

Abstract
Bile acids are required for proper absorption of dietary lipids, including fat-soluble vitamins. Here, we show that the dietary vitamins A and D inhibit bile acid synthesis by repressing hepatic expression of the rate-limiting enzyme CYP7A1. Receptors for vitamin A and D induced expression of Fgf15, an intestine-derived hormone that acts on liver to inhibit Cyp7a1. These effects were mediated through distinct cis-acting response elements in the promoter and intron of Fgf15. Interestingly, transactivation of both response elements appears to be required to maintain basal Fgf15 expression levels in vivo. Furthermore, whereas induction of Fgf15 by vitamin D is mediated through its receptor, the induction of Fgf15 by vitamin A is mediated through the retinoid X receptor/farnesoid X receptor heterodimer and is independent of bile acids, suggesting that this heterodimer functions as a distinct dietary vitamin A sensor. Notably, vitamin A treatment reversed the effects of the bile acid sequestrant cholestyramine on Fgf15, Shp, and Cyp7a1 expression, suggesting a potential therapeutic benefit of vitamin A under conditions of bile acid malabsorption. These results reveal an unexpected link between the intake of fat-soluble vitamins A and D and bile acid metabolism, which may have evolved as a means for these dietary vitamins to regulate their own absorption.
Keywords: Bile Acid, Intestine, Liver, Nuclear Receptors, Vitamin A, Vitamin D, FGF15, FXR, RXR, VDR
Introduction
Lipid-soluble vitamins are essential for human health and must be obtained from the environment. They exist in four major groups (A, D, E, and K), each of which comprises a series of structurally related compounds with multiple biological functions. Members of the nuclear receptor family of transcription factors are central to the mechanism of action of the lipid-soluble vitamins A and D. Metabolites of vitamin A regulate gene transcription by binding to and activating the retinoic acid receptors (RARs4; NR1B) and the retinoid X receptors (RXRs; NR2B) (1). Similarly, the genomic actions of vitamin D are mediated by the vitamin D receptor (VDR; NR1I1) (2).
Retinol, the basic form of vitamin A, is obtained from foods of animal origin, where it is present in the form of retinyl esters. Vitamin A can also be synthesized in the liver from β-carotene, which is present in foods of plant origin. In the body, retinol is converted to several metabolites that function as signaling molecules in various biological processes, including vision, development, growth, metabolism, and cell differentiation (3–5). Most of the transcriptional actions of vitamin A in vivo have been shown to require the RXR/RAR heterodimer (3).
Vitamin D is synthesized in the skin from 7-dehydrocholesterol through a process that requires sunlight, or it can be obtained directly from the diet. Dietary sources include cholecalciferol (vitamin D3, animal origin) and ergocalciferol (vitamin D2, plant origin). Whether obtained from the diet or photoactivation in the skin, vitamin D must be converted to its bioactive form, 1α,25-dihydroxyvitamin D3, by the action of cytochrome P450 enzymes in the liver and kidney (6). Vitamin D is best known for its essential role in regulating calcium and phosphate homeostasis (7). Recently, the repertoire of physiological systems regulated by vitamin D and its receptor has been expanded to include both innate and adaptive immunity and bile acid detoxification (8–10).
The absorption of lipid-soluble vitamins from the diet requires the detergent actions of bile acids. Bile acids are amphipathic sterols synthesized from cholesterol in the liver and secreted into the intestine, where, when present at high concentrations, they function to emulsify dietary lipids (11). Cholesterol 7α-hydroxylase (CYP7A1), which catalyzes the rate-limiting step in bile acid biosynthesis, is tightly regulated at the transcriptional level by bile acids and other signaling molecules (12). Positive transcriptional regulators of Cyp7a1 include orphan nuclear receptors, LRH-1 (liver-related homologue-1; NR5A2) and hepatocyte nuclear factor 4α (NR2A1) (13, 14). Negative feedback regulation of Cyp7a1 in vivo involves two complementary mechanisms. First, bile acids activate the nuclear bile acid receptor (farnesoid X receptor (FXR); NR1H4) in the intestine to induce expression of Fgf15 (fibroblast growth factor 15; FGF19 in humans), which signals from the intestine to repress hepatic Cyp7a1 through a mechanism that involves the atypical nuclear receptor SHP (small heterodimer partner) and the membrane receptor FGFR4 (fibroblast growth factor receptor 4) (15). Second, bile acids activate FXR in the liver to induce transcription of SHP, which subsequently binds to LRH-1 and hepatocyte nuclear factor 4α, resulting in the repression of Cyp7a1 (16–18). How these two FXR-regulated pathways interact to control Cyp7a1 expression is not clear, albeit both are required for the FXR-mediated feedback repression of bile acid biosynthesis (19).
Although it is well established that bile acids are essential for the absorption of lipid-soluble vitamins, it is not known whether lipid-soluble vitamins affect bile acid biosynthesis. In this study, we demonstrate that vitamins A and D regulate bile acid synthesis by overlapping but distinct mechanisms. As expected, the action of vitamin D on bile acid homeostasis occurs through activation of VDR. Interestingly, the action of vitamin A is mediated through the RXR/FXR heterodimer. This latter finding supports a novel role for the RXR/FXR heterodimer as a dietary sensor for vitamin A.
EXPERIMENTAL PROCEDURES
Animals and Animal Husbandry
Male C57BL/6 mice were purchased from Charles River Laboratories and used for all experiments involving only wild-type animals. VDR+/+ and VDR−/− mice were obtained from heterozygous breeders on a pure 129T2 background. FXR+/+ and FXR−/− mice were obtained from heterozygous breeders on a pure 129S background. FGF15+/+ and FGF15−/− mice were obtained from homozygous breeders on a mixed C57BL/6;129S background. SHP+/+ and SHP−/− mice were obtained from homozygous breeders on a pure 129S background. All animals were housed in the same specific pathogen-free facility. Animals were maintained under a temperature-controlled environment with 12-h light/dark cycles with ad libitum access to water and irradiated rodent chow (TD.2916, Harlan-Teklad). The expression of metabolic genes analyzed in this study is affected by circadian and feeding cycles; therefore, the following steps were taken to ensure a synchronous feeding cycle. On the day prior to death, chow was removed 10 h before the dark cycle, replaced at the onset of the dark cycle, and removed 4 h after the onset of the dark cycle. The change in body weight between the beginning and end of the dark cycle was <5% for all animals. All experiments were done with 3–6-month-old (age-matched) male mice killed between 4 and 6 h after the beginning of the light cycle. Mice were killed by isoflurane inhalation and exanguinated via the descending vena cava prior to tissue collection.
Animal Treatments
Vitamin D (1α,25-dihydroxycholecalciferol; Sigma) in sterile saline was administered by intraperitoneal injection at the end of the dark cycle and 4 h before death. Vitamin A (retinyl palmitate, ∼1800 USP units/mg; Sigma) and vitamin E (d-α-tocopherol acetate, ∼1360 IU/g; Sigma) in 1% Tween 80, 1% methylcellulose, and 25 mm HEPES were administered by oral gavage at the beginning and end of the dark cycle (16 and 4 h before death). Cholestyramine was admixed in the diet (custom diet TD.07658, Harlan-Teklad). GW4064 (GlaxoSmithKline) and TTNPB (Sigma) were admixed in the diet (500 and 0.25 mg/kg diet, respectively) and provided ad libitum for 12 h before death. LG268 in 0.25% Tween 80 and 1% methylcellulose was administered by oral gavage 12 h before death at a dose of 30 mg/kg. In all cases, animals in the vehicle group received the appropriate vehicle solutions and diets in a manner identical to the treatment groups.
Mouse Ileum Explant Culture
Following death, 5 cm of the terminal ileum was collected and flushed with phosphate-buffered saline to remove the contents. Segments (1 × 3 mm) of the ileum were cultured at 37 °C and 95% oxygen for 6 h in Dulbecco's modified Eagle's medium (containing 4 g/liter glucose and l-glutamine; Invitrogen) supplemented with 10% charcoal-stripped heat-inactivated fetal bovine serum, 25 mm HEPES, 100 units/ml penicillin, 100 μg/ml streptomycin, and either vitamin D or 0.1% ethanol (vehicle). Each condition contained four 1 × 3-mm segments of ileum, from which total RNA was extracted at the end of the 6-h culture period.
Bile Acid Extraction and Analysis by Liquid Chromatography/Mass Spectrometry
Bile acid pool size measurements were performed as described previously with minor modifications (20). The following is a brief description of the protocol, including modified parameters. Liver, gallbladder, intestines, and attached mesentery were removed en bloc and homogenized in ethanol. Homogenates were heating to boiling, cooled, and then filtered. This step was repeated twice. Filtrates were combined and adjusted to a constant volume. Bile acids were resolved by reverse-phase liquid chromatography (C8 precolumn, C18 analytical column) and quantified by mass spectrometry with electrospray ionization in negative ion mode. The following unconjugated and taurine-conjugated bile acids were used as calibration standards: cholic acid (5β-cholanic acid-3α,7α,12α-triol), α-muricholic acid (5β-cholanic acid-3α,6β,7α-triol), β-muricholic acid (5β-cholanic acid-3α,6β,7β-triol), ω-muricholic acid (5β-cholanic acid-3α,6α,7β-triol), hyocholic acid (5β-cholanic acid-3α,6α,7α-triol), chenodeoxycholic acid (5β-cholanic acid-3α,7α-diol), deoxycholic acid (5β-cholanic acid-3α,12α-diol), hyodeoxycholic acid (5β-cholanic acid-3α,6α-diol), ursodeoxycholic acid (5β-cholanic acid-3α,7β-diol), murocholic acid (5β-cholanic acid-3α,6β-diol), lithocholic acid (5β-cholanic acid-3α-ol), taurocholic acid (5β-cholanic acid-3α,7α,12α-triol-N-(2-sulfoethyl)amide), tauro-α-muricholic acid (5β-cholanic acid-3α,6β,7α-triol-N-(2-sulfoethyl)amide), tauro-β-muricholic acid (5β-cholanic acid-3α,6β,7β-triol-N-(2-sulfoethyl)amide), tauro-ω-muricholic acid (5β-cholanic acid-3α,6α,7β-triol-N-(2-sulfoethyl)amide), taurohyocholic acid (5β-cholanic acid-3α,6α,7α-triol-N-(2-sulfoethyl)amide), taurochenodeoxycholic acid (5β-cholanic acid-3α,7α-diol-N-(2-sulfoethyl)amide), taurodeoxycholic acid (5β-cholanic acid-3α,12α-diol-N-(2-sulfoethyl)amide), tauroursodeoxycholic acid (5β-cholanic acid-3α,7β-diol-N-(2-sulfoethyl)amide), taurohyodeoxycholic acid (5β-cholanic acid-3α,6α-diol-N-(2-sulfoethyl)amide), taurolithocholic acid (5β-cholanic acid-3α-ol-N-(2-sulfoethyl)amide), and taurolithocholic acid 3-sulfate (5β-cholanic acid-3α-ol-N-(2-sulfoethyl)amide 3-sulfate). Deuterium-labeled cholic acid (5β-cholanic acid-3α,7α,12α-triol-2,2,4,4-d4) and chenodeoxycholic acid (5β-cholanic acid-3α,7α-diol-2,2,4,4-d4) were used as recovery controls. Norcholic acid (23-nor-5β-cholanic acid-3α,7α,12α-triol) was used as the sample loading control.
RNA Extraction and Quantitative Reverse Transcription (RT)-PCR
Following death, the left lateral lobe of the liver and distal ileum (5 cm proximal to the ileocecal junction, flushed with phosphate-buffered saline) was collected and frozen immediately in liquid nitrogen. Total RNA was extracted using RNA STAT-60TM (IsoTex Diagnostics). RNA (4 μg) from each sample was DNase-treated and reverse-transcribed using random hexamers. The resulting cDNA was analyzed by quantitative RT-PCR using a protocol described previously (21). Briefly, quantitative PCRs containing 25 ng of cDNA, 150 nmol of each primer, and SYBR® GreenERTM PCR Master Mix (Invitrogen) were carried out in triplicate in 384-well format using an ABI PRISM® 7900HT instrument (Applied Biosystems). Relative mRNA levels were calculated using the comparative CT method normalized to U36b4. The primer sequences used for gene expression analyses are listed in supplemental Table 1. They were designed using Primer Express® software (Applied Biosystems) and validated as described previously (21).
Chromatin Immunoprecipitation (ChIP) Analysis
Following death, the ileum was collected, flushed with phosphate-buffered saline, and frozen immediately in liquid nitrogen. Samples from four to five mice were pulverized and pooled. 300 mg from each pool was fixed in phosphate-buffered saline with 1% formaldehyde for 10 min and quenched with glycine for 5 min. Samples were then Dounce-homogenized in hypotonic buffer (10 mm HEPES (pH 7.9), 1.5 mm MgCl2, 10 mm KCl, 0.2% Nonidet P-40, 1 mm EDTA, and 5% sucrose) and layered over cushion buffer (10 mm Tris-HCl (pH 7.5), 15 mm NaCl, 60 mm KCl, 1 mm EDTA, and 10% sucrose), followed by centrifugation at 200 × g to collect the crude nuclear pellet. Subsequent ChIP steps were done using the ChIP EZ kit (Upstate) materials and methods. 3 μg of protein from sonicated chromatin was used for pulldown assay with rabbit IgG (Santa Cruz Biotechnology), anti-VDR (Santa Cruz Biotechnology C-20x), or anti-RXR (Santa Cruz Biotechnology ΔN-197x). Primers scanning −1500 to +3000 of the Fgf15 locus were designed and used for analyzing VDR and RXR binding by quantitative PCR as described (21). Primer sequences are shown in supplemental Table 2.
Expression Plasmids
pCMX-hFXR, pCMX-hVDR, and pCMX-hRXRα expressing full-length nuclear receptors under the control of the constitutive cytomegalovirus promoter have been described previously (18, 22, 23).
Fgf15 Promoter and Intron 2 Cloning
Total DNA was isolated from mouse colon using the DNeasy Blood & Tissue kit (Qiagen) and protocol. A 2338-nucleotide region of the Fgf15 promoter including six nucleotides downstream of the transcriptional start site was amplified using the following primers containing restriction endonuclease sites (underlined): CCTAAACCAAGCTTCTGGCCATCTG (forward) and AGAGTTACTGCGTCGACAGTGG (reverse). This product was inserted upstream of the thymidine kinase promoter sequence in the pTk-LUC plasmid (24) to generate p2300Fgf15+TATA_TkLUC. This plasmid was used to amplify shorter regions of the Fgf15 promoter, which were likewise inserted into pTk-LUC. Promoter regions were amplified using distinct forward primers (CTGCGAAAGCTTGCTAAAGGAGAG for p400Fgf15+TATA_TkLUC, CCACCAAGCTTCTGTGCATTGAAC for p230Fgf15+TATA_TkLUC, and CCTGTCGACGCATCAAGTCTCC for p100Fgf15+TATA_TkLUC) and a common reverse primer (GGATCCTCTAGAGTCGACAGTGG). p230Fgf15+TATA_TkLUC was amplified using the following overlapping primers containing two altered nucleotides (underlined): GGCCTGGGCGGGACCACTGGGTTGGGG (forward) and CCCAACCCAGTGGTCCCGCCCAGGCC (reverse). The Fgf15 promoter region containing the two-nucleotide mutation was reinserted into pTk-LUC to generate p230(mut-145)Fgf15+TATA_TkLUC. A 2460-nucleotide region encompassing the second intron of Fgf15 was amplified using the following primers containing restriction endonuclease sites (underlined): GAGCGCGGTCGACAAGATATACG (forward) and AAGGTACAGTCGACCTCCGAGTAG (reverse). The product was inserted in reverse orientation upstream of the thymidine kinase promoter sequence in pTk-LUC to generate pFgf15intron2rev_TkLUC. The modified regions of all plasmids were verified by DNA sequencing at the McDermott Sequencing Core of the University of Texas Southwestern Medical Center.
Cotransfection and Luciferase Assay
HEK293 cells were grown at 37 °C and 5% CO2 in 96-well plates in Dulbecco's modified Eagle's medium (containing 4 g/liter glucose and l-glutamine) supplemented with 10% charcoal-stripped heat-inactivated fetal bovine serum and transfected by calcium phosphate co-precipitation as described previously (23). Following a 16-h treatment with GW4064 and vitamin D, luciferase and β-galactosidase activities were measured as described previously (18). Luciferase activity was normalized for transfection efficiency using β-galactosidase activity and expressed as relative luciferase units. For each experiment, all conditions were tested in triplicate, and all experiments were repeated three times. Data shown are the mean ± S.D. of triplicate assays from one representative experiment.
Statistical Analysis
Data are presented as the mean ± S.E. and were analyzed by two-tailed unpaired Student's t test. p values <0.05 were considered significant.
RESULTS
Bile Acid Metabolism Is Dysregulated in VDR−/− Mice
Activation of VDR in the intestine by 1α,25-dihydroxyvitamin D3 or lithocholic acid results in the induction of bile acid-detoxifying genes such as CYP3A4 (22, 25, 26). To determine the extent of VDR contribution to bile acid metabolism in vivo, we measured the bile acid pool size and composition in VDR−/− mice. Surprisingly, we found that VDR−/− mice had an ∼30% larger bile acid pool size at 3 months of age and more than twice the amount of bile acids at 6 months of age as their wild-type littermates (Fig. 1A). The loss of VDR also increased the hydrophobicity of the bile acid pool due to a greater increase in taurocholic acid relative to tauromuricholic acids (Fig. 1A).
FIGURE 1.

Bile acid metabolism is dysregulated in VDR−/− mice. A, total bile acids were extracted from the gallbladder, liver, intestine, and portal blood and quantitated by liquid chromatography/mass spectrometry. The results shown are bile acid pool size and composition in 3-month-old (n = 7) and 6-month-old (n = 4) male mice of the indicated genotype. Taurocholate (tCA) and tauromuricholates (tMCA; includes α-, β-, and ω-muricholate) compose the majority of bile acids in the mouse. See “Experimental Procedures” for a complete list of bile acids grouped as “others.” Data represent the mean ± S.E. B and C, total RNA was extracted from the liver (B) and ileum (C) and analyzed by quantitative RT-PCR. The level of mRNA expression was normalized to U36b4 and graphed relative to the wild-type control. Data represent the mean ± S.E. of seven animals/group. For all panels, the p value was determined by Student's t test.
To determine the mechanism underlying the perturbation of bile acid homeostasis caused by the loss of VDR, we analyzed the expression of genes in the liver known to play a role in bile acid metabolism. In humans and rodents, bile acids are synthesized from cholesterol in the liver via two pathways (27). CYP7A1 catalyzes the first and rate-limiting step in the classic (neutral) pathway of bile acid synthesis, whereas CYP27A1 catalyzes the first step in the alternative (acidic) pathway. Under normal physiological conditions, the majority of bile acids are produced via the classic pathway. Consistent with an increased rate of bile acid synthesis, we found that VDR−/− mice had 3-fold higher expression of Cyp7a1 (Fig. 1B). In addition, the enzyme that catalyzes the final step in the synthesis of cholic acid, Cyp8b1, was increased (Fig. 1B). These changes were accompanied by a 2-fold decrease in Shp mRNA and more modest changes in FXR, hepatocyte nuclear factor 4α, and Lrh-1 expression (Fig. 1B and supplemental Fig. 1A). Expression analysis of the major bile acid transporters revealed decreased expression of the canalicular bile acid export pump Abcb11, a small increase in the phospholipid exporter Abcb4, and no change in the expression of the sinusoidal sodium taurocholate import pump Slc10a1 (supplemental Fig. 1B). In contrast to enzymes of the classic bile acid synthesis pathway, no change was seen in expression of genes encoding enzymes of the alternative bile acid synthesis pathway (supplemental Fig. 1C). These alterations in gene expression in the livers of VDR−/− mice were somewhat surprising given that VDR is not expressed in hepatocytes and suggested that the changes either were secondary to alterations in the bile acid pool size and composition or were caused by perturbation of vitamin D signaling outside the hepatocyte.
We next examined gene expression in the terminal small intestine (ileum), which is the major site of bile acid reabsorption and thus plays an integral role in maintaining bile acid homeostasis. Here, bile acid activation of FXR induces expression of bile acid transporters and Fgf15 (15, 28). As described previously, FGF15 is essential for FXR-mediated feedback repression of Cyp7a1 and bile acid synthesis. Surprisingly, despite normal levels of FXR and bile acid transporters, the expression of Fgf15 was decreased markedly in VDR−/− mice (Fig. 1C and supplemental Fig. 1D).
Vitamin D Induces Fgf15 to Suppress Cyp7a1
Because VDR is expressed in enterocytes and Fgf15 expression was lower in VDR−/− mice, we hypothesized that VDR might regulate Fgf15 at the transcriptional level. Indeed, treatment of wild-type but not VDR−/− mice with 1α,25-dihydroxyvitamin D3 (hereafter referred to as vitamin D) for 4 h increased Fgf15 expression in the intestine and decreased Cyp7a1 in the liver (Fig. 2A). Intestinal bile acid transporters were also decreased, whereas the gene encoding the ileal bile acid-binding protein (an FXR target gene also known as Fabp6) was not changed (supplemental Fig. 2). Importantly, the repression of Cyp7a1 by vitamin D was absent in FGF15-null (FGF15−/−) mice, demonstrating that FGF15 is required for vitamin D-dependent suppression of Cyp7a1 (Fig. 2B).
FIGURE 2.

VDR induces Fgf15 to suppress Cyp7a1. A–C, mice of the indicated genotype were treated for 4 h by intraperitoneal injection of 75 μg/kg (A) or 50 μg/kg (B and C) 1α,25-dihydroxyvitamin D3. mRNA expression in the ileum (Fgf15) and liver (Cyp7a1 and Shp) was determined by quantitative RT-PCR, normalized to U36b4, and graphed relative to the wild-type vehicle-treated control. Data represent the mean ± S.E. of five to six animals/group. *, p < 0.05 compared with the vehicle of the same genotype; #, p < 0.05 compared with the wild type of the same treatment group. D, ileum explants were treated for 6 h with the indicated concentrations of 1α,25-dihydroxyvitamin D3. Total RNA was extracted and analyzed by quantitative RT-PCR. The level of mRNA expression was normalized to U36b4 and graphed relative to the highest treatment dose. Data represent the mean ± S.D. of two independent experiments. E, pooled ilea from three to four VDR+/+ and VDR−/− mice treated for 2 h with 50 μg/kg 1α,25-dihydroxyvitamin D3 were analyzed by ChIP using specific antibodies for VDR and RXR and an isotype-matched IgG control antibody. Bound DNA was quantitated by quantitative PCR and normalized to the input. Data represent the mean ± S.E. of four independent experiments. Numbers indicate nucleotide position relative to the transcriptional start site. The location and sequence of the IR1 FXR/RXR response element and the putative DR3 VDR/RXR response element are shown below the graph. Half-sites are in boldface, and nucleotides mutated in the promoter analysis shown in F are underlined. F, the promoter (−2332 to +6; white boxes) and intron 2 (+673 to +3133; gray boxes) of FGF15 were cloned upstream of the thymidine kinase minimal promoter (Tk) and luciferase gene as shown. HEK293 cells were cotransfected with the indicated reporter constructs, β-galactosidase, human RXR, and either human FXR or human VDR. Following treatment with 1 μm GW4064 (FXR agonist), 100 nm 1α,25-dihydroxyvitamin D3, or vehicle, luciferase activity was quantitated and normalized to β-galactosidase activity. Data are graphed relative to the first data point and represent the mean ± S.D. of three replicates. X indicates the mutated DR3 site. RLU, relative luciferase units.
To determine whether physiological levels of vitamin D contribute to the regulation of Fgf15, we compared the induction of Fgf15 with that of the gene encoding Calbindin-D9k (a known VDR target gene) in ileum explants. Notably, both genes were induced by vitamin D in a comparable dose-dependent manner (Fig. 2D). Taken together with the finding that VDR is required for normal expression of Fgf15, these data provide evidence that the transcriptional regulation of Fgf15 by vitamin D is physiologically relevant.
Previous work has shown that FXR directly regulates Fgf15 expression by binding to an FXR/RXR response element in the second intron of the gene (15). Given the rapid effect of vitamin D treatment on Fgf15 expression, it seemed likely that the regulation of Fgf15 by VDR was also direct. ChIP analysis was performed to identify VDR/RXR-binding sites in the regulatory regions of Fgf15. As expected, a strong RXR-binding site was detected in the second intron, and an additional binding site was found in the proximal promoter near the transcriptional start site (Fig. 2E). VDR binding was also detected at both sites (Fig. 2E). Interestingly, analysis of the Fgf15 regulatory regions by reporter gene assay showed that VDR transactivation occurred exclusively through the proximal promoter site, whereas FXR transactivation occurred exclusively at the second intron (Fig. 2F). Promoter truncations localized the vitamin D-responsive cis-acting element to a region between 100 and 200 nucleotides upstream of the transcriptional start site (Fig. 2F). Analysis of this region revealed a DR3 site with similarity to known VDR response elements, and transactivation of the Fgf15 promoter by VDR was eliminated by mutating two nucleotides within this site (Fig. 2F). These results demonstrate that VDR binds directly to the Fgf15 promoter and regulates Fgf15 expression through a direct transcriptional mechanism.
Given that both VDR and FXR induce Fgf15, we sought to determine whether VDR could substitute for FXR in mediating repression of bile acid synthesis. Although the -fold induction of Fgf15 by vitamin D was similar in wild-type and FXR−/− mice, the absolute levels of Fgf15 in vitamin D-treated FXR−/− mice remained below those in vehicle-treated wild-type mice (Fig. 2C). Notably, the repression of Cyp7a1 by vitamin D was absent in FXR−/− mice (Fig. 2C). These data indicate that the transcriptional regulation of Fgf15 by VDR is not dependent on FXR; however, FXR is required for the induction of Fgf15 to levels that suppress Cyp7a1.
Vitamin A Induces Fgf15 and Shp to Suppress Cyp7a1
Given the effects of vitamin D on Fgf15 and Cyp7a1 regulation, we sought to determine whether other lipid-soluble vitamins also cause repression of bile acid synthesis. Although d-α-tocopherol (i.e. vitamin E) had no effect on Cyp7a1 or any other genes analyzed, retinyl palmitate (i.e. vitamin A) dramatically increased Fgf15 expression in the intestine and suppressed Cyp7a1 in the liver (Fig. 3A). Furthermore, vitamin A increased expression of Shp in the liver (Fig. 3A).
FIGURE 3.

Vitamin A induces Fgf15 and Shp to suppress Cyp7a1. A, wild-type mice were treated for 16 h by oral gavage with 500 mg/kg d-α-tocopherol (vitamin E) or 100 mg/kg retinyl palmitate (vitamin A). mRNA expression in the ileum (Fgf15) and liver (Cyp7a1 and Shp) was determined by quantitative RT-PCR, normalized to U36b4, and graphed relative to the vehicle-treated control. B, wild-type mice were treated for 1 day with diets containing the indicated synthetic ligands. The target nuclear receptor is shown in parentheses. Data analysis was as described for A. C and D, mice of the indicated genotype were treated for 16 h by oral gavage with 100 mg/kg retinyl palmitate (vitamin A). mRNA expression in the ileum (Fgf15) and liver (Cyp7a1 and Shp) was determined by quantitative RT-PCR, normalized to U36b4, and plotted relative to the wild-type vehicle-treated control. For all panels, data represent the mean ± S.E. of five animals/group. *, p < 0.05 compared with the vehicle of the same genotype; #, p < 0.05 compared with the wild type of the same treatment group.
In vivo, vitamin A may be metabolized to all-trans-retinoic acid, which activates RAR, and 9-cis-retinoic acid, which activates both RAR and RXR. To determine which nuclear receptor was mediating the transcriptional effects of vitamin A on Fgf15, Cyp7a1, and Shp, we treated wild-type mice with the synthetic ligands TTNPB (selective for RAR) and LG268 (selective for RXR). Both RAR and RXR induced Shp and suppressed Cyp7a1; however, Fgf15 was induced only by RXR (Fig. 3B). Because the VDR/RXR heterodimer is not activated by RXR ligands (29), we hypothesized that the induction of Fgf15 by RXR ligand occurred through activation of the RXR/FXR heterodimer. To test this idea, we treated FXR−/− mice with vitamin A and found that, in the absence of FXR, vitamin A had no effect on Fgf15 expression (Fig. 3C). Taken together, these data suggest that Fgf15 is induced by vitamin A through the RXR partner of the RXR/FXR heterodimer complex.
Interestingly, despite the absence of Fgf15 induction, vitamin A still efficiently repressed Cyp7a1 in FXR−/− mice, indicating that additional vitamin A-dependent mechanisms exist to suppress Cyp7a1 (Fig. 3C). Further analysis of hepatic gene expression demonstrated that, despite lower basal levels of Shp in FXR−/− mice, FXR was not required for induction of Shp by vitamin A (Fig. 3C), suggesting that Shp induction contributed to Cyp7a1 repression by vitamin A. In support of this idea, we found that repression of Cyp7a1 by vitamin A was greatly reduced in the absence of SHP (Fig. 3D).
Vitamin A Rescues Fgf15 and Shp Expression and Suppresses Cyp7a1 under Conditions of Impaired Bile Acid Feedback Repression
Our finding that vitamin A could repress Cyp7a1 expression by inducing bile acid feedback regulatory genes in both the liver and intestine suggested that vitamin A analogs might be useful therapeutically in pathological conditions in which feedback repression of bile acid synthesis is interrupted. One such condition is bile acid malabsorption, which is characterized by decreased intestinal bile acid reabsorption, resulting in low FGF19 levels (human ortholog of FGF15) and excessive bile acid synthesis by the liver (30). The effects of bile acid malabsorption on expression of bile acid feedback regulatory genes can be mimicked by administering the bile acid-binding resin, cholestyramine. As expected, cholestyramine treatment of wild-type mice dramatically decreased Fgf15 and Shp expression and increased Cyp7a1 expression (Fig. 4). Amazingly, vitamin A completely rescued Fgf15 and Shp expression and reversed the derepression of Cyp7a1 caused by interrupted bile acid reabsorption in cholestyramine-treated animals (Fig. 4). Consistent with our previous results, in the absence of bile acids, vitamin D modestly induced Fgf15 but did not change Shp or Cyp7a1 expression (Fig. 4). These data highlight the potential of vitamin A analogs to correct disrupted feedback repression of bile acid biosynthesis.
FIGURE 4.
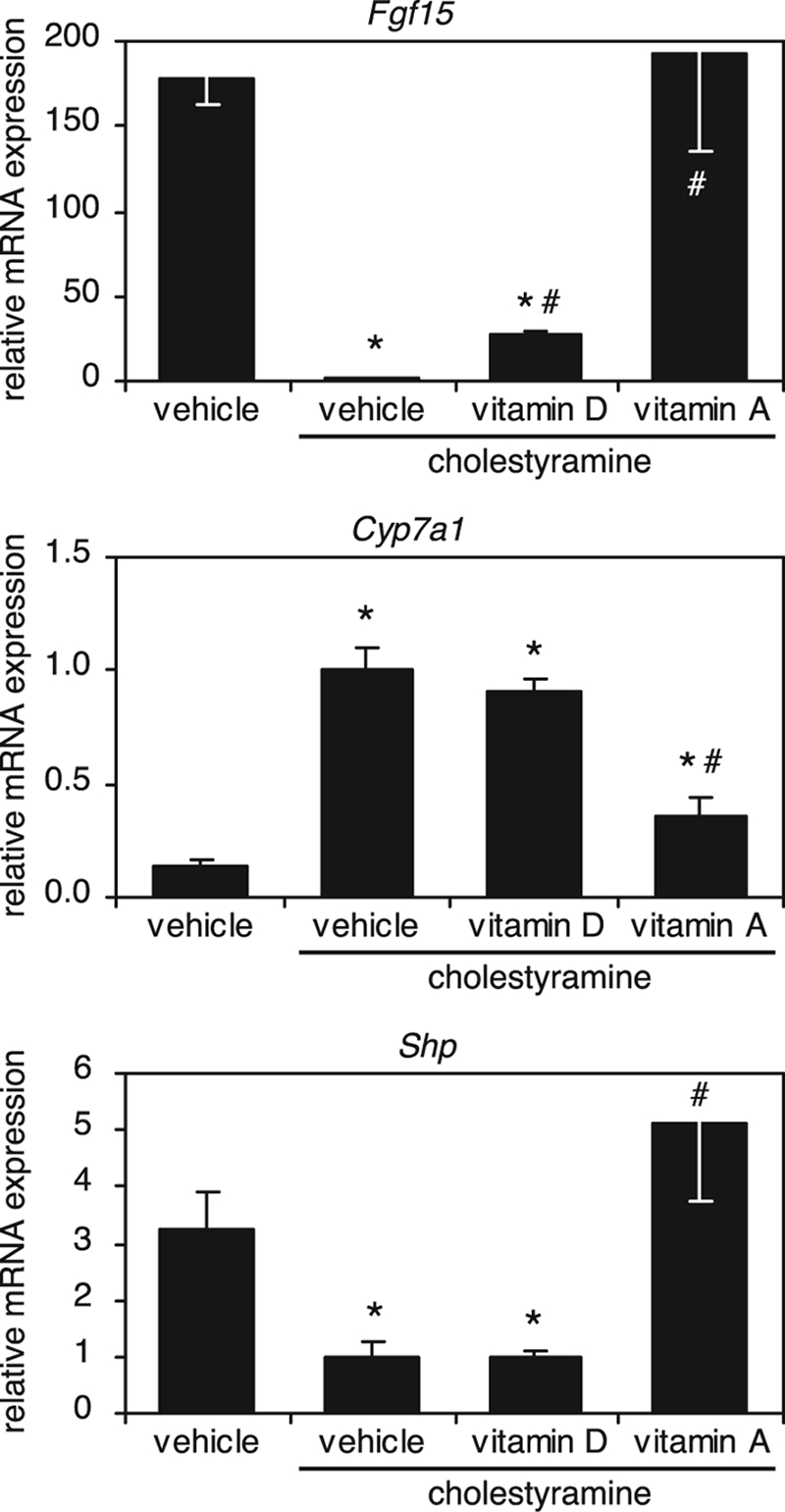
Vitamin A rescues Fgf15 and Shp expression and suppresses Cyp7a1 under conditions of impaired bile acid feedback repression. Wild-type mice were treated for 2 days with a 2% (w/w) cholestyramine diet and either 50 μg/kg 1α,25-dihydroxyvitamin D3 (vitamin D) or 100 mg/kg retinyl palmitate (vitamin A). Vitamin D and A treatments were administered as described in the legends to Figs. 2 and 3, respectively. The control group received standard chow and the appropriate vehicle treatments. mRNA expression in the ileum (Fgf15) and liver (Cyp7a1 and Shp) was determined by quantitative RT-PCR, normalized to U36b4, and plotted relative to vehicle-treated control. Data represent the mean ± S.E. of three animals/group. *, p < 0.05 compared with the control; #, p < 0.05 for vitamin treatment compared with cholestyramine alone.
DISCUSSION
Because of their unique physicochemical properties, bile acids are essential structural components of lipid micelles (11). In this capacity, bile acids promote the intestinal absorption of lipids and lipid-soluble vitamins. Here, we have revealed an unexpected link between lipid-soluble vitamins and bile acid biosynthesis. Surprisingly, vitamins A and D exert negative feedback on bile acid synthesis in vivo by decreasing Cyp7a1 expression. FGF15 and SHP play a central role in the feedback regulation of bile acid synthesis by bile acids and FXR (31). Our study shows that FGF15 is integral to the mechanism of Cyp7a1 regulation by vitamin D and that both FGF15 and SHP are important for the regulation of bile acid synthesis by vitamin A.
We found that VDR transcriptionally regulated Fgf15 in the intestine and that this pathway was essential for the repression of bile acid synthesis by vitamin D. Surprisingly, VDR was required to maintain normal Fgf15 expression and bile acids levels in vivo. Together, these results demonstrate that vitamin D and its receptor contribute to the feedback regulation of bile acid synthesis by controlling expression of the endocrine hormone FGF15. We note that VDR expression has been reported in non-parenchymal cells in the liver (32), leaving open the possibility that paracrine signals might also contribute to VDR-dependent regulation of bile acid synthesis.
Interestingly, Cyp7a1 repression by vitamin D required an intact FXR signaling pathway, indicating that VDR activation alone is not sufficient to suppress bile acid synthesis. There are at least two possible explanations for this finding. First, activation of both VDR and FXR may be required to induce Fgf15 to the level required for Cyp7a1 suppression. Second, as has recently been shown for FXR (19), feedback repression of bile acid synthesis may require corresponding signals in the intestine and liver, the latter of which does not occur upon activation of VDR alone.
Vitamin A repressed Cyp7a1 through both FXR-dependent and FXR-independent mechanisms. Ligands for RXR, but not RAR, induced Fgf15, and FXR was required for the induction of Fgf15 by vitamin A. These results indicate that induction of Fgf15 expression by vitamin A occurs through RXR as the obligate heterodimeric partner of the RXR/FXR heterodimer complex. This finding provides evidence that RXR functions as a vitamin A receptor in vivo and demonstrates that the RXR/FXR heterodimer can serve as a sensor for dietary vitamin A. In contrast to its effects on FGF15 expression, vitamin A-dependent induction of Shp expression did not require FXR. Interestingly, we found that the RAR ligand TTNPB induced Shp and suppressed Cyp7a1, suggesting that Shp induction by RAR may be an FXR-independent mechanism whereby vitamin A suppresses Cyp7a1. Taken together, these results point to two distinct nuclear receptor-mediated mechanisms by which vitamin A regulates bile acid synthesis.
Under normal physiological conditions, bile acids are efficiently reabsorbed in the ileum (11). Bile acid malabsorption is a pathological condition often seen in patients with Crohn disease or ileal resection and is characterized by reduced ileal bile acid reabsorption and delivery of large quantities of bile acids to the colon (30). Increased luminal concentrations of bile acids in the colon induce fluid secretion, resulting in cholerheic enteropathy and the characteristic symptom of watery diarrhea. Bile acid sequestrants are currently the primary therapy and provide symptomatic relief but do not correct bile acid overproduction and hypersecretion by the liver. The exciting finding that vitamin A induces Fgf15 and suppresses bile acid synthesis under conditions of interrupted bile acid reabsorption suggests that vitamin A analogs may provide therapeutic benefit to patients with bile acid malabsorption and increased hepatic bile acid synthesis. Interestingly, a subset of patients with bile acid malabsorption have increased hepatic bile acid synthesis despite normal ileal bile acid transport. This condition, termed idiopathic bile acid malabsorption, has recently been shown to be associated with decreased plasma levels of FGF19 (33). It is not known why FGF19 levels are abnormally low in these patients. Our data suggest that vitamins A and D may be useful tools to examine the underlying cause of low FGF19 levels in these patients.
Elevated levels of bile acid in the colon may promote colon cancer, whereas vitamin D is associated with reduced risk of colon cancer (34–37). We showed previously that VDR is activated by lithocholic acid and induces enzymes that detoxify bile acids in the colon (22). The present study suggests that vitamin D-dependent regulation of bile acid synthesis may be an additional mechanism by which vitamin D protects against the tumor-promoting effects of toxic bile acids.
With regard to the mechanism underlying the hormonal effects of vitamin D, an interesting parallel emerges between the regulation of bile acid synthesis by FGF15 and the role of FGF23 in renal phosphate metabolism. Previous studies have shown that vitamin D induces Fgf23 in bone and that FGF23 signals in a bone-kidney axis to control phosphate absorption and vitamin D metabolism in the kidney (38). In this study, we have shown that vitamin D induces Fgf15 in the intestine, which signals in an intestine-liver axis to regulate bile acid synthesis in the liver. Thus, a paradigm emerges in which endocrine fibroblast growth factors function as downstream messengers to mediate the homeostatic effects of vitamin D and coordinate vitamin D signaling between organ systems.
In summary, our findings highlight the importance of nuclear receptors in the regulation of bile acid metabolism and provide mechanistic insight into the elegant signaling pathways involving FGF15 and SHP that govern feedback repression of bile acid biosynthesis. We speculate that the mechanisms allowing vitamins A and D to control feedback repression of bile acid synthesis may have evolved to protect the organism from exposure to potentially toxic levels of lipid-soluble vitamins in the diet.
Supplementary Material
Acknowledgments
We thank Dr. Carolyn Cummins for assistance in developing liquid chromatography/mass spectrometry methods, Eva Borowicz for primer validation, and Hannah Perkins and Kevin Vale for assistance with animal experiments and breeding. We also thank Drs. Mark Valasek and Joyce Repa for advice on mouse intestinal explant culture.
This work was supported, in whole or in part, by National Institutes of Health Grants U19DK62434 (to D. J. M.), GM07062 (to D. R. S. and A. L. B.), and CA114109 (to S. A. K.). This work was also supported by the Howard Hughes Medical Institute and Robert A. Welch Foundation Grants I-1275 (to D. J. M.) and I-1558 (to S. A. K.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2 and Tables 1 and 2.
- RAR
- retinoic acid receptor
- RXR
- retinoid X receptor
- VDR
- vitamin D receptor
- FXR
- farnesoid X receptor
- RT
- reverse transcription
- ChIP
- chromatin immunoprecipitation
- TTNPB
- 4-((E)-2(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl)benzoic acid.
REFERENCES
- 1.Mangelsdorf D. J., Thummel C., Beato M., Herrlich P., Schütz G., Umesono K., Blumberg B., Kastner P., Mark M., Chambon P., Evans R. M. (1995) Cell 83, 835–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haussler M. R., Haussler C. A., Jurutka P. W., Thompson P. D., Hsieh J. C., Remus L. S., Selznick S. H., Whitfield G. K. (1997) J. Endocrinol. 154, S57–S73 [PubMed] [Google Scholar]
- 3.Mark M., Ghyselinck N. B., Chambon P. (2009) Nucl. Recept. Signal. 7, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ross A. C. (1993) J. Nutr. 123, 346–350 [DOI] [PubMed] [Google Scholar]
- 5.Zile M. H. (1998) J. Nutr. 128, 455S–458S [DOI] [PubMed] [Google Scholar]
- 6.Prosser D. E., Jones G. (2004) Trends Biochem. Sci. 29, 664–673 [DOI] [PubMed] [Google Scholar]
- 7.DeLuca H. F. (2004) Am. J. Clin. Nutr. 80, 1689S–1696S [DOI] [PubMed] [Google Scholar]
- 8.Liu P. T., Stenger S., Li H., Wenzel L., Tan B. H., Krutzik S. R., Ochoa M. T., Schauber J., Wu K., Meinken C., Kamen D. L., Wagner M., Bals R., Steinmeyer A., Zügel U., Gallo R. L., Eisenberg D., Hewison M., Hollis B. W., Adams J. S., Bloom B. R., Modlin R. L. (2006) Science 311, 1770–1773 [DOI] [PubMed] [Google Scholar]
- 9.Mangelsdorf D. J., Motola D. L. (2005) in Vitamin D (Feldman D. ed) p. 863, Elsevier Academic Press, London [Google Scholar]
- 10.Moro J. R., Iwata M., von Andriano U. H. (2008) Nat. Rev. Immunol. 8, 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hofmann A. F. (1999) News Physiol. Sci. 14, 24–29 [DOI] [PubMed] [Google Scholar]
- 12.Gilardi F., Mitro N., Godio C., Scotti E., Caruso D., Crestani M., De Fabiani E. (2007) Pharmacol. Ther. 116, 449–472 [DOI] [PubMed] [Google Scholar]
- 13.Crestani M., Sadeghpour A., Stroup D., Galli G., Chiang J. Y. (1998) J. Lipid Res. 39, 2192–2200 [PubMed] [Google Scholar]
- 14.Nitta M., Ku S., Brown C., Okamoto A. Y., Shan B. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 6660–6665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inagaki T., Choi M., Moschetta A., Peng L., Cummins C. L., McDonald J. G., Luo G., Jones S. A., Goodwin B., Richardson J. A., Gerard R. D., Repa J. J., Mangelsdorf D. J., Kliewer S. A. (2005) Cell Metab. 2, 217–225 [DOI] [PubMed] [Google Scholar]
- 16.Goodwin B., Jones S. A., Price R. R., Watson M. A., McKee D. D., Moore L. B., Galardi C., Wilson J. G., Lewis M. C., Roth M. E., Maloney P. R., Willson T. M., Kliewer S. A. (2000) Mol. Cell 6, 517–526 [DOI] [PubMed] [Google Scholar]
- 17.Lee Y. K., Dell H., Dowhan D. H., Hadzopoulou-Cladaras M., Moore D. D. (2000) Mol. Cell. Biol. 20, 187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu T. T., Makishima M., Repa J. J., Schoonjans K., Kerr T. A., Auwerx J., Mangelsdorf D. J. (2000) Mol. Cell 6, 507–515 [DOI] [PubMed] [Google Scholar]
- 19.Kim I., Ahn S. H., Inagaki T., Choi M., Ito S., Guo G. L., Kliewer S. A., Gonzalez F. J. (2007) J. Lipid Res. 48, 2664–2672 [DOI] [PubMed] [Google Scholar]
- 20.Lee Y. K., Schmidt D. R., Cummins C. L., Choi M., Peng L., Zhang Y., Goodwin B., Hammer R. E., Mangelsdorf D. J., Kliewer S. A. (2008) Mol. Endocrinol. 22, 1345–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bookout A. L., Mangelsdorf D. J. (2003) Nucl. Recept. Signal. 1, e012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makishima M., Lu T. T., Xie W., Whitfield G. K., Domoto H., Evans R. M., Haussler M. R., Mangelsdorf D. J. (2002) Science 296, 1313–1316 [DOI] [PubMed] [Google Scholar]
- 23.Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., Shan B. (1999) Science 284, 1362–1365 [DOI] [PubMed] [Google Scholar]
- 24.Willy P. J., Umesono K., Ong E. S., Evans R. M., Heyman R. A., Mangelsdorf D. J. (1995) Genes Dev. 9, 1033–1045 [DOI] [PubMed] [Google Scholar]
- 25.Jurutka P. W., Thompson P. D., Whitfield G. K., Eichhorst K. R., Hall N., Dominguez C. E., Hsieh J. C., Haussler C. A., Haussler M. R. (2005) J. Cell. Biochem. 94, 917–943 [DOI] [PubMed] [Google Scholar]
- 26.Nehring J. A., Zierold C., DeLuca H. F. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 10006–10009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Russell D. W. (2003) Annu. Rev. Biochem. 72, 137–174 [DOI] [PubMed] [Google Scholar]
- 28.Dawson P. A., Lan T., Rao A. (2009) J. Lipid Res. 50, 2340–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shulman A. I., Larson C., Mangelsdorf D. J., Ranganathan R. (2004) Cell 116, 417–429 [DOI] [PubMed] [Google Scholar]
- 30.Hofmann A. F., Mangelsdorf D. J., Kliewer S. A. (2009) Clin. Gastroenterol. Hepatol. 7, 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang J. Y. (2009) J. Lipid Res. 50, 1955–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gascon-Barré M., Demers C., Mirshahi A., Néron S., Zalzal S., Nanci A. (2003) Hepatology 37, 1034–1042 [DOI] [PubMed] [Google Scholar]
- 33.Walters J. R., Tasleem A. M., Omer O. S., Brydon W. G., Dew T., le Roux C. W. (2009) Clin. Gastroenterol. Hepatol. 7, 1189–1194 [DOI] [PubMed] [Google Scholar]
- 34.Garland C. F., Gorham E. D., Mohr S. B., Garland F. C. (2009) Ann. Epidemiol. 19, 468–483 [DOI] [PubMed] [Google Scholar]
- 35.Lamprecht S. A., Lipkin M. (2003) Nat. Rev. Cancer 3, 601–614 [DOI] [PubMed] [Google Scholar]
- 36.Nagengast F. M., Grubben M. J., van Munster I. P. (1995) Eur. J. Cancer 31A, 1067–1070 [DOI] [PubMed] [Google Scholar]
- 37.Yin L., Grandi N., Raum E., Haug U., Arndt V., Brenner H. (2009) Aliment. Pharmacol. Ther. 30, 113–125 [DOI] [PubMed] [Google Scholar]
- 38.Liu S., Quarles L. D. (2007) J. Am. Soc. Nephrol. 18, 1637–1647 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.