

Abstract
It has previously been shown that the acylphosphatase from Sulfolobus solfataricus is capable of forming amyloid-like aggregates under conditions in which the native structure is maintained and via the transient formation of native-like aggregates. Based on the previously determined NMR structure of the native protein, showing a ferredoxin-like fold and the peculiar presence of an unstructured N-terminal segment, we show here, at a molecular level using NMR spectroscopy, that indeed S. solfataricus acylphosphatase remains in a native-like conformation when placed in aggregating conditions and that such a native-like structure persists when the protein forms the initial aggregates, at least within the low molecular weight species. The analysis carried out under different solution conditions, based on the measurement of the combined 1H and 15N chemical shifts and hydrogen/deuterium exchange rates, enabled the most significant conformational changes to be monitored upon transfer of the monomeric state into aggregating conditions and upon formation of the initial native-like aggregates. Important increases of the hydrogen/deuterium exchange rates throughout the native protein, accompanied by small and localized structural changes, in the monomeric protein were observed. The results also allow the identification of the intermolecular interaction regions within the native-like aggregates, that involve, in particular, the N-terminal unstructured segment, the apical region including strands S4 and S5 with the connecting loop, and the opposite active site.
Keywords: Biophysics, Diseases/Amyloid, Exchange, Protein/Molecular Dynamics, Protein/Self-assembly, Protein/Structure
Introduction
Proteins and peptides have a generic propensity to form well organized aggregates characterized by a fibrillar morphology and an extended cross-β structure, generally referred to as amyloid-like fibrils (1, 2). This process is important for a number of reasons. From a physicochemical perspective, it represents an essential feature of the behavior of polypeptide chains that need to be fully understood for a thorough characterization of the nature of proteins (3). From a more biological perspective, formation of amyloid fibrils, or intracellular inclusions with structurally related characteristics, is associated with over 40 pathological conditions in humans (4, 5). It is also a major problem in biotechnology because the large scale expression of proteins potentially interesting to the market often results in their self-assembly in inclusion bodies with amyloid-like structural features (4).
A large body of experimental data supports the hypothesis that globular proteins, normally adopting a well defined three-dimensional fold, need to unfold, at least partially, to become aggregation-competent (2, 5). However, it is also becoming increasingly evident that some normally folded proteins can aggregate from native-like states directly accessible through thermal fluctuations of the native state and with no need of transitions across the energy barrier for unfolding (reviewed in Ref. 6). One of the proteins shown to have an ability to aggregate directly from the native-like state is the acylphosphatase from Sulfolobus solfataricus (Sso AcP).3 The structure of native Sso AcP has been solved with both NMR and x-ray crystallography (7). Similarly to other acylphosphatases so far characterized, Sso AcP has a ferredoxin-like fold and thus consists of two parallel α-helices packed against a five-stranded β-sheet. However, by contrast with other acylphosphatases of known structure, Sso AcP contains an 11-residue, unstructured segment at the N terminus (7). In the presence of 15–25% (v/v) 2,2,2-trifluoroethanol (TFE), 50 mm acetate buffer, pH 5.5, 25 °C, Sso AcP has been shown to aggregate within 30 min into structured spherical and chain-like protofibrils (8). Protofibril formation is also observed in the absence of TFE under the same conditions of pH and temperature, although the process requires weeks in this case (8). The addition of inorganic phosphate, a competitive inhibitor binding to the active site of the enzyme, stabilizes the native fold of the protein and inhibits protofibril formation (9).
Under conditions in which protofibril formation occurs more rapidly (50 mm acetate buffer, 20% (v/v) TFE, pH 5.5, 25 °C), it has been shown that Sso AcP adopts initially, before aggregation starts, an essentially native fold (8, 10). Two sequential phases have been detected in the aggregation process (11). In a first rapid phase, the native protein aggregates within 2 min into oligomers in which the individual molecules still possess considerable enzymatic activity. These oligomeric species do not bind Congo red, thioflavin T (ThT), or 1-anilino-8-naphtalenesulfonic acid and are not yet characterized by the high content of β-sheet structure typical of amyloid aggregates (11). These initial native-like aggregates convert directly, within 30 min, into spherical and chain-like protofibrils. These latter species have a diameter of 3–5 nm; have no enzymatic activity; bind Congo red, ThT, and 1-anilino-8-naphtalenesulfonic acid; and have a large β-sheet structure content (11). Importantly, both phases of aggregation are more rapid than the unfolding reaction determined for the monomeric, native state under the same conditions, indicating that unfolding is not a necessary step in the process.
A combination of limited proteolysis experiments (10), protein engineering (10, 12, 13), and specific kinetic tests with peptides resembling the N-terminal segment (12) have put forward a model in which the rate-determining step for the first aggregation phase consists in the collision of the N-terminal segment with the globular unit of Sso AcP, probably with β-strand 4 (12). By contrast, the further conversion of the native-like assemblies into protofibrils appears to involve a cooperative and position-independent conversion of the protein molecules present in the initial aggregates (10). Importantly, however, the rates of both aggregation phases correlate with the free energy change of unfolding in the groups of mutants investigated, indicating that cooperative fluctuations are required for the formation of both the initial aggregates and the subsequent protofibrils (10).
Because aggregation of Sso AcP into amyloid-like protofibrils involves the formation, as a first detectable step, of assemblies in which the individual protein molecules retain a native-like conformation, it is important to explore the structure and dynamics of the native protein under conditions in which protofibril formation occurs, with an aim of comparing these characteristics with those determined under conditions in which aggregation does not occur. In this work we have used NMR spectroscopy to detect possible structural changes in six different conditions, chosen on the basis of their ability to stabilize the native state, populate the monomeric, native-like state that is prone to aggregation, or facilitate the detection of the native-like structure within the early aggregates.
MATERIALS AND METHODS
Expression and Purification of Sso AcP
Sso AcP was expressed and purified as previously described (14). The uniformly 15N-labeled protein was produced and purified by ASLA (Riga, Latvia) according to the protocol previously optimized (14). The resulting sequence was GSMKKWSDTEVFEMLKRMYARVYGLVQGVGFRKFVQIHAIRLGIKGYAKNLPDGSVEVVAEGYEEALSKLLERIKQGPPAAEVEKVDYSFSEYKGEFEDFETY. The initial GS dipeptide (labeled Gly−1 and Ser0) does not belong to the protein and is due to the gene cloning into the pGEX-2T plasmid. UV absorption was employed to determine Sso AcP concentration using an extinction coefficient at 280 nm (ϵ280) of 1.246 ml mg−1 cm−1.
Aggregation Kinetics of Sso AcP
Aggregation kinetics of Sso AcP were studied by ThT fluorescence under different experimental conditions. In brief, the reaction was started by diluting a solution containing 10 mm Tris, pH 8.0, and 10 mg ml−1 native Sso AcP to reach a final protein concentration equal to 0.4 mg ml−1. Four different final conditions were analyzed: (A) 50 mm NaCl, 50 mm phosphate buffer, pH 5.7; (B) 50 mm NaCl, 50 mm phosphate buffer, 20% (v/v) TFE, pH 5.7; (C) 50 mm NaCl, 50 mm acetate buffer, pH 5.7; and (D) 50 mm NaCl, 50 mm acetate buffer, 20% (v/v) TFE, pH 5.7. Conditions from A to D will be named in the text as phosphate, phosphate-TFE, acetate, and acetate-TFE, respectively. All of the samples were incubated at 25 °C. At several times, 60 μl of the sample were added to 440 ml of 25 μm ThT, 25 mm phosphate buffer, pH 6.0, at 25 °C. The resulting fluorescence was measured with a PerkinElmer LS-55 fluorimeter (Wellesley, MA) thermostatted with a Thermo-HAAKE F8 bath (Karlsruhe, Germany). Excitation and emission wavelengths were 440 and 485 nm, respectively. The resulting plot of ThT fluorescence (expressed as ratio between the observed signal and ThT fluorescence in the absence of protein) versus time was fitted to a single exponential equation of the following form,
where Fl(t) is the fluorescence observed at time t, A is the fluorescence at the apparent equilibrium, B is the change of fluorescence during the exponential phase, k is the apparent rate constant, and t is the time. Before starting the experiment, the sample was centrifuged at 16,000 × g for 5 min, and the protein concentration was measured.
NMR Experimental Conditions and Chemical Shift Analysis
In addition to the four experimental conditions aforementioned (conditions A–D), NMR experiments were also performed in the following: (E) 50 mm acetate buffer, 3 mm phosphate, and (F) 50 mm acetate buffer, 3 mm phosphate, 20% (v/v) TFE. 50 mm NaCl was present in all samples; the pH was adjusted to 5.7. These conditions will be referred to as “pseudoacetate” and “pseudoacetate-TFE,” respectively.
In every experimental condition listed, we acquired [1H,15N]HSQC spectra, which can be considered as the fingerprint of every protein. NMR experiments were acquired on a Bruker Avance 500 spectrometer on 0.4 mm Sso AcP samples. For the assignment conventional proton 15N decoupled homonuclear two-dimensional total correlation spectroscopy and nuclear Overhauser effect spectroscopy, two-dimensional [1H,15N]HSQC, HSQC-total correlation spectroscopy, and three-dimensional [1H,15N]HSQC-nuclear Overhauser effect spectroscopy at 37 and 25 °C were recorded. Typically a sweep width of 12 and 35 ppm, 1024–2048 × 256–512 data points were used at proton and nitrogen frequency, respectively. Data acquired and processed using Topspin (Bruker Biospin) were apodized with a squared sinebell shifted by 90° and polynomial base-line correction. The assignment was performed into the Felix (Accelrys, San Diego, CA) and Sparky (T. D. Goddard and D. G. Kneller, SPARKY3, University of California, San Francisco) framework. The combined 1H,15N chemical shift changes of backbone amides and Asn, Gln side chains amines, and Arg side chains amides in the different buffering conditions versus phosphate were analyzed.
NMR Exchange Measurements
3 mg of protonated Sso AcP were dissolved in 500 μl of deuterated buffer at pH* 5.7. (pH* refers to the pH measured in a D2O solution, uncorrected for the deuterium isotope effect.) A series of [1H,15N]HSQC experiments with 1k × 60 data points, 8 or 16 scans (480 or 960 s/spectrum) were recorded at 25 °C to follow amide decaying signals over time in the six previously mentioned conditions. The total duration of the experiments ranges from 3 to 4 days. To relatively quantify protein concentration in solution one-dimensional spectra using the ERETICTM (Bruker Biospin) were interleaved between HSQC experiments. The ERETICTM technique (electronic reference to access in vivo concentration) is a quantification experiment that uses an electronic signal as a reference.
The data were processed with NMRPipe (15) using a 90° shifted sinebell apodization function and standard linear prediction and analyzed with NMRViewJ 7.0.8 (16). The lag time between Sso AcP dissolution and the starting of the first measurement was ∼13 min in phosphate and 10 min in both pseudoacetate and acetate buffer, respectively; therefore fast exchanging amides were not detected, and a lower limit for their kinetic constant kex was estimated to be ∼10−3 s−1. We will refer to these amides as fast. In the opposite time limit very slow exchanging amide protons were still detectable after several days, and an upper limit for the kinetic constant was evaluated to be ∼10−9 s−1. We will refer to these amides as slow. For the remaining HN, referred in the text as intermediates, an exponential fitting permitted reliable evaluation of the kex, and the values are reported as a logarithm of protection factor (Log(PF)). To obtain the experimental exchange rates, kex, we measured, inside the NMRViewJ framework, the volumes of peaks at different deuteration times and fitted the data to an exponential function through a nonlinear fitting procedure. Protection factors PF were calculated as PF = kint/kex, where kint is the intrinsic rate, observed in unstructured peptides (17).
Diffusion Ordered NMR Spectroscopy (DOSY) Experiments
To measure the diffusion coefficient of Sso AcP in solution at 37 °C, the convection compensated two-dimensional double stimulated echo sequence (18) was used, and a matrix of 2k in the F2 dimension by 80 points in F1 where the gradient strength varies linearly from 2 to 95% of its maximum value (61.1 gauss/cm). The length of the diffusion gradient and the stimulated gradient were optimized for each sample to give a total decay of the signal of ∼95%. The water suppression was achieved with the addition of a sculpting module (19). The self-diffusion coefficient D can be evaluated from the diffusion decay of the protein signal according to the Stejskal and Tanner equation (20) I = I0e−D(γgδ)2(Δ-δ/3-τ/2), where γ is the gyromagnetic ratio, g is the gradient strength, δ is the applied gradient duration, τ is the correction rephasing time, and Δ is the echo time. The data were processed using a 90° sinesquare function in F2, and the analysis along F1 was realized using the Bruker Dosy software and fitting the signal decay with a single exponential.
In the presence of TFE, massive precipitation prevents the measurement of the diffusion coefficient. In fact during the experiment the signal intensity decays not only because of the strength of the gradient but also because of sample precipitation. To overcome the problem the DOSY was acquired as a series of 80 one-dimensional double stimulated echo experiments at different gradient strengths interleaved with one-dimensional spectra with ERETICTM. A precipitation index, obtained from the ratio between the protein and the ERETIC signal, was used to scale the DOSY spectra that were subsequently transformed in a two-dimensional matrix and processed as described.
The experiments were carried out in the above reported different buffering and cosolvent conditions, except for the acetate case, where only 15% (v/v) TFE was added to prevent a too massive signal loss forbidding the direct result comparison. Moreover, a normalization was performed measuring the trimethylsilylpropionic acid self-diffusion coefficient in each of the different experimental conditions that exhibit different viscosity (data reported in supplemental Table S1). Experimental uncertainties were estimated by comparing the variation of measured decay constants between individual recorded frequencies (21).
Molecular Dynamics Simulations
A molecular dynamics simulation of Sso AcP has been performed starting from the NMR structure (first model in the entry with Protein Data Bank code 1Y9O). The structure was protonated when needed and neutralized as described elsewhere (22). Sodium phosphate molecules were added by generating first the phosphate ion and then adding the sodium as described for the protein. The single molecule was then randomly rotated and placed at 18 Å spaced points in a cubic grid of 90 × 90 × 90 Å3 surrounding the protein. The system was then relaxed by 200 conjugate gradients minimization steps. The dielectric was set to 10.0 to minimize the effect of missing solvent, and the cut-off was 12.0 Å. The minimized system was solvated using the module solvate in the VMD software package (23) in a box with margins at 4.0 Å distance from any solute atom. The systems contained ∼65000 atoms. The solvent molecules were relaxed, keeping the solute (including the ion) fixed, by molecular dynamics simulation (22). The system without restraints was energy minimized by 300 conjugate gradients minimization steps. The system was then heated to 27 °C in 2 ps, and a further 118 ps of simulation was run to let the system equilibrate, and finally production run could start. The system was then simulated for 5 ns.
In all molecular dynamics simulation, the temperature was kept constant through a simple velocity rescaling procedure, whereas the pressure was controlled through a Berendsen bath (24) using a relaxation time of 100 fs. The resulting concentrations of phosphate and protein were 309 and 2.6 mm, respectively. All of the structural analyses, in particular root mean square deviations, secondary structure, and angular order parameter analyses, have been performed using the program MOLMOL (25). The analysis of solute phosphate contacts was performed with home-written C programs. The definition for a contact was a distance between heavy atoms less than the sum of van der Waal's radii plus 1 Å.
RESULTS
Sso AcP Does Not Aggregate in Phosphate Buffer
In a first preliminary experiment, the time course of Sso AcP aggregation was studied by means of ThT fluorescence in phosphate and acetate buffer, both in the absence and in the presence of 20% (v/v) TFE. Sso AcP induces an 11-fold increase in ThT fluorescence only in acetate buffer supplemented with 20% (v/v) TFE (Fig. 1). Such an increase follows apparent single exponential kinetics and was previously shown to be related to amyloid-like aggregation, particularly protofibril formation (8). The observed rate constant k in these conditions is (1.4 ± 0.2) × 10−3 s−1. Under the other three conditions, namely acetate buffer without TFE and phosphate buffer with and without 20% (v/v) TFE, Sso AcP does not induce any significant increase in ThT fluorescence, confirming that no amyloid-like aggregates form in these conditions on the studied time scale (Fig. 1).
FIGURE 1.

Aggregation time courses, monitored using ThT fluorescence, of four samples containing 0.4 mg ml−1 Sso AcP and different experimental conditions. The conditions were: 50 mm NaCl, 50 mm acetate buffer, 20% (v/v) TFE, pH 5.7 (●); 50 mm NaCl, 50 mm acetate buffer, pH 5.7 (○); 50 mm NaCl, 50 mm phosphate buffer, 20% (v/v) TFE, pH 5.7 (■); and 50 mm NaCl, 50 mm phosphate buffer, pH 5.7 (□). The continuous line represents the best fit to Equation 1 (see “Materials and Methods”) of the data collected in acetate buffer with TFE.
Sso AcP Retains a Native-like Structure in the Initial Small Aggregates as Revealed by the Combination of Pulse Field Gradient NMR Diffusion and Heterocorrelated NMR Spectra Analysis
Under conditions promoting amyloid-like aggregation, that is, acetate buffer with 20% (v/v) TFE, samples containing 0.3 mm Sso AcP prepared for NMR analysis showed a massive precipitation. Therefore, a crucial point was to clarify the actual aggregation state of the fraction of Sso AcP that was still present in solution after precipitation and that gave rise to the NMR signals. A series of pulse field gradient diffusion experiments were carried out in four different experimental conditions, acetate and phosphate buffers, both with and without 20% (v/v) TFE (experimental conditions A–D, as described under “Materials and Methods”). Fig. 2 shows two representative DOSY spectra obtained in acetate buffer and 20% (v/v) TFE (blue) and in phosphate buffer without TFE (black). From the diffusion coefficient values (D) obtained in each of these experiments, a value of apparent hydrodynamic radius (r) was obtained according to the Stokes-Einstein equation r = kT/6πηD, where k is the Boltzmann constant, T is the absolute temperature, and η is the viscosity coefficient (26, 27). r values of 15.3 ± 0.3 and 15.8 ± 0.5 Å were obtained in phosphate buffer without TFE and with 20% (v/v) TFE, respectively. These values are in agreement with that of monomeric, native Sso AcP, as determined previously by NMR (7). A higher value of 18.0 ± 0.3 Å was determined for r in acetate buffer in the absence of TFE, still in agreement with that of a monomeric, native Sso AcP molecule. The DOSY spectra obtained in acetate buffer and 15% (v/v) TFE revealed a wide range of D values (Fig. 2). Application of the Stokes-Einstein equation to the observed range of D values leads to r values spanning from 24 to 75 Å. In these experiments the TFE concentration was kept to 15% (v/v), rather than 20% (v/v), to avoid a prohibitive loss of signal. Precipitation of Sso AcP prevented the use of standard two-dimensional DOSY experiments and required the definition of a specific protocol described under “Materials and Methods.” Such DOSY experiments indicate the concomitant occurrence of species with different oligomeric states, ranging from low molecular weight species to aggregates composed of many molecules.
