

Abstract
Techniques for analyzing the membrane diffusion of molecules are the most promising methods for investigating the compartmentalization of G-protein-coupled receptors, particularly as relevant to receptor signaling processes. Here, we report fluorescence recovery after photobleaching (FRAP) measurements performed at variable spot radius for human mu opioid (hMOP) receptors on SH-SY5Y neuroblastoma cells in the presence of ligands. Although an antagonist did not affect the behavior of the receptors compared with the basal state, two different agonists, DAMGO and morphine, caused markedly different changes to receptor diffusion. Like receptors in the absence of ligand, receptors bound to morphine exhibited diffusion confined to joined semipermeable domains, but with smaller domain size and diffusion coefficient. This effect was inhibited by pertussis toxin, strongly suggesting that this dynamic behavior is associated with early steps of signaling. In the presence of DAMGO, half of the receptors displayed free long-range diffusion and the other half were confined to smaller isolated domains. Hypertonic sucrose buffer suppressed this effect, which we attribute to receptor entry into clathrin-coated pits. It is likely that the observation of distinct receptor dynamics in the presence of DAMGO and morphine involves the agonist-selective phosphorylation of the receptor.
Keywords: Biophysics, G Proteins/Coupled Receptors (GPCR), Lipid/Raft, Membrane/Biophysics, Membrane/Function, Receptors/Membrane, Lateral Diffusion, Membrane Domains
Introduction
G-protein-coupled receptors (GPCRs)5 constitute the largest family of plasma membrane receptors and are involved in numerous cell processes (1–3). Understanding the molecular basis of the signal transduction mechanism they mediate, notably the initial membrane steps, is thus of fundamental importance. Two principal membrane events after agonist binding to a receptor have been clearly described. Signal transduction following activation of the heterotrimeric G-protein and effectors drives the cell response. The second event, receptor internalization, proceeding predominantly by the clathrin-coated pit pathway, leads to signal cessation (4, 5). GPCR are remarkably efficient and rapid at transmitting the signal despite the involvement of interactions with numerous partners. Thus, compartmentalization of the receptors with their partners, G-proteins and effectors, may explain the signal transduction properties (6–8).
The dynamics of GPCR in the plasma membrane and its relationship with receptor function are central to the actively developing field of research that is the functional and structural organization of cell membranes (9–11). Numerous experiments involving fluorescence recovery after photobleaching (FRAP) or single molecule tracking and concerning GPCR have been reported. These studies all indicate that GPCR diffusion is restricted to domains. However, the results reported are conflicting, such that no consensus has been reached about the origin of the confinement domains and their role in signal transduction (12).
Investigations of the effects of agonist binding on receptor organization and dynamics may clarify this issue. Most available diffusion data reveal a decrease of the diffusion coefficient with decreasing domain size (12). Some studies convincingly attribute this to the internalization of the bound receptors through the clathrin-coated pit route (13–15). According to the model in which membrane lipid microdomains act as signaling platforms, the so-called rafts are another potential source of receptor confinement (16, 17). Analyses of GPCR enrichment in detergent-resistant membranes has given variable results, and no consensus has yet emerged (18–20).
We previously used the high resolution FRAP approach at variable radius to study T7-EGFP-hMOP receptor diffusion in the plasma membrane of SH-SY5Y neuronal cells: we showed that diffusion of receptors in the basal state is confined to joined permeable domains (21). Here, using the same method, we analyze the influence of the binding of one antagonist and two agonist ligands on the dynamic organization of the receptors. To study the origins and links to functional processes of receptor organization and dynamics, we examined the effects after agonist binding of the inhibition of two phenomena: clathrin recruitment and G-protein activation. Interestingly, we found two distinct patterns of compartmentalization, each associated with one of the major events occurring at the plasma membrane after receptor activation.
EXPERIMENTAL PROCEDURES
Cell Culture
Cells from the transfected neuroblastoma cell line SH-SY5Y stably expressing T7-EGFP-hMOP receptor (21) were grown at 37 °C, under 5% CO2 in air, in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 1% glutamine, 0.05 mg/ml gentamicin, and 0.4 mg/ml neomycin (G418, Invitrogen) to maintain selection. Two days before the experiment cells were plated on coverslips (22 × 22 mm2).
