma membrane (1, 2). The activation/inhibition of the Na-K-2Cl cotransporter is directly associated to the phosphorylation state of the protein (3, 4). In previous studies, we demonstrated that two Ste20-like serine/threonine kinases, SPAK and OSR1 (oxidative stress response 1), physically associate with the N-terminal tail of the cotransporter at a putative RFX(V/I) binding motif (5, 6). In vitro phosphorylation experiments with glutathione S-transferase (GST) fusion proteins and in vivo ion flux experiments with heterologously expressed cRNA in Xenopus laevis oocytes have also shown that SPAK/OSR1 phosphorylates and activates the cotransporter (7, 8). In 2007, we reported that overexpression of an apoptosis-associated tyrosine kinase (AATYK) inhibited NKCC1 activity. This effect was not mediated by the catalytic activity of the kinase, but was due to the scaffolding of protein phosphatase 1 (PP1) in proximity to SPAK (9). These data suggested an intriguing possibility that PP1 inhibited NKCC1 activity by also dephosphorylating the activating kinase. Many protein phosphatases have a conserved hydrophobic groove that exhibits low affinity binding for small degenerate sequence motifs. Protein phosphatase 1, for instance, has been shown to dock at three possible motifs: the RVXF, MyPhoNE, and SILK motifs (10). In addition to the two putative SPAK binding motifs, the N-terminal tail of NKCC1 also contains a RVXF docking motif for PP1 (11). Although Darman and Forbush (11) demonstrated that mutations of this motif affected NKCC1 activity, the overlap between the PP1 motif and the second SPAK binding motif made it unclear whether the effect of the phosphatase on cotransporter function was solely due to NKCC1 dephosphorylation.
In this study, we used in vitro phosphorylation assays and in vivo ion flux experiments to demonstrate that both NKCC1 and SPAK are dephosphorylated by PP1. We also show that dephosphorylation of SPAK is significantly greater when the kinase physically interacts with the N-terminal tail of NKCC1 to scaffold the phosphatase and kinase in proximity to one another. In summary, our results are consistent with the N-terminal tail of NKCC1 scaffolding a regulatory phosphatase (PP1) in proximity to an activating kinase (SPAK), and thus directly and indirectly participating in its own inactivation.
EXPERIMENTAL PROCEDURES
Construction of Mouse PP1 Mutants
In this study, we moved the full-length PP1 cDNA previously cloned into the mammalian expression vector (pCDNA3 (9)) into the X. laevis oocyte expression vector (pBF). We also used the full-length PP1 cDNA previously cloned into pBluescript (9) as a template for QuikChange Mutagenesis (Stratagene, La Jolla, CA) with complementary sense and antisense oligonucleotides to mutate a histidine residue (His-248) into a lysine residue to create a catalytically inactive PP1 (12, 13). The mutation was verified by DNA sequencing and the full-length mutated PP1 was moved back into pBF with EcoRI-XhoI restriction enzymes.
Assessment of Mouse NKCC1 Expression in Oocyte Plasma Membranes
The surface expression of NKCC1 in the oocyte plasma membrane was measured by fluorescence using an enhanced green fluorescent protein (EGFP)-NKCC1 construct (7). Individual oocytes were monitored for EGFP fluorescence using Zeiss confocal laser-scanning microscope LSM510 (Plan-Apochromat ×5 objective, 0.16 numerical aperture lens). Excitation wavelength was set at 488 nm and emission signals were collected using a 505-nm bandpass filter. Gain and offset were manually adjusted to contain the EGFP fluorescence signal within the 0–215 intensity range of the 8-bit gray density scale. We captured a Z-stack of 6 optical slices near the middle of the oocyte and chose a single optical section with the largest diameter, indicative of the equatorial center of the oocyte. These settings were used to qualitatively assess fluorescence of EGFP-NKCC1 in the presence and absence of PP1.
Surface Expression of NKCC1 in X. laevis Oocytes
Groups of 50 oocytes were injected on day 2 after isolation with 50 nl of water containing 18.5 ng of NKCC1 cRNA and on day 3 with 50 nl of water alone or with 10 ng of PP1 cRNA. On day 5, the oocytes were washed with isosmotic flux solution and gently rotated every 10 min on ice with 2.5 ml of isotonic flux solution containing 1 mg/ml of EZ-link sulfo-NHS biotin (Pierce) for 2 h. Following biotin labeling, oocytes were washed 3 times with an isotonic solution containing 100 mm glycine, then placed in lysis buffer (20 μl/oocyte) containing 150 mm NaCl, 10 mm Tris-Cl (pH 7.4), 1 mm EDTA, 1% Triton X-100, and protease inhibitors (Roche Applied Science) on ice for 15 min. Oocytes were homogenized by trituration with a 200-μl pipette, rotated at 4 °C for 1 h, then centrifuged for 15 min at 10,000 × g. Supernatants were rotated overnight at 4 °C with 75 μl of streptavidin bead slurry (50% in lysate buffer). Samples were centrifuged for 2 min at 8,000 × g and the streptavidin beads were washed 3 times with lysis buffer prior to the addition of ×4 sample buffer (0.26 m Tris, pH 6.8, 4.8% SDS, 40% glycerol, 0.002% bromphenol blue). After 20 min denaturation at 60 °C, the samples were loaded and separated on 7.5% polyacrylamide gels, and transferred to polyvinylidene difluoride membranes (ThermoFisher Scientific). Membranes were blocked for 2 h at room temperature with 5% nonfat dry milk in TBS-T (150 mm NaCl, 10 mm Tris HCl, 0.5% Tween 20 (polyoxyethylene sorbitan monolaurate)), then probed overnight at 4 °C with a mouse anti-NKCC1 C-terminal polyclonal antibody (1:1000) (14). Membranes were rinsed several times in TBS-T before a 1-h incubation with an anti-mouse horseradish peroxidase-conjugated secondary antibody (1:4000) in 5% nonfat dry milk in TBS-T at room temperature. The membranes were again rinsed several times in TBS-T, then finally subjected to enhanced chemiluminescence (ECL) detection (Amersham Biosciences). Densitometry analysis was performed using the Image J imaging software (NIH, Bethesda, MD).
cRNA Synthesis
All cDNA clones in pBF were linearized with MluI and transcribed into cRNA using the mMESSAGE mMACHINE SP6 transcription system (Ambion, Austin, TX). RNA quality was verified by gel electrophoresis (1% agarose, 0.693% formaldehyde) and quantitated by measurement of absorbance at 260 nm.
Isolation of X. laevis Oocytes
Stages V-VI X. laevis oocytes were isolated from 8 different frogs as previously described (7) and maintained at 16 °C in modified L15 medium (Leibovitz L15 solution diluted with water to a final osmolarity of 195–200 mosmol and supplemented with 10 mm HEPES and 44 μg of gentamicin sulfate). Oocytes were injected on day 2 with 50 nl of water containing 15 ng of wild-type or mutant NKCC1 cRNA and on day 3 with 50 nl of water containing 10 ng of each kinase or phosphatase cRNA. Control oocytes were injected with 50 nl of water. 86Rb uptakes were performed on day 5 post-isolation.
K+ Uptakes in X. laevis Oocytes
Groups of 20 oocytes in a 35-mm dish were washed once with 3 ml of isosmotic saline (96 mm NaCl, 4 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 5 mm HEPES buffered to pH 7.4) and preincubated for 15 min in 1 ml of the same isosmotic saline containing 1 mm ouabain. The solution was then aspirated and replaced with 1 ml of isosmotic flux solution containing 5 μCi of 86Rb. Two 5-μl aliquots of flux solution were sampled at the beginning of each 86Rb uptake period and used as standards. After 1 h uptake, the radioactive solution was aspirated and the oocytes were washed 4 times with 3 ml of ice-cold isosmotic solution. Single oocytes were transferred into glass vials, lysed for 1 h with 200 μl of 0.25 n NaOH, neutralized with 100 μl of glacial acetic acid, and 86Rb tracer activity was measured by β-scintillation counting. NKCC1 flux was expressed in nanomoles of K+/oocyte/h.
In Vitro Phosphatase Assays
First, kinase reactions were performed as previously reported (15). Briefly, kinase reactions (15 μl) containing wild-type and mutant forms of GST-NKCC1 and wild-type GST-SPAK fusion proteins, 20 mm HEPES (pH 7.4), 4 mm MnCl2, 5 mm dithiothreitol, 1 μm cold NaATP, and 8 μCi of [γ-32P]ATP were incubated at 37 °C for 45 min. Next, individual reactions were combined with recombinant protein phosphatase I (New England Biolabs, Beverly, MA) in a phosphatase reaction buffer (15 μl) containing 20 mm HEPES (pH 7.4), 4 mm MnCl2, 5 mm dithiothreitol, 1 mm EGTA, 0.025% Tween 20 and incubated at 37 °C. Results were similar whether or not staurosporine (20 μm) was added prior to the phosphatase reaction to inhibit residual kinase activity. Reactions were then stopped by adding 30 μl of 4× SDS sample buffer, followed by denaturing at 70 °C for 15 min. Reactions were separated by SDS-PAGE on 9–12% polyacrylamide gels. Gels were washed 3 times for 10 min each in a buffer containing 1% Na-pyrophosphate and 5% trichloroacetic acid, dried at 60 °C for 2 h, and phosphorylated products were visualized by autoradiography.
Western Blot Analysis
Wild-type and SPAK binding-deficient GST-NKCC1 fusion proteins (∼10 μg) were resolved by 10% SDS-PAGE and transferred to polyvinylidene difluoride membranes. Membranes were blocked for 2 h at room temperature with 5% nonfat dry milk in TBS-T. Membranes were then subjected to polyclonal anti-GST antibody (1:2000) in 5% nonfat dry milk in TBS-T overnight at 4 °C, washed extensively in TBS-T, and incubated for 1 h with horseradish peroxidase-conjugated secondary antibody (1:5000) in 5% nonfat dry milk in TBS-T. After extensive washing, protein bands were visualized by enhanced chemiluminescence.
Statistical Analyses
Differences between 86Rb uptake groups were tested by one-way analysis of variance followed by multiple comparison using Student-Newman-Keuls, Bonferroni, and Tukey post tests. Difference between relative densitometries of biotinylated samples was tested using paired t test. p < 0.001 was considered to be very significant.
RESULTS
The catalytic subunit of PP1 binds to a RVXF motif present in the N terminus of NKCC1 and is functionally important in regulating the phosphorylation state and activity of the cotransporter in mammalian cells (16). This putative motif overlaps with one of two binding motifs (RFX(V/I)) for SPAK, the kinase which phosphorylates and activates the cotransporter (5, 7, 17). To confirm the role of PP1 in modulating NKCC1 function in X. laevis oocytes, we co-injected mouse PP1 with a cotransporter tagged with EGFP. Fig. 1A shows a significant reduction in NKCC1 function under both isosmotic and hyperosmotic conditions. Note that the levels of uptake obtained with the tagged NKCC1 at both osmolarities is identical to the levels of uptake observed with wild-type NKCC1 (see Fig. 2), indicating that the N-terminal addition of EGFP does not affect cotransporter function. As the phosphatase was injected 2 days prior to the flux, we needed to confirm that PP1 did not prevent trafficking of the cotransporter to the plasma membrane. Confocal microscopy analysis of EGFP fluorescence revealed similar membrane expression of NKCC1 in the absence or presence of the phosphatase (Fig. 1B). Densitometry analysis of surface biotinylation/streptavidin pulldown demonstrates that co-expression of PP1 does not affect membrane expression of the cotransporter (Fig. 1, C and D).
FIGURE 1.

PP1 inhibits the activity of Na-K-2Cl cotransporters located at the plasma membrane. A, K+ uptake of X. laevis oocytes expressing 15 ng of fluorescently tagged cotransporter (EGFP-NKCC1) in the presence or absence of 10 ng of wild-type PP1 measured under isosmotic (195 mosmol, white bars) and hyperosmotic (260 mosmol, black bars) conditions. Bars represent mean ± S.E. (n = 20 oocytes). Uptake experiments were repeated twice with similar results. Asterisk denotes statistical significance (p < 0.001). B, confocal laser microscopy of membrane fluorescence in oocytes 3 days after injection with 15 ng of EGFP-NKCC1 cRNA and 10 ng of PP1 cRNA. C, representative surface biotinylation of oocytes injected with 18.5 ng of wild-type NKCC1 cRNA in the presence or absence of 10 ng of PP1 cRNA. Samples were immunoblotted with a polyclonal antibody raised against a portion of the C-terminal domain of NKCC1. Open arrow indicates NKCC1-specific signal. D, densitometry analysis of cotransporter surface expression from three separate biotinylation experiments. Paired t tests found no statistical difference (p = 0.38, n = 3).
FIGURE 2.

Catalytic activity and binding of PP1 are necessary for NKCC1 inhibition. K+ uptake of X. laevis oocytes expressing 15 ng of wild-type cotransporter (NKCC1 (wt)) or PP1 binding-deficient cotransporter (NKCC1 (bd)) in the presence or absence of 10 ng of wild-type phosphatase (PP1 (wt)) or catalytically inactive phosphatase (PP1 (ci)) under isosmotic (195 mosmol, white bars) and hyperosmotic (260 mosmol, black bars) conditions. All conditions were repeated twice with similar results. Bars represent mean ± S.E. (n = 20 oocytes). Asterisk denotes statistical significance (p < 0.001).
To verify that the catalytic activity of the phosphatase was responsible for the inactivation of the cotransporter, we mutated a histidine residue into a lysine (H248K), an alteration known to catalytically inactivate the phosphatase (12, 13). Co-expression of the catalytically inactive PP1 resulted in less inhibition of wild-type NKCC1 under both isosmotic and hyperosmotic conditions (Fig. 2). This data suggests both a catalytic-dependent and an -independent effect of PP1. Next, to determine whether PP1 exerts its inhibitory effect on NKCC1 activity by binding to the cotransporter, we utilized a “binding-deficient” mutant of NKCC1 where we removed amino acids 117–162, which contain the second SPAK binding motif (RFRV) and the putative PP1 binding motif (RVNF) (8). Interestingly, there was no significant difference in NKCC1 activity when either wild-type or catalytically inactive PP1 were co-expressed with the PP1 binding-deficient form of the cotransporter under isosmotic or hyperosmotic conditions (Fig. 2). Therefore, our results indicate that PP1 reduces NKCC1 function in a catalytic- and binding-dependent manner, but also, somehow, reduces cotransporter function in a catalytic- and binding-independent manner.
The simplest explanation for the catalytic- and binding-dependent effect of PP1 is that the phosphatase dephosphorylates and inactivates the cotransporter. However, there is also the possibility that PP1 dephosphorylates and inactivates the kinase, whereas some other phosphatase inactivates NKCC1. Therefore, we wanted to demonstrate that PP1 inactivates the cotransporter independently of the kinase. To do so, we utilized a constitutively active form of SPAK (T243E, S383A (18)) and a cotransporter mutant in which the first SPAK binding motif (F79A) is inactivated (8). The residues that render SPAK constitutively active are those targeted by WNK4 and WNK1 phosphorylation (19, 20). The use of the first SPAK binding-deficient NKCC1 mutant was to avoid the possibility that the constitutively active kinase might overwhelm the reaction and prevent us from observing any effect of PP1 on the cotransporter. Thus, active SPAK and PP1 must compete at the second overlapping motif and if PP1 dephosphorylates NKCC1, we should observe inactivation of the cotransporter. As shown in Fig. 3, mutant NKCC1 activity is similar in the presence of wild-type SPAK and WNK4 kinases or the constitutively active SPAK. In addition, expression of PP1 inhibits cotransporter activity regardless of the kinases used. To confirm that PP1 is not simply outcompeting SPAK at the overlapping binding site and thus effectively inhibit cotransporter activity, we repeated the PP1 effect with constitutively active SPAK and wild-type NKCC1. We obtained similar results indicating the prevalence of PP1.
FIGURE 3.
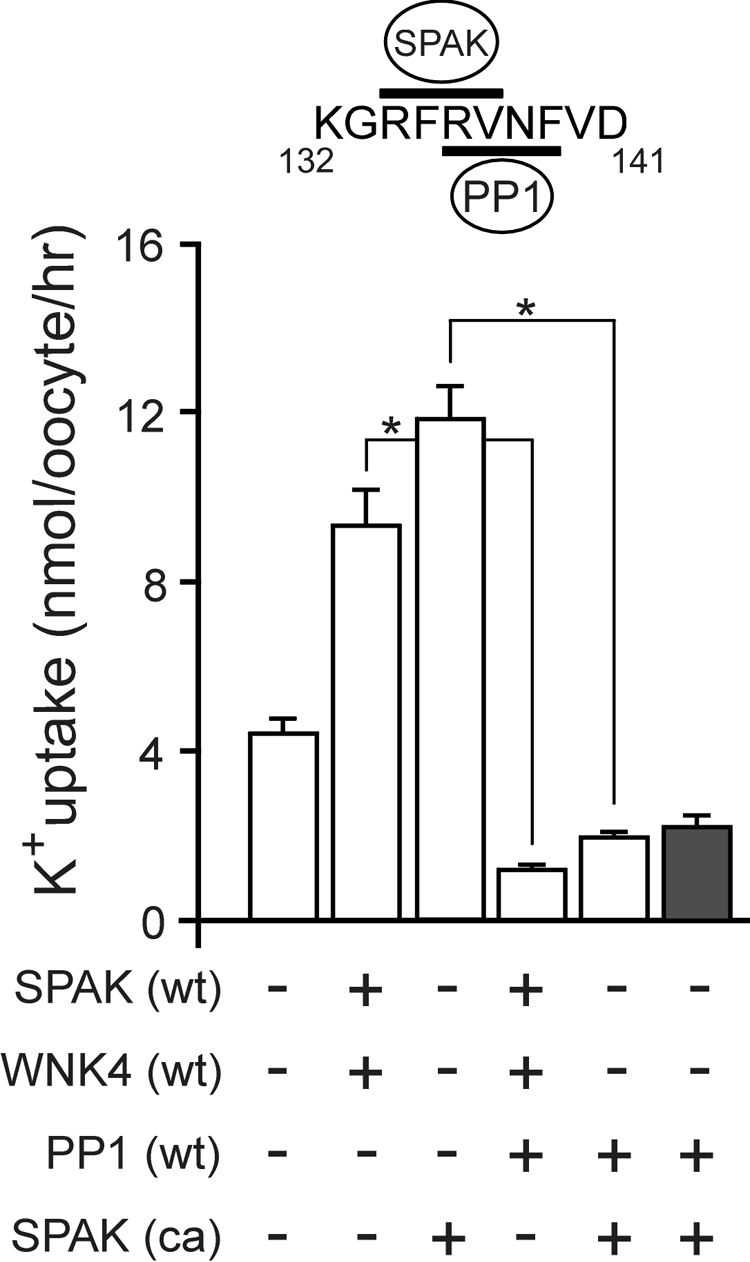
PP1 mediates direct inhibition of NKCC1. K+ uptake of X. laevis oocytes expressing 15 ng of mutant NKCC1 (F79A, white bars) or wild-type NKCC1 (gray bar) in the presence or absence of 10 ng of wild-type SPAK (SPAK (wt)), wild-type WNK4 (WNK4 (wt)), wild-type PP1 (PP1 (wt)), and constitutively active SPAK (SPAK (ca)) under isosmotic (195 mosmol) conditions. All conditions were repeated twice with similar results. Bars represent mean ± S.E. (n = 20 oocytes). Asterisk denotes statistical significance (p < 0.001). Inset, amino acid sequence of mouse NKCC1 (residues 132–141) with overlapping binding sites for SPAK and PP1 highlighted by horizontal bars.
The possibility that PP1 also inhibits NKCC1 activity by dephosphorylating and inactivating SPAK is based on an observation from an earlier study where we found that apoptosis-associated tyrosine kinase (AATYK1) significantly inhibited cotransporter activity when co-expressed with NKCC1 (9). We determined that the inhibitory effect of AATYK1 was a result of scaffolding of endogenous PP1 in proximity to SPAK, thus allowing for phosphatase dephosphorylation of the kinase. However, because NKCC1 is a direct target of PP1 dephosphorylation, we cannot use the cotransporter as both the scaffold and the “reporter” of the functional assay. Therefore, we utilized AATYK1 as an alternative scaffolding molecule to determine whether the observed PP1 inhibition of cotransporter activity also involves SPAK dephosphorylation. As seen in Fig. 4, addition of AATYK1 reduces the level of uptake promoted by wild-type SPAK and wild-type WNK4 expression (a), but not when promoted by constitutively active SPAK (b). Therefore, the difference between the level of K+ uptake with SPAK + WNK4 versus constitutively active SPAK (c), in the presence of AATYK1, represents the inactivating effect of PP1 on SPAK.
FIGURE 4.

AATYK inhibition of NKCC1 activity is prevented by a constitutively active SPAK. K+ uptake of X. laevis oocytes expressing 15 ng of wild-type NKCC1 in the presence or absence of 10 ng of wild-type SPAK (SPAK (wt)), wild-type WNK4 (WNK4 (wt)), wild-type AATYK (AATYK (wt)), and constitutively active SPAK (SPAK (ca)) under isosmotic (195 mosmol) conditions. All conditions were repeated twice with similar results. Bars represent mean ± S.E. (n = 20 oocytes). Asterisk denotes statistical significance (p < 0.001). Letters a, b, and c indicate specific comparisons discussed in the text. Inset, amino acid sequence of mouse AATYK (residues 1170–1178 and 1277–1294) with the one PP1 and two SPAK binding sites highlighted by horizontal bars.
Although, the use of phospho-specific antibodies (16) and 86Rb tracer fluxes in X. laevis oocytes (7, 15) are established experimental methods for investigating the regulation of NKCC1 activity, they are still indirect assays. Using GST fusion proteins of wild-type and mutant SPAK and NKCC1 in in vitro 32P assays, we have demonstrated autophosphorylation of SPAK and SPAK phosphorylation of specific threonine residues in the N-terminal domain of NKCC1 (15). To confirm that PP1 dephosphorylates both SPAK and NKCC1, we initially performed 32P experiments with SPAK and NKCC1 fusion proteins, followed by addition of recombinant PP1 for increasing amounts of time. As shown in Fig. 5, A and B, although PP1 dephosphorylates SPAK in a time-dependent manner (open triangles), addition of the N-terminal domain of NKCC1 as a scaffold significantly increases the rate of kinase dephosphorylation (open squares). Note that PP1 completely dephosphorylates NKCC1 within 5 min (closed circles).
FIGURE 5.

Scaffolding of SPAK and PP1 increases the rate of kinase dephosphorylation. A, autoradiograph of a 45-min kinase reaction involving wild-type SPAK and wild-type NKCC1, followed by the addition of 2.5 units of recombinant PP1 for 5–30 min. One unit of PP1 hydrolyzes 1 nmol of substrate in 1 min. Upper panel, SPAK signal from reaction containing wild-type SPAK only; middle panel, SPAK signal from reaction containing both wild-type SPAK and wild-type NKCC1; bottom panel, NKCC1 signal from reaction containing both wild-type SPAK and wild-type NKCC1. B, densitometry analysis of the upper panel A (open triangles), middle panel A (open squares), and bottom panel A (filled circles). C, autoradiograph of a 45-min kinase reaction involving wild-type SPAK, wild-type NKCC1, and SPAK binding-deficient NKCC1 (F79A and F135A), followed by the addition of specified units of recombinant PP1 for 10 min. Upper panel, SPAK signal from the reaction containing both wild-type SPAK and SPAK binding-deficient NKCC1; middle panel, SPAK signal from reaction containing both wild-type SPAK and wild-type NKCC1; bottom panel, NKCC1 signal from reaction containing both wild-type SPAK and wild-type NKCC1. D, densitometry analysis of upper panel C (open diamonds), middle panel C (open squares), and bottom panel C (filled circles). Western blot analysis demonstrates that both wild-type NKCC1 (wt) and SPAK binding-deficient NKCC1 (bd) were present in sufficient amounts in their respective experiments (D, inset). Assays were repeated twice with similar results.
Given the speed of dephosphorylation observed in this experiment, we next titrated the amount of recombinant phosphatase from 0 to 12.8 × 10−2 units, using a single time point of 10 min. As a negative control for the scaffolding effect of the N terminus of NKCC1 on SPAK dephosphorylation, we utilized a mutant form of the N terminus that cannot bind SPAK. As previously shown (8), alanine substitution of key phenylalanine residues (F79A and F135A) in the two SPAK binding motifs of NKCC1 prevents both in vitro phosphorylation and in vivo activation of the cotransporter. Fig. 5, C and D, shows that PP1 concentrations capable of dephosphorylating SPAK (open squares) when the phosphatase is scaffolded with the kinase do not affect the level of SPAK phosphorylation when the SPAK binding-deficient form of the cotransporter is added (open diamonds). Note that the SPAK binding-deficient form of the cotransporter was also not phosphorylated by the kinase (data not shown). To verify that the lack of phosphatase activity on SPAK in the presence of the binding-deficient form of NKCC1 was not a result of protein levels in the in vitro reaction, we performed a Western blot analysis of equivalent amounts of wild-type and binding-deficient NKCC1 fusion proteins used in our reactions with an anti-GST antibody. The inset of Fig. 5D clearly demonstrates that both wild-type and binding-deficient forms of the cotransporter were present in sufficient amounts in their respective experiments. Interestingly, the level of SPAK autophosphorylation observed in the absence of the cotransporter or in the presence of a mutant cotransporter that cannot scaffold SPAK is significantly greater than the level of SPAK autophosphorylation observed in the presence of a cotransporter that can bind and be phosphorylated by the kinase. This difference is not due to a limiting amount of ATP (or 32P), as it is also observed in kinase reactions of shorter durations (data not shown).
DISCUSSION
Many proteins are functionally regulated by signaling cascades that involve both kinase and phosphatase activity. Pharmacological studies over the past 20 years have demonstrated that the functional activity of NKCC1 is determined by its state of phosphorylation (3, 4). Application of staurosporine or K252a, serine/threonine kinase inhibitors, has been shown to shift the “regulatory balance” of NKCC1 toward dephosphorylation and inactivation (21–23). Conversely, addition of calyculin A, which inhibits both PP1 and PP2A (24) has been shown to result in an increase in NKCC1 function (4, 21, 25).
Molecular biology advances over the past 16 years have allowed the identification of the gene that encodes for NKCC1 (26, 27), as well as, the identification of unique peptide sequences that serve as binding motifs for the serine/threonine kinases SPAK/OSR1 (5, 6), and for the phosphatase PP1 (11). In this study, we wanted to precisely determine how PP1 exerts its inhibitory effect on NKCC1 function, through dephosphorylation of the cotransporter, dephosphorylation of upstream signaling molecules, or through some combination of both.
As anticipated, overexpression of PP1 in X. laevis oocytes results in a significant reduction in NKCC1 function. Similar to the activating effect of SPAK phosphorylation (7, 8), PP1 dephosphorylation does not involve trafficking of the cotransporter to or from the plasma membrane. Indeed, confocal microscopy of individual oocytes co-expressing PP1 and EGFP-tagged NKCC1 exhibited similar levels of fluorescence at or near the plasma membrane as that of oocytes expressing EGFP-tagged NKCC1 alone, and biotinylation experiments confirmed that co-expression of PP1 did not alter membrane insertion of NKCC1.
When a catalytically inactive mutant of PP1 (H248K) was co-expressed with wild-type NKCC1, we observed significantly less inhibition of both isosmotic and hyperosmotic cotransporter activity, indicating that the phosphatase activity of PP1 is important to the functional regulation of the cotransporter. However, we did not observe a complete recovery under isosmotic or hyperosmotic conditions. Because the H248K mutation is known to completely abrogate PP1 function (12, 13), our data suggest a catalytically independent effect of PP1 on NKCC1 activity. Alternatively, the catalytic activity of the phosphatase might have opposing mechanistic effects on NKCC1 function, one that inhibits the cotransporter, and another that promotes cotransporter function. Overexpression of catalytically active and inactive PP1 might therefore have differential effects on the balance between cotransporter inhibition and activation.
To further sort out these effects, we utilized a deletion mutant of NKCC1 that lacks the putative PP1 binding motif. This mutant form of the cotransporter also lacks the second binding site for SPAK, but we knew from previous studies that one site was enough for activation of the cotransporter (8). In fact, SPAK binding and activation were confirmed in our experiments by the characteristic activation of the mutant NKCC1 under hyperosmotic conditions, a property affected when overexpressing catalytically inactive SPAK (7) or when the two SPAK binding domains are removed or mutated (8). As wild-type PP1 was unable to bind to NKCC1, the phosphatase was unable to fully inhibit cotransporter function, as with wild-type cotransporter. However, we still observed a decrease in cotransporter activity under both isosmotic and hyperosmotic conditions to the same level observed with catalytically inactive phosphatase, confirming the existence of another pathway of PP1 regulation. At this point, we do not have an explanation for this “undefined” effect of PP1.
Separating the effects of PP1 on the cotransporter and on the kinase was not a trivial task. To demonstrate dephosphorylation of NKCC1, we needed to make sure that the phosphatase could not deactivate the kinase, nor the phosphatase reaction be overwhelmed by the kinase reaction. Through the use of a constitutively active SPAK (T243E, S383A), which cannot be de-activated by the phosphatase and a mutant form of NKCC1 (F79A), which forces SPAK and PP1 to compete at an overlapping docking motif (134RFRVNFVD141), we observed a significant decrease in NKCC1 activity, demonstrating that it is the cotransporter that is being dephosphorylated and inactivated. On the other hand, to demonstrate that PP1 dephosphorylates SPAK in addition to dephosphorylating the cotransporter, we needed to use a protein that scaffolded both PP1 and SPAK, which is different from the protein that reports the functional effects of phosphorylation/dephosphorylation. To this end, we co-expressed AATYK1 with wild-type SPAK or constitutively active SPAK. Indeed, we previously demonstrated that expression of AATYK1 significantly inhibited heterologously expressed mouse NKCC1 activity in X. laevis oocytes (9). In that study, we determined that the catalytic activity of the tyrosine kinase was not a factor, but that AATYK1 served as an alternative scaffolding molecule for the regulatory kinase and phosphatase. Furthermore, AATYK1 scaffolding allowed endogenous levels of PP1 to significantly inhibit NKCC1 activity. However, the physiological role of AATYK is still unknown and has yet to be fully addressed. In this study, co-expression of AATYK1 with SPAK significantly decreased NKCC1 function, whereas co-expression of AATYK1 with the constitutively active SPAK resulted in NKCC1 activity similar to that observed in the absence of the scaffold. These data indicate that endogenous PP1 can dephosphorylate wild-type SPAK, but not constitutively active SPAK, when scaffolded in proximity of the kinase. Because these data are indirect demonstrations of PP1 dephosphorylation of NKCC1 and SPAK, we used GST fusion proteins of SPAK, NKCC1, and recombinant PP1 to demonstrate phosphatase dephosphorylation of NKCC1 and SPAK. The use of a SPAK binding-deficient form of NKCC1 (F79A, F135A) also demonstrates that PP1 dephosphorylation of SPAK requires the presence of scaffold (i.e. N-terminal domain of the cotransporter).
Altogether, our data indicate that PP1 inhibits NKCC1 activity at a minimum of three different levels: one that we called undefined because it did not require the catalytic activity of the phosphatase, a second that is through dephosphorylation and inactivation of SPAK, and a third that involves dephosphorylation and inactivation of the cotransporter. The work presented here is consistent with the concept that phosphatases might inhibit kinases or activate other phosphatases as part of signaling pathways that modulate cellular activities. Additional studies will be required to unveil the mechanism underlying the inhibitory effect of the catalytically inactive phosphatase.
Acknowledgments
We thank Ghali Abdelmessih for help in the husbandry and surgery of the X. laevis frogs and Kerri Rios for technical assistance in creating the mutants and preparing the DNA for cRNA transcription.
This work was supported, in whole or in part, by National Institutes of Health Grant GM074771 (to E. D.).
- NKCC1
- Na-K-2Cl cotransporter 1
- EGFP
- enhanced green fluorescent protein
- GST
- glutathione S-transferase
- AATYK
- apoptosis-associated tyrosine kinase
- SPAK
- Ste20-related proline/alanine-rich kinase
- PP1
- protein phosphatase 1.
REFERENCES
- 1.Russell J. M. (2000) Physiol. Rev. 80, 211–276 [DOI] [PubMed] [Google Scholar]
- 2.Gamba G. (2005) Physiol. Rev. 85, 423–493 [DOI] [PubMed] [Google Scholar]
- 3.Lytle C., Forbush B., 3rd (1992) J. Biol. Chem. 267, 25438–25443 [PubMed] [Google Scholar]
- 4.Lytle C. (1997) J. Biol. Chem. 272, 15069–15077 [DOI] [PubMed] [Google Scholar]
- 5.Piechotta K., Lu J., Delpire E. (2002) J. Biol. Chem. 277, 50812–50819 [DOI] [PubMed] [Google Scholar]
- 6.Piechotta K., Garbarini N., England R., Delpire E. (2003) J. Biol. Chem. 278, 52848–52856 [DOI] [PubMed] [Google Scholar]
- 7.Gagnon K. B., England R., Delpire E. (2006) Am. J. Physiol. Cell Physiol. 290, C134–C142 [DOI] [PubMed] [Google Scholar]
- 8.Gagnon K. B., England R., Delpire E. (2007) Cell Physiol. Biochem. 20, 131–142 [DOI] [PubMed] [Google Scholar]
- 9.Gagnon K. B., England R., Diehl L., Delpire E. (2007) Am. J. Physiol. Cell Physiol. 292, C1809–C1815 [DOI] [PubMed] [Google Scholar]
- 10.Roy J., Cyert M. S. (2009) Sci. Signal. 2, re9. [DOI] [PubMed] [Google Scholar]
- 11.Darman R. B., Forbush B. (2002) J. Biol. Chem. 277, 37542–37550 [DOI] [PubMed] [Google Scholar]
- 12.Zhang J., Zhang Z., Brew K., Lee E. Y. (1996) Biochemistry 35, 6276–6282 [DOI] [PubMed] [Google Scholar]
- 13.Traweger A., Wiggin G., Taylor L., Tate S. A., Metalnikov P., Pawson T. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 10402–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaplan M. R., Plotkin M. D., Brown D., Hebert S. C., Delpire E. (1996) J. Clin. Invest. 98, 723–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagnon K. B., England R., Delpire E. (2006) Mol. Cell. Biol. 26, 689–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darman R. B., Flemmer A., Forbush B. I. (2001) J. Biol. Chem. 276, 34359–34362 [DOI] [PubMed] [Google Scholar]
- 17.Dowd B. F., Forbush B. (2003) J. Biol. Chem. 278, 27347–27353 [DOI] [PubMed] [Google Scholar]
- 18.Gagnon K. B., Delpire E. (2010) FASEB J. 24, 609.3 (abstr.) [Google Scholar]
- 19.Vitari A. C., Thastrup J., Rafiqi F. H., Deak M., Morrice N. A., Karlsson H. K., Alessi D. R. (2006) Biochem. J. 397, 223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moriguchi T., Urushiyama S., Hisamoto N., Iemura S., Uchida S., Natsume T., Matsumoto K., Shibuya H. (2005) J. Biol. Chem. 280, 42685–42693 [DOI] [PubMed] [Google Scholar]
- 21.Palfrey H. C., Pewitt E. B. (1993) Pflugers Arch. 425, 321–328 [DOI] [PubMed] [Google Scholar]
- 22.O'Donnell M. E., Martinez A., Sun D. (1995) Am. J. Physiol. Cell Physiol. 269, C1513–C1523 [DOI] [PubMed] [Google Scholar]
- 23.Muzyamba M. C., Cossins A. R., Gibson J. S. (1999) J. Physiol. 517, 421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fagerholm A. E., Habrant D., Koskinen A. M. (2010) Mar. Drugs 8, 122–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lytle C., Forbush B. I. (1996) Am. J. Physiol. Cell Physiol. 270, C437–C448 [DOI] [PubMed] [Google Scholar]
- 26.Xu J. C., Lytle C., Zhu T. T., Payne J. A., Benz E., Jr, Forbush B., 3rd (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 2201–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delpire E., Rauchman M. I., Beier D. R., Hebert S. C., Gullans S. R. (1994) J. Biol. Chem. 269, 25677–25683 [PubMed] [Google Scholar]