FIGURE 2.

Diffusion coefficient values for Sso AcP in aggregating and nonaggregating conditions. DOSY signals for Sso AcP in phosphate without TFE (black) and in acetate with 15% (v/v) TFE (blue). A very noisy trace was obtained for the latter condition because of a low signal to noise ratio. The paucity of the signal prevented a fine analysis by means of deconvolution techniques, and each column of the DOSY spectrum was processed with a single exponential function. It is, however, evident that different oligomeric components are present in acetate and TFE.
Fig. 3 shows [1H,15N]HSQC spectra acquired in acetate buffer, in which aggregation does not occur on the time investigated in this work, and in acetate supplemented with 20% (v/v) TFE, where aggregation does occur. The spectra indicate that in both conditions Sso AcP adopts a fully folded state. The resonance peaks obtained in aggregating conditions originate from the low molecular weight aggregates, in exchange with the larger oligomers present in solution. Overall, the native-like appearance of the HSQC spectrum and the large D values in the DOSY spectra indicate that, in acetate and TFE, the individual protein molecules adopt a native-like conformation and are in intermolecular exchange within the aggregates.
FIGURE 3.

Two-dimensional [1H,15N]HSQC spectra of Sso AcP acquired in acetate in the absence (A) and presence (B) of 20% (v/v) TFE. The assignments of backbone amide groups are labeled. The sc labels indicate side chain peaks from tryptophan, asparagine, glutamine, or arginine residues. The latter ones are folded downfield by 33.55 ppm. Unassigned peaks are indicated by asterisks.
Changes of Structure and Dynamics of Sso AcP under Aggregating Conditions: Methodology Set-up
The ThT fluorescence and diffusion NMR data described above show that the addition of 20% (v/v) TFE to a sample containing Sso AcP in acetate buffer triggers aggregation. On the other hand the same data show that Sso AcP remains soluble in phosphate buffer, independently of the presence of TFE in solution. The persistent solubility of Sso AcP in phosphate buffer upon the addition of TFE is due to the fact that phosphate acts as a competitive inhibitor of acylphosphatases (28) with a Kd of 1.12 ± 0.10 mm in the case of Sso AcP at pH 5.5 (12). The binding of the phosphate ion to Sso AcP causes a dramatic increase in conformational stability and structural rigidity of the native state, inhibiting aggregation, which by contrast requires structural fluctuations within the native state (6). This scenario is ideal to investigate the effect of TFE on the monomeric state of Sso AcP. Indeed, by comparing the dynamics and structure of Sso AcP in phosphate buffer with and without TFE, the effect of the fluoroalcohol on the monomeric state of Sso AcP can be investigated.
A further condition was also investigated in which phosphate ions are in 6-fold molar excess with respect to Sso AcP in an acetate buffer. Under these conditions, which will be referred to as “pseudoacetate” buffer, Sso AcP aggregation is partially inhibited, and phosphate ions are present in the active site of Sso AcP. Therefore, a comparison between the consequences of adding TFE in phosphate and pseudoacetate buffer will give the possibility of distinguishing between the chaotropic and local effects of phosphate. On the other hand, the comparison between the dynamics and structure of Sso AcP in acetate buffer with and without TFE allows the consequences of the aggregation process to be determined directly.
Structure and Dynamics of Monomeric Sso AcP in Phosphate and Phosphate-TFE
As a first analysis we titrated the protein in phosphate buffer with TFE concentrations ranging from 0 to 20% (v/v) and acquiring [1H,15N]HSQC spectra at 25 °C for each of the progressive TFE additions. Supplemental Fig. S1 depicts the HSQC spectra acquired at 0, 10, and 20% (v/v) TFE. The 1H and 15N chemical shifts of the amide peaks are consistent with the maintenance of a structured protein up to 20% (v/v) TFE. The comparison of the combined 1H and 15N chemical shift changes in the presence and absence of 20% (v/v) TFE is reported in Fig. 4A. The major differences are in the N-terminal segment (residues Phe10–Met12), in loop L1 (residue Leu23), in helix H1 (residues Phe32–Gln34 and Arg39), in strand S2 (residue Gly44), in strand S3 (residues Ser53 and Val54), in loop L4 (residue Glu62), in helix H2 (residues Glu70–Ile72), in loop L5 (residue Ala78), in strand S4 (residue Lys83), in the long loop L6 (residue Glu90), and in strand S5 (residue Tyr101). The cosolvent addition causes a widespread but moderate effect in terms of chemical shift deviations.
FIGURE 4.

Combined HN and N chemical shift changes determined for the various residues according to Mulder et al. (30) (where Δδ is the square root of (ΔδHN)2 + (ΔδN/6.51)2) between 0 and 20% (v/v) TFE, in phosphate buffer (A), in pseudoacetate buffer (B), and in acetate buffer (C). Charts color code: aquamarine for N-HN Δδ; blue for the Arg, Asn, and Gln side chains Δδ. The mean combined chemical shift change (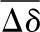 ) is indicated by the horizontal tick line; the + σ value, where σ is the standard deviation for the Δδ value, is indicated by the dashed line; the + 2σ value is indicated by the dotted line. In all panels, insets report the Δδ values on the native structure of Sso AcP according to the following color code: green for residues with Δδ ≤ , yellow for residues with < Δδ < + σ, orange for residues with σ < Δδ < + 2σ, red for residues with Δδ > + 2σ; and gray for proline and for nondefined residues. Orange cylinders and blue arrows above each panel refer to α-helices and β-strands, respectively, and facilitate the association between the Δδ values reported in the main panel and their localization in the native structure of Sso AcP.
) is indicated by the horizontal tick line; the + σ value, where σ is the standard deviation for the Δδ value, is indicated by the dashed line; the + 2σ value is indicated by the dotted line. In all panels, insets report the Δδ values on the native structure of Sso AcP according to the following color code: green for residues with Δδ ≤ , yellow for residues with < Δδ < + σ, orange for residues with σ < Δδ < + 2σ, red for residues with Δδ > + 2σ; and gray for proline and for nondefined residues. Orange cylinders and blue arrows above each panel refer to α-helices and β-strands, respectively, and facilitate the association between the Δδ values reported in the main panel and their localization in the native structure of Sso AcP.
As a second analysis the rates of H/D exchange of the amide hydrogen atoms were compared in phosphate buffer in the presence and absence of 20% (v/v) TFE. Supplemental Fig. S2A reports the PF measured in the two conditions for all the non-proline amides. The high stability of Sso AcP, ultimately caused by its hyperthermophilic origin, determines a high structural rigidity where the hydrogen atoms belonging to the core of the protein (∼⅓ of the total NHs) do not exchange in a time frame of a few days. On the other hand, another third of NHs are solvent-exposed and exchange before our measurements are started. Nevertheless it was possible to qualitatively describe the differences in protein dynamics in the six experimental conditions studied here.
We can first focus on the condition in the absence of TFE and Fig. 5A depicts the structure of Sso AcP color-coded to show the differences of protection factors among the various residues in these conditions. 33 of 100 non-proline amide peaks did not exchange during the 71 h of experimental acquisition. This percentage of slow exchange amides is much higher than usually observed for mesophilic proteins but seems to be consistent with the hyperthermophilic nature of Sso AcP. The overall behavior highlights that the paired strands S1, S3, and S2, located in the central part of the sheet, do not exchange even after 3 days in deuterated solution except for Tyr45 located in the center of S2 strand, which is in the fast exchange regime. The two edge strands S4 and S5 show a more “patchy” behavior; in particular in strand S4 all amides pointing outward (Val84, Tyr86, and Phe88) are in the fast exchange regime, whereas those pointing inward and forming hydrogen bonds with strand S1 exchange at intermediate (Lys83, Asp85, and Ser89) or slow (Ser87) rates. Strand S5 is characterized by intermediate exchange rate except for the fast Phe98 amide, which points outward. Amides of residues Thr99 and Arg101 are hydrogen-bonded to residues of strand S2, whereas residue Thr100 points toward the bulk. It is thus striking to observe an intermediate exchange rate for the latter.
FIGURE 5.

Protection factors of amide hydrogen atoms in phosphate buffer in the absence (A) and in the presence (B) of 20% (v/v) TFE; in pseudoacetate buffer in the absence (C) and in the presence (D) of 20% (v/v) TFE; and in acetate buffer in the absence (E) and in the presence (F) of 20% (v/v) TFE. The color code indicates the exchange regime of the amide residues: fast (red), intermediate (green), and slow (blue).
In both α-helices all residues of the protein core exchange slowly except for the two N-terminal residues Phe29 and Arg30 in helix H1 that are in the fast exchange mode. The residues exposed to the solvent show intermediate exchange rates, except for the fast exchange of Glu62, Glu63, and Ala64 at the N terminus of helix H2 and the slow exchange of Ile35 in helix H1. As expected, the N-terminal segment of the protein and all loops are in fast or intermediate exchange regime except for the slow amides of residues Ile42 (loop L2) and Tyr61 (loop L4), both located at the N-terminal apex of the β-sheet.
The presence of 20% (v/v) TFE in phosphate buffer induces a general decrease in protection factor values, except for strand S1, remaining in the slow exchange regime, and for the facing Lys92 in L6 and Gly41 in L2 loops, which instead increase their PF values (Fig. 5B). The most remarkable variations are in strands S2 and S3 and in the facing helix H1. Residues Leu69 and Lys73 in helix H2 and Ser87 in strand S4 are also affected. To facilitate the comparison between the two sets of conditions in phosphate buffer (with and without 20% (v/v) TFE), Fig. 6A reports the differences of protection factor regimes, viewed on both the structure and along the sequence of Sso AcP.
FIGURE 6.

Protection factors changes, reported as differences of Log(PF) values, between the absence and the presence of 20% (v/v) TFE in phosphate (A), pseudoacetate (B), and acetate (C) buffer. The ΔLog(PF) value is arbitrarily given as 1 when the residue changes its protection factor from slow to intermediate and from intermediate to fast, as 2 from slow to fast, and as −1 from fast to intermediate and from intermediate to slow. The color code reflects the exchange regime of the residue in the presence of 20% (v/v) TFE (red for residues in fast exchange regime, green for intermediate, and blue for slow exchange regime). Protection factors changes are also reported on the native structure of Sso AcP, with the following color code: red for residues going from slow to fast regime; orange from intermediate to fast and from slow to intermediate regime; and blue from fast to intermediate regime. Orange cylinders and blue arrows below each panel refer to α-helices and β-strands, respectively, and facilitate the association between the ΔLog(PF) values reported in the main panel and their localization in the native structure of Sso AcP.
Structure and Dynamics of Monomeric Sso AcP in Pseudoacetate and Pseudoacetate-TFE
We next titrated the protein in pseudoacetate buffer with TFE concentration ranging from 0% (v/v) to 20% (v/v) by acquiring [1H,15N]HSQC spectra at 25 °C for each of the progressive TFE additions. The combined chemical shift changes of the amide peaks, shown in Fig. 4B, are similar to those determined in phosphate buffer: N-terminal segment (residues Phe10, Glu11, and Leu13), strand S1 (residue Arg15), loop L1 (residue Leu23), helix H1 (residues Phe32, Val33, Ile35, and Arg39), loop L4 (residue Glu62), helix H2 (residue Glu63), strand S4 (residue Tyr86), and strand S5 (residue Tyr101). Hence, similarly to the previously described analysis in the phosphate buffer, in the pseudoacetate buffer the cosolvent addition causes a moderate effect involving the entire protein.
The rates of H/D exchange of the amide hydrogen atoms were then determined in pseudoacetate buffer in both the presence and absence of 20% (v/v) TFE (supplemental Fig. S2B). Similarly to with the phosphate condition, in pseudoacetate buffer without TFE the protein dynamics is characterized by a very rigid core, comprising the central part of the β-sheet and the inner portion of the facing helices (Fig. 5C).
A striking loosening of the conformational rigidity of Sso AcP occurs upon the addition of 20% (v/v) TFE in pseudoacetate buffer (Fig. 5D). No slow exchanging amide survives even in the central strands. In contrast, the amides of eight residues, located on the sheet surface (residues Leu13, Gly44, Asn48, Gly60, and Thr100) and in loop L1 (residue Val24) and loop L6 (residues Tyr91 and Lys92), switch from fast to intermediate exchange regime (Fig. 6B). It is worth noting that the molecular flexibility in phosphate and pseudoacetate are rather similar, whereas TFE increases internal motions more effectively in pseudoacetate than in phosphate, indicating a protective chaotropic effect of phosphate (Fig. 5, compare D with B).
Structure and Dynamics of Sso AcP in Acetate and Acetate-TFE
We finally titrated the protein in acetate buffer with increasing TFE concentrations. As described above, the comparison between the NMR data obtained in acetate buffer with and without TFE allows the consequences of amyloid-like aggregation to be determined directly. The combined chemical shift changes of the amide peaks following the addition of 20% (v/v) TFE indicate that TFE produces a more specific structural effect (Fig. 4C). Robust deviations are observed for residues Phe10, Glu11, Leu13 (N-terminal segment); Gln25 (backbone and side chain amides, loop L1); Glu63 (helix H2); Tyr86 (strand S4); Lys92 and Gly93 (loop L6); and Phe98 (strand S5). It is also evident that the regions of the sequence encompassing approximately the proximal portion of the N-terminal segment (residues Glu8–Glu11) and the regions involving the strand S4, loop L6, and strand S5 (residues Ala79–Phe98) contain the highest density of residues with Δδ values higher than the mean, indicating that such regions undergo statistically significant structural perturbations. A further analysis was conducted by subtracting from the combined chemical shift changes in acetate (acetate in the presence and in the absence of TFE) the combined chemical shift changes in phosphate (phosphate in the presence and in the absence of TFE), thus removing the structural contribution caused by TFE addition on the monomeric protein (supplemental Fig. S4). The analysis shows that the net structural changes caused by the aggregation event involve the N-terminal segment (Trp4 and Glu8–Leu13), the N-terminal portion of helix H2 (Glu63), residue Tyr86 belonging to strand S4, loop L6 (Lys92 and Gly93), and Phe98 and Tyr101 of strand S5.
A global increase of the H/D exchange rate following TFE addition was observed in acetate buffer, although not as marked as in pseudoacetate buffer (supplemental Fig. S2C; Fig. 5, compare F with D). In fact, the central strand S1 remains mainly slow exchanging. Moreover 12 residues increase their protection factors: Lys14 and Tyr21 (strand S1), Val24 (loop L1), Arg39 (helix H1), Gly41 (loop L2), Ala64 and Ser66 (helix H2), Ala79(loop L5), Tyr91 and Lys92 (loop L6), and Thr100 and Tyr101 (strand S5) (Fig. 6C).
The Phosphate Ion Modifies the Cradle Conformation of the Sso AcP Active Site and Influences the Protein Dynamics
Variations in structure and dynamics are caused by different phosphate concentrations as inferred from comparison of the data collected in experimental conditions C (acetate) and E (pseudoacetate) with those obtained in condition A (phosphate). Upon decreasing the phosphate concentration (experimental conditions A, C, and E), the main chemical shift deviations are located, as expected (28), in the active site cradle (residues Gln25–Phe32 and Lys47–Gly52; supplemental Fig. S3). In particular, the Δδ extent is much larger in acetate than in pseudoacetate. This is probably due to an active site conformational variation and to the deshielding effect of the phosphate anion oxygen. The phosphate ions exchange between the free and bound position in the active site of Sso AcP in an intermediate exchange regime, with respect to the NMR time scale (data not shown) and interact preferably with Gly26 and Gly28 backbone amides. A 5-ns molecular dynamics simulation was performed starting from the AcP phosphate-free form (2.6 mm) (NMR solution structure 1Y9O) in the presence of 309 mm phosphate ions, as described under “Materials and Methods.” The snapshots at 0, 1.1, and 5 ns are shown in Fig. 7A. Interestingly, a phosphate ion after 2.4 ns faces exactly Gly26, Gly28, and Arg30. The phosphate anion appears to be driven by electrostatic forces from the bulk to the active site. This simulation may be regarded as a refinement of the NMR structure in the L1 and L3 loops that contain the active site cradle. In fact the geometry of Gly26 and Gly28 changes, and the two amide hydrogens, highly shifted in the acetate NMR spectra, rotate to improve their interaction with the phosphate anion (Fig. 7B).
FIGURE 7.

Molecular dynamics simulation in the presence of phosphate anions. A phosphate molecule reaches the Sso AcP catalytic cradle, which undergoes significant conformational changes. A, snapshots of the Sso AcP molecular dynamics trajectory in the presence of phosphate anions at the beginning, after 1.1 and at 5 ns. B, backbone superimposition of the initial (green) and final (light blue) structure: Gly26 and Gly28 backbone HN, and the side chain of the two catalytic residues (Arg30 and Asn48) are highlighted by sticks. The distances in Å between the HN of the two glycines and a phosphate oxygen are shown.
The backbone dynamics variations of Sso AcP, occurring in pseudoacetate, can be highlighted by comparing panels C and A of Fig. 5. The dynamics of the β-sheet portion formed by S4-S1-S3 strands do not change, with the exception of residue Gly60 (C-terminal portion of strand S3). On the other edge more changes occur; the amides of residues Gly44, Asn48 (strand S2), and Thr100 (strand S5) increase their mobility, whereas the amide of residue Gly99 (strand S5) becomes slow exchanging. Four residues belonging to the helices, namely residues Gln34, Ile35, Arg39 (helix H1), and Leu65 (helix H2) untie. The only change in the loops is the loosening of Val24 (loop L1).
A mild overall protection factor decrease is observed in acetate (Fig. 5, A and E), more pronounced at the beginning of strand S3 (Val54) consistently with the absence of bound phosphate, in the following L4 loop (Tyr61) and in H2 helix (Leu65). The absence of the protective chaotropic phosphate has an effect limited to the C-terminal edge of the protein (S5, S2, and H2), whereas when the phosphate is completely absent the increased H/D exchange rate is spread throughout the whole molecule.
DISCUSSION
It has been reported that the addition of 15–25% (v/v) TFE to solutions containing Sso AcP at pH 5.5 induces formation of native-like, enzymatically active aggregates, which subsequently reorganize, with no need of dissolution and reaggregation, into amyloid-like protofibrils (8, 10–12). The aim of our work was to investigate, at a molecular level using NMR spectroscopy, the structural changes induced by TFE on the monomeric state of Sso AcP, before aggregation occurs to any significant extent, and on the structure adopted by the protein within the initial native-like aggregates. The first aim was achieved by adding TFE to solutions of Sso AcP in buffers containing high or small concentrations of phosphate and exploiting the capability of this ion to bind to the active site of the protein, stabilize the native state, and thus inhibit aggregation. The second aim was achieved by studying the protein directly in acetate buffer in the absence of phosphate, i.e. under conditions in which aggregation occurs.
In the presence of phosphate, the radius of gyration of the protein, measured from DOSY experiments, remains compatible with that of native-like, monomeric Sso AcP when TFE is added. This confirms the effective inhibition of Sso AcP aggregation under these conditions upon TFE addition. By contrast, in acetate buffer the measured radius of gyration is compatible with that of native, monomeric Sso AcP only in the absence of TFE, with larger aggregated species forming in its presence. [1H,15N]HSQC spectra obtained in the latter conditions highlight an essentially native-like structure for Sso AcP (Fig. 3), indicating that (i) the protein retains a compact native-like state and (ii) a highly dynamic character occurs within the aggregated species. It was just the dynamic nature of the intermolecular interactions within the early aggregates that allowed the protein to be probed within the aggregates.
In phosphate buffer, the addition of 20% (v/v) TFE does not induce global structural changes on Sso AcP. The protein maintains an essentially folded, native-like state, as determined from the analysis of [1H,15N]HSQC spectra. Changes of the combined 1H and 15N chemical shifts are detected only for some residues and are generally small to moderate (Fig. 4A). The observation that such changes involve different portions of the sequence and of the three-dimensional structure of Sso AcP suggests that the structural modification following TFE addition, albeit moderate, is widespread throughout the protein fold and also involves the unstructured N-terminal region. The analysis performed in pseudoacetate buffer, at a phosphate concentration that is sufficient to inhibit aggregation but with phosphate exchanging very rapidly between the free and bound state and leaving ∼30% of the Sso AcP molecules unbound to the ion, confirms that native Sso AcP undergoes a very moderate distortion in different areas of the three-dimensional fold (Fig. 4B). The only significant difference between the structural distortions observed upon TFE addition in phosphate and pseudoacetate involves strand S3, which appears to undergo a lower degree of variation in pseudoacetate.
In phosphate buffer native Sso AcP becomes more dynamic following TFE addition, as determined by measurements of H/D exchange rates (Fig. 5, compare B with A). With the exception of strand S1, all other portions of the molecule undergo increased exchange rates, indicating a loosening of the whole structure. This behavior is more marked in pseudoacete buffer (Fig. 5, compare D with C), where even strand S1 becomes more dynamic upon TFE addition. Overall, the NMR analysis indicates that following the addition of TFE Sso AcP maintains, in its monomeric state, a native-like structure that appears only marginally distorted, but markedly less rigid. Nevertheless, the higher structural lability does not suffice to induce fibril formation.
A different picture holds in acetate buffer, where TFE addition causes the conversion of the native protein into aggregates. [1H,15N]HSQC spectra obtained in the presence of TFE show that Sso AcP retains an essentially native-like structure in the aggregates (Fig. 3B). The peak line width observed in [1H,15N]HSQC spectra obtained in acetate buffer in the presence of TFE is considerably increased relative to all other nonaggregating conditions. This observation is consistent with the presence of a molecular weight distribution of oligomers of Sso AcP molecules, in fast intermolecular exchange, ranging from tetramers up to large oligomers of approximately a hundred molecules, when a protein density of 1.42 × 104 kg/m3 is used (29). It can be stated that the aggregates observed with NMR are the native-like aggregated species detected early in the aggregation process rather than the protofibrils forming later. Indeed, the latter have high molecular weights and stable β-sheet packing leading to prohibitive relaxation rates for NMR observation.
The analysis carried out with the combined 1H and 15N chemical shifts reveals important differences between the monomeric, native state in acetate buffer in the absence of TFE and the native-like species in the protein aggregates populated upon TFE addition. Although the structural changes are globally of small entity, more pronounced perturbations occur in the N-terminal region and in the nearby apical region involving strand S4, strand S5, and loop L6 connecting them and in the opposite active site region. These perturbations are possibly induced by intermolecular contacts (supplemental Fig. S4).
The H/D exchange rate data are more challenging to interpret in this case. Indeed, the loosening of the native structure caused by TFE addition brings about an increase of the exchange rates, whereas structure packing as a consequence of protein aggregation causes the opposite effect. The observed change of the H/D exchange rate for a given residue consists in the net result of these two opposite tendencies, which complicates the analysis. The fact that the intermolecular interactions occurring within the aggregates may be highly dynamic represents a further complicating factor. Despite these difficulties the observation that residues from different portions of the structure decrease their exchange rates as a consequence of aggregation, particularly in the two apical regions of the Sso AcP native structure (Fig. 6C), indicates again the involvement of different regions of the sequence in the formation of the native-like aggregates. Similar findings were also observed by simulation experiments for other small amyloidogenic globular proteins (22).
The six experimental conditions used here permit the filtering of the effects of the cosolvents and the different buffers on the structure and dynamics of Sso AcP. In particular, it is evident that the protective effect of the phosphate ion against aggregation is mainly due to its binding to the active site but also by a widespread chaotropic effect.
The experimental approach adopted here is applicable only to amyloidogenic proteins that do not unfold when placed in amyloidogenic conditions. It is well known, in fact, that for many amyloidogenic proteins the process of fibril formation requires misfolded monomeric intermediates. In such cases a similar structural study in aggregating conditions would be prevented by the unfolding and eventually precipitation of the protein.
CONCLUSIONS
The data presented here illustrate, for the first time at a molecular level, that Sso AcP maintains a native-like structure in its monomeric state under conditions that promote aggregation rather than undergoing a process of partial unfolding. This confirms previous observations obtained with kinetic analysis of folding and unfolding, enzymatic activity measurements and CD spectroscopy (8, 10). Our data also indicate that TFE-induced aggregation of Sso AcP does not arise from the potential ability of TFE to promote a partial or global unfolding of the compact native state of the protein. Instead, the fluoroalcohol increases the internal dynamics within the native state, thus facilitating the structural fluctuations that are necessary to reach a proper intermolecular matching and initiate aggregation. Structural flexibility seems a necessary prerequisite for Sso AcP aggregation and requires the total absence of phosphate ions, which prevent aggregation by binding to the active site and limiting the structural fluctuations. The structural perturbations of Sso AcP are only moderate and localized under aggregating conditions, whereas the increase of its internal dynamics is substantial and seems to involve the whole structure. The inverse correlation found in a group of mutants between the conformational stability of the native state and the rate of formation of the native-like aggregates supports this view, particularly when considering that the mutations were located in different regions of the Sso AcP structure (10).
In addition to providing information on the structure and dynamics of the monomeric state populated under conditions promoting aggregation, the NMR analysis presented here also provides information on the structure adopted by Sso AcP in the initial aggregates. The data show that the native-like fold is maintained in the initial aggregates, which seem to be characterized by loose and dynamic intermolecular interactions between native-like Sso AcP molecules. The combined chemical shift analysis suggests that the regions undergoing the most significant changes upon formation of the native-like aggregates include the N-terminal unstructured segment, the apical region encompassing strand S4, loop L6, and strand S5, and the active site region. The native-like and dynamic character of such aggregates explains why such species possess enzymatic activity, have a secondary structure content similar to that of the native state, and do not bind dyes diagnostic for amyloid, such as ThT and Congo red (11). The involvement of the N-terminal and apical region involving strands S4-S5 and the interconnecting loop is also in agreement with the finding that the N-terminal region and strand S4 are solvent-exposed and flexible, as detected using limited proteolysis (10), and that mutations affecting such regions influence the conversion of the native-like state into initial aggregates more effectively than other mutations (10, 13).
Supplementary Material
This work was supported by MIUR Grants PRIN 2007 XY59ZJ, PRIN M3E2T2, FIRB RBNE03PX83, and FIRB RBRN07BMCT and European Union EURAMY Project LSHM-CT-2005-037525.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S4.
- Sso AcP
- S. solfataricus acylphosphatase
- TFE
- 2,2,2-trifluoroethanol
- CR
- congo red
- ThT
- thioflavin T
- PF
- protection factor
- DOSY
- diffusion ordered NMR spectroscopy
- H/D
- hydrogen/deuterium
- HSQC
- heteronuclear single quantum coherence.
REFERENCES
- 1.Stefani M., Dobson C. M. (2003) J. Mol. Med. 81, 678–699 [DOI] [PubMed] [Google Scholar]
- 2.Uversky V. N., Fink A. L. (2004) Biochim. Biophys. Acta 1698, 131–153 [DOI] [PubMed] [Google Scholar]
- 3.Jahn T. R., Radford S. E. (2005) FEBS J. 272, 5962–5970 [DOI] [PubMed] [Google Scholar]
- 4.Ventura S., Villaverde A. (2006) Trends Biotechnol. 24, 179–185 [DOI] [PubMed] [Google Scholar]
- 5.Chiti F., Dobson C. M. (2006) Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 6.Chiti F., Dobson C. M. (2009) Nat. Chem. Biol. 5, 15–22 [DOI] [PubMed] [Google Scholar]
- 7.Corazza A., Rosano C., Pagano K., Alverdi V., Esposito G., Capanni C., Bemporad F., Plakoutsi G., Stefani M., Chiti F., Zuccotti S., Bolognesi M., Viglino P. (2006) Proteins 62, 64–79 [DOI] [PubMed] [Google Scholar]
- 8.Plakoutsi G., Taddei N., Stefani M., Chiti F. (2004) J. Biol. Chem. 279, 14111–14119 [DOI] [PubMed] [Google Scholar]
- 9.Soldi G., Plakoutsi G., Taddei N., Chiti F. (2006) J. Med. Chem. 49, 6057–6064 [DOI] [PubMed] [Google Scholar]
- 10.Plakoutsi G., Bemporad F., Monti M., Pagnozzi D., Pucci P., Chiti F. (2006) Structure 14, 993–1001 [DOI] [PubMed] [Google Scholar]
- 11.Plakoutsi G., Bemporad F., Calamai M., Taddei N., Dobson C. M., Chiti F. (2005) J. Mol. Biol. 351, 910–922 [DOI] [PubMed] [Google Scholar]
- 12.Bemporad F., Vannocci T., Varela L., Azuaga A. I., Chiti F. (2008) Biochim. Biophys. Acta 1784, 1986–1996 [DOI] [PubMed] [Google Scholar]
- 13.Soldi G., Bemporad F., Chiti F. (2008) J. Am. Chem. Soc. 130, 4295–4302 [DOI] [PubMed] [Google Scholar]
- 14.Modesti A., Taddei N., Bucciantini M., Stefani M., Colombini B., Raugei G., Ramponi G. (1995) Protein Expr. Purif. 6, 799–805 [DOI] [PubMed] [Google Scholar]
- 15.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 16.Johnson B. A., Blevins R. A. (1994) J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 17.Bai Y., Milne J. S., Mayne L., Englander S. W. (1993) Proteins Struct. Funct. Genet. 17, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jerschow A., Müller N. (1997) J. Magn Res. 125, 372–375 [Google Scholar]
- 19.Hwang T. L., Shaka A. J. (1995) J. Magn Res. 112, 275–279 [Google Scholar]
- 20.Stejskal E. O., Tanner J. E. (1965) J. Chem. Phys. 42, 288–292 [Google Scholar]
- 21.Baldwin A. J., Anthony-Cahill S. J., Knowles T. P., Lippens G., Christodoulou J., Barker P. D., Dobson C. M. (2008) Angew Chem. Int. Ed. Engl. 47, 3385–3387 [DOI] [PubMed] [Google Scholar]
- 22.Fogolari F., Corazza A., Viglino P., Zuccato P., Pieri L., Faccioli P., Bellotti V., Esposito G. (2007) Biophys. J. 92, 1673–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Humphrey W., Dalke A., Schulten K. (1996) J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 24.Berendsen H. J., Postma J. P., Van Gunsteren W. F., Dinola A., Haak J. R. (1984) J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 25.Koradi R., Billeter M., Wuthrich K. (1996) J. Mol. Graph. 14, 51–55 [DOI] [PubMed] [Google Scholar]
- 26.Einstein A. (1956) Investigations on the Theory of Brownian Movement, Dover Publication Inc., New York [Google Scholar]
- 27.Poling B. E., Prausnitz J. M., O'Connel J. P. (2001) The Properties of Gases and Liquids, McGraw-Hill Professional, New York [Google Scholar]
- 28.Stefani M., Taddei N., Ramponi G. (1997) Cell Mol. Life Sci. 53, 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer H., Polikarpov I., Craievich A. F. (2004) Protein Sci. 13, 2825–2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulder F. A., Schipper D., Bott R., Boelens R. (1999) J. Mol. Biol. 292, 111–123 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.