Preparation for Microscopic Observations
The cells were carefully rinsed with PBS (Eurobio), and the coverslip was placed in a homemade steel base designed in such a way that it constituted one face of an observation chamber of 30-μm depth; the chamber was filled with PBS. Except when specified, all FRAP measurements were performed at room temperature (20 ± 1 °C). To ensure good cell viability, the observations were limited to a duration of 30 min per preparation.
For analyses in the presence of agonists or antagonist, ligands were diluted in PBS to a final concentration of 1 μm. Cells on the coverslip were incubated at room temperature with 60 μl of this solution for 10 min ([d-Ala2, N-Me-Phe4, Gly-ol5]enkephalin, DAMGO) or 30 min (morphine and d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2, CTAP). Samples were not additionally rinsed before the coverslip was put in the observation chamber; the cells were thus maintained in the same conditions for the experiments.
The effects of sucrose and pertussis toxin (PTX) were investigated. For sucrose, cells were preincubated with 0.5 m sucrose in PBS for 15 min at 37 °C, and the ligand was diluted in PBS 0.5 m sucrose. For pertussis toxin, PTX (Sigma) was added to the culture medium of the cells plated on a coverslip to a final concentration of 100 ng/ml the day before the experiment.
Variable Radius FRAP Experiments and Data Analysis
FRAP measurements were carried out in uniform disk illumination conditions using a homemade device based on an epifluorescence microscope (13). The bleaching times were set between 20 and 40 ms, and the fluorescence recovery was recorded at a sampling rate of 2 ms over 30 s. The radius of the observation area R was varied between 1.40 and 3.45 μm.
For each experimental condition tested, about 30 recovery curves were accumulated on different cells per R value and individually fitted using a suitable diffusion equation (22) assuming one diffusing population. The recovery curves leading to unfounded values of the mobile fraction (M > 110%), diffusion coefficient (D > 1 μm2/s) or normalized fluorescence intensity just after bleaching (negative values) were rejected and not further considered. The valid recovery curves were further averaged (Fig. 1a) and the diffusion coefficient D and the mobile fraction M determined from this mean curve by a single population fit with a confidence interval of 95% (22). Importantly at this step, we verified that these values of M and D are consistent with the averages values calculated from the results of the fit of the individual recovery curves (see supplemental Fig. S1). This procedure is carried out at least in triplicate.
FIGURE 1.

Recovery curves of FRAP measurements on T7-EGFP-hMOP receptors with an observation radius r = 3. 45 μm: raw, averaged data, and fit. Fluorescence intensities, plotted as a function of time after bleaching, are normalized with respect to the extent of the decrease of the intensity just after the bleaching pulse. a, raw data for a single measurement (one cell: gray squares), average of a set of about 30 measurements on distinct cells (1 day: empty squares) and average of 4 sets of measurements (4 days: black full squares). b, fit of this recovery curve, average over 4 sets of measurements (4 days), by a diffusion equation (see text) assuming two-diffusing populations (2D fit) minimizes the sum of the quadratic deviations compared with that assuming a single population (1D fit).
The membrane compartmentalization is characterized by analyzing the variations of M and D with R as follows (23, 24): 1) In the absence of domains, M and D are independent of R and the measured diffusion coefficient corresponds to the real values. 2) If the membrane is compartmentalized then M and D are dependent on R as a function of the domain size r. For R < r, the mobile fraction approaches a limit value as 1/R increases which can be roughly estimated from the value of the mobile fraction at R ∼ r. For R > r, the following relationship in Equation 1 is expected,
where MP is the permanent mobile fraction. Depending on the MP value, two cases can be considered. 1) If Mp ≈ 0, there is a single population of tracers confined within closed domains. The measured diffusion coefficient is an apparent one (Dapp), and the true diffusion coefficient inside the domains Dconf can be calculated using Equation 2.
This analysis requires verification that Dconf is invariant with R. 2) If MP > 0, the tracers are either inside joined but semi-permeable domains or distributed in both inside and outside isolated domains. Here, it is necessary to establish whether two populations exist or not by examining the quality of the fit obtained assuming two populations of tracers. Whereas no difference was noted for averaged single sets of measurements, an additional averaging over several sets (see Fig. 1) allowed us to observe marked improvement of the quality of the fit assuming two populations for several conditions among those studied.
If no improvement is observed, a single population of tracers inside joined but semi-permeable domains is assumed. D, Mp, and r are determined as the means (± S.D.) of the values calculated for each set of measurements.
If the two-population fit is of better quality, the analysis is pursued for each population. The measured diffusion coefficient of one population (confined tracers with Dconf) varies according to Equation 2; the diffusion coefficient Dfree of the second population (tracers with free long range diffusion) is invariant with R. If Dfree > Dconf, domains are isolated with a long range diffusion taking place around them. Note that for accurate analysis of the two diffusion coefficients, the relative proportion of the free diffusion population should be in the same range as MP. Dfree and Dconf are determined by the two-population fit with a confidence interval of 95%, Mp and r are estimated according to a least square fit of M versus 1/R with an error given by the standard deviation. All FRAP data were exploited by this procedure.
DRM Preparation
Cell membranes were purified as described previously (21) and incubated with or without agonists (1 μm) for 30 min at 25 °C. An aliquot of 1 ml of membrane preparation (containing 7 mg of proteins) was then incubated at 4 °C under gentle agitation for 30 min with Triton X-100 (0.35%, v/v) and added to an equal volume of a 70% (w/v) sucrose solution. This mixture was placed at the bottom of a centrifuge tube and successively overlaid with 4 ml of 30% (w/v) sucrose and 4 ml of 5% (w/v) sucrose. The tubes were centrifuged for 18 h at 200,000 × g in a Beckman rotor type SW 41, and eight fractions of 1.2 ml each were collected from the top; the remaining content was fraction 9. We measured the turbidity by an absorbance measurement at 620 nm, the sucrose concentration and the EGFP fluorescence intensity (25) of each fraction. According to their high content of alcaline phosphatase and large ratio of cholesterol to lipid, the fractions 3–5 corresponded to the DRM. Fraction 9 was the pellet, and the other fractions (1–2, 6–8) were solubilized fractions.
RESULTS
Agonist, but Not Antagonist, Binding Changes the Dynamic Lateral Organization of T7-EGFP-hMOP Receptor
We used high resolution FRAP at variable radius (23, 24) to study the changes in the dynamic lateral organization of T7-EGFP-hMOP receptor caused by the presence of ligands at a saturating concentration.
The stably transfected neuronal cell line SH-SY5Y expressing T7-EGFP-hMOP receptor used has been described previously (21). We verified that the binding of the two agonists, DAMGO and morphine, to T7-EGFP-hMOP receptor inhibited cAMP production by adenylyl cyclase and induced receptor internalization, this last one being slower for morphine than for DAMGO. Binding of the antagonist CTAP did not induce either signal transduction or internalization of the receptors. We also studied the diffusion of T7-EGFP-hMOP receptor in the basal state in this cell line by high-resolution FRAP at variable radius. T7-EGFP-hMOP receptor was found distributed into juxtaposed domains of about 700 nm radius, inside which it diffuses. However, long range diffusion was detected, indicating that escape to adjacent domains is possible (21).
Thus, in the same experimental conditions as the previous study, we observed similar behavior except, systematically, that the domains were larger (1.2 ± 0.2 μm and Table 1). To avoid artifacts associated with daily variations, we systematically compared the results obtained in each condition tested to those for the basal state measured on the same day.
TABLE 1.
Lateral diffusion characteristics of T7-EGFP-hMOP receptors
Diffusion coefficients and domain sizes are reported for the receptor in the presence and in the absence of the agonists, and with and without PTX pretreatment and hypertonic sucrose buffer. Reported values are the results of the analysis of means of at least three independent series of 30 measurements on distinct cells when single population is observed and values estimated from the analysis of the average of several sets of measurements when two populations are evidenced (see “Experimental Procedures”). Dconf is the diffusion coefficient for the receptors with diffusion restricted to domains of size r (r and Dconf are calculated according to Eq. 1 and 2, respectively). Dfree is the diffusion coefficient of receptors with long-range free diffusion. Mp is the permanent mobile fraction corresponding to the y-intercept on the graph M = f(1/R). (#) Because of day-to-day variability, this result has been normalized to the set of measurements for the basal condition collected the same day. (*, **) The difference with the result in the basal state is statistically significant according to the paired Student's t-test (*, p < 0.05; **, p < 0.01).
| Experimental conditions | Domain size | Dconf | Dfree | % Free | Mp |
|---|---|---|---|---|---|
| μm | 10−2 μm2/s | 10−2 μm2/s | % | % | |
| Basal | 1.2 ± 0.2 | 8 ± 3 | – | – | 48 ± 5 |
| Basal 14 °C # | 1.0 ± 0.4 | 7 ± 3 | – | – | 44 ± 4 |
| PTX | 1.2 ± 0.4 | 8 ± 7 | – | – | 49 ± 13 |
| Sucrose | 1.4 ± 0.6 | 9 ± 8 | – | – | 24 ± 14 |
| DAMGO (14 °C) | 0.6 ± 0.3** | 8 ± 6 | 24 ± 17 | 53 ± 10 | 50 ± 9 |
| Sucrose + DAMGO | 1.1 ± 0.4 | 6 ± 5 | – | – | 32 ± 11 |
| PTX + DAMGO (14 °C) | 0.5 ± 0.2** | 5 ± 4 | 43 ± 23 | 45 ± 25 | 52 ± 4 |
| Morphine | 0.6 ± 0.2* | 2 ± 1 | – | – | 64 ± 3 |
| Sucrose + Morphine # | 0.6 ± 0.5* | 2 ± 3 | – | – | 50 ± 19 |
| PTX + Morphine | 1.2 ± 0.3 | 8 ± 4 | – | – | 46 ± 8 |
The binding of the antagonist CTAP did not perturb the dynamic lateral organization of the receptors. Neither the domain radius (r = 1.1 ± 0.4 μm versus 1.2 ± 0.2 μm) nor the diffusion coefficient (Dconf = (5 ± 4) 10−2 μm2/s versus (8 ± 2) 10−2 μm2/s) were significantly different in its presence and absence.
In contrast, agonist binding resulted in significant changes to the dynamic membrane organization of T7-EGFP-hMOP receptor. Single population analysis revealed a marked decrease of the slope, by a factor of about 2, of the mobile fraction plotted against the reciprocal of the observation radius for both agonists (Fig. 2). This decrease indicates that, in both cases, the receptors were confined in domains that were about 50% smaller than those in the basal state (Table 1).
FIGURE 2.

FRAP analysis of the dynamic organization of T7-EGFP-hMOP receptors in the presence of DAMGO and morphine. Plots of the mobile fraction M against the reciprocal of the observation radius R. Each data set corresponds to a single run of measurements. For each ligand, the reference measurements in the basal state were performed the same day at the same temperature. T7-EGFP-hMOP receptor diffusion was analyzed in the presence of agonists at a concentration of 1 μm: DAMGO (■) and basal state (□) at 14 °C, and morphine (■) and basal state (□) at room temperature.
An analysis assuming two diffusing populations appeared to better fit the data obtained in the presence of DAMGO: one population confined in closed domains, and the other freely diffusing outside these domains. About half of the receptors diffused in domains of 0.6 ± 0.3 μm radius with a diffusion coefficient Dconf = (8 ± 6) 10−2 μm2/s. The other half of the receptors, corresponding to the permanent mobile fraction Mp, diffused freely around these domains with a larger diffusion coefficient Dfree = (24 ± 17)10−2 μm2/s (Table 1).
The experiments in the presence of DAMGO were performed at 14 °C to block the process of vesicle endocytosis (26). Although endocytosis was substantially inhibited at this temperature (data not shown), the dynamic lateral organization of the receptors in the basal state was not substantially altered, other than a 5% decrease in the Mp value (Table 1).
The dynamic organization of the receptor observed after morphine binding was different from that following DAMGO binding. A two-diffusing population model does not lead to reliable description of the results in the presence of morphine, and only a single population of receptors could be confirmed. The receptors are organized in small (0.6 ± 0.2 μm radius) semi-permeable joined domains allowing long range diffusion. The receptor diffusion coefficient was lower ((2 ± 1)10−2 μm2/s) than for the receptors in the basal state ((8 ± 3)10−2 μm2/s) and the proportion of receptors able to diffuse at long range was substantially higher (from 48 to 64% and Table 1). Two different types of organization are thus observed with the two agonists tested. We investigated the causes of these distinct lateral organizations.
T7-EGFP-hMOP Receptor Membrane Confinement Is Not Related to DRM Domains
Various possible origins for membrane domains, and lipid microdomains have been extensively studied (17, 19, 27). To test whether the domains identified by FRAP were rafts, DRM were isolated from transfected SH-SY5Y membranes by cold Triton X-100 extraction, and sucrose gradient fractionation. The pellet (bottom fraction, 9), the DRM (non-solubilized fractions, 3–5), and the solubilized fractions (fractions 1–2 plus 6–8) were tested for the presence of T7-EGFP-hMOPr by EGFP fluorescence measurement (25).
In the absence of incubation of the cell membrane preparation with ligand, more than 95% of the receptors were found in the solubilized fractions (Fig. 3). None of the agonists tested displaced the receptors from the solubilized to DRM fractions (Fig. 3). Thus, T7-EGFP-hMOP receptor domains identified by FRAP are not DRM domains in any of the conditions tested (basal, morphine, DAMGO).
FIGURE 3.
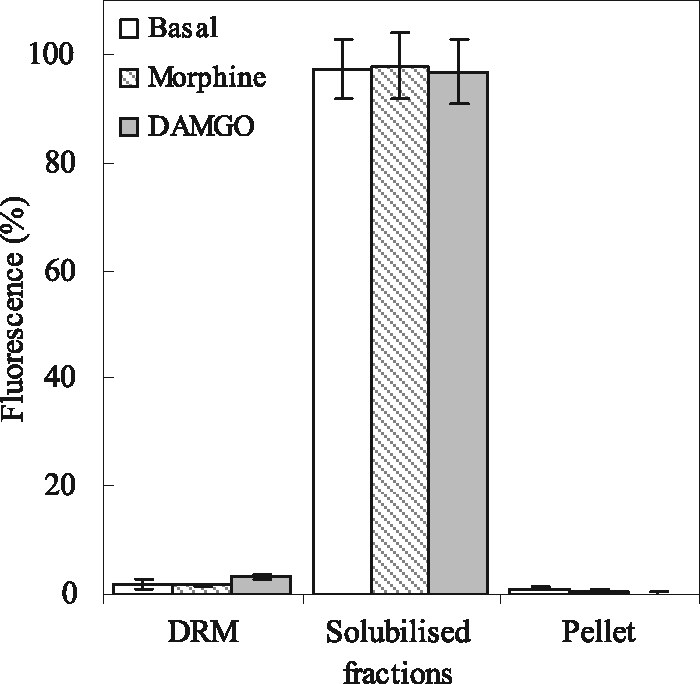
Distribution of T7-EGFP-hMOP receptors in fractions of SH-SY5Y membranes separated on sucrose gradients. Membranes without treatment (open bars) and after exposure to morphine (hatched bars) or DAMGO (filled bars) were solubilized in 0.35% of Triton X-100 at 4 °C and separated on a sucrose density gradient. EGFP fluorescence was measured in DRM (fractions 3–5), pellet (fraction 9), and solubilized fractions (fractions 1–2 and 6–8). The data are expressed as relative percentages of the total fluorescence obtained from two independent experiments.
Changes Induced by the Binding of DAMGO Are Caused by Receptor Internalization
Agonist binding causes internalization of GPCR mostly through the clathrin-coated pit pathway (5, 28). This phenomenon has been suggested to be responsible for the receptor confinement observed after agonist binding (13, 14, 29). Indeed, DAMGO binding is followed by internalization of T7-EGFP-hMOP receptor (21) proceeding via clathrin-coated pits as demonstrated by the colocalization of endocytic T7-EGFP-hMOP receptors with transferrin receptors (supplemental Fig. S2). Although the endocytosis promoted by DAMGO is impeded at 14 °C (the temperature used in our experiments), lateral reorganization of the receptors in the membrane may still occur.
Hypertonic sucrose prevents the formation of coated pits by rendering clathrin unavailable (28, 30). After checking that it blocks the internalization of transferrin but also T7-EGFP-hMOP receptor internalization (supplemental Fig. S2), we performed FRAP experiments with DAMGO in hypertonic sucrose buffer. In the absence of ligand, hypertonic sucrose did not affect the dynamic organization of T7-EGFP-hMOP receptor (Table 1), other than to decrease the size of the permanent mobile fraction (from 48 to 24%). This decrease may be explained by a side effect of hypertonic sucrose such as a partial immobilization of the membrane molecules (31) and also in part by larger variability (error bar) for the experiments performed in the presence of sucrose.
The presence of hypertonic sucrose abolished the effect of DAMGO on receptor domain size; M versus 1/R was unchanged (Fig. 4). Furthermore, as in the basal state, only a single population of receptors was observed, diffusing with Dconf = (6 ± 5)10−2 μm2/s in large (1.1 ± 0.4 μm) permeable (Mp = 32 ± 11%) domains (Table 1). This indicates that inhibition of the formation of coated pits abolishes the effects of DAMGO on the dynamic organization of T7-EGFP-hMOP receptor.
FIGURE 4.

FRAP analysis of the dynamic organization of T7-EGFP-hMOP receptors in the presence of DAMGO and hypertonic sucrose. Plots of the mobile fraction M against the reciprocal of the observation radius R. Each data set corresponds to a single run of measurements, all performed on the same day. At room temperature, T7-EGFP-hMOP receptors bound to DAMGO in the presence of hypertonic sucrose (▴) have a behavior similar to that in the basal state in the presence of hypertonic sucrose (□). The presence of DAMGO in the absence of sucrose is accompanied by the expected dynamic change (■) at 14 °C (see Fig. 2).
Conversely, internalization processes cannot explain the reduced domain size in the presence of morphine because there was no receptor internalization at room temperature, even after 1 h of exposure. Nevertheless we tested the effect of morphine in the presence of hypertonic sucrose. The decrease of the domain size (from 1.2 to 0.6 μm) and slowdown of the receptors (Dconf = (2 ± 3)10−2 μm2/s) relative to basal state were similar to those in the absence of hypertonic sucrose. So, we explored another possible cause of the confinement in the presence of morphine: signal transduction and G-protein activation.
The Interaction of Receptors with G-proteins Is Responsible for the Changes Induced by the Presence of Morphine
PTX inhibits Gi/o-protein activation and consequently the transduction signal involving these proteins (32). We verified that PTX pretreatment reduced adenylyl cyclase inhibition following agonist binding to hMOPr in our cell line: the reduction was more than 85%, consistent with previous findings for the SH-SY5Y neuroblastoma cell line (33).
PTX pretreatment did not modify the diffusional behavior of T7-EGFP-hMOP receptor in the basal state (Table 1). However, the changes induced by the binding of morphine were inhibited by PTX pretreatment (Fig. 5): the dynamic organization of the receptors remained similar to that in the basal state, with comparable domain size and long-range mobile fraction (Table 1). The diffusion coefficients of receptors in the basal state, Dconf = (8.0 ± 0.4)10−2 μm2/s and in the presence of morphine, Dconf = (8.0 ± 0.3) 10−2 μm2/s were similar (Table 1). This indicates that the activation of Gi/o proteins is required for the effects of morphine on receptor organization.
FIGURE 5.

FRAP analysis of the dynamic organization of T7-EGFP-hMOP receptor in the presence of morphine and PTX. Plots of the mobile fraction M against the reciprocal of the observation radius R. At room temperature, T7-EGFP-hMOP receptors bound to morphine preincubated with PTX (▴) have a behavior similar to that in the basal state (□). The presence of morphine in the absence of pretreatment by PTX is accompanied by the expected dynamic change (■) (see Fig. 2).
Pretreatment with PTX did not modify the effects of DAMGO on the dynamic organization of the receptor (Table 1). However, PTX pretreatment did not prevent receptor internalization after exposure to agonists that normally induce internalization (data not shown). These findings are consistent with the notion that the internalization process is involved in the confinement of the receptors in the presence of DAMGO.
DISCUSSION
Binding of agonists induces conformational changes of the receptors followed by the successive coupling of the receptor to a set of proteins partners. The details of this cascade are regularly up-dated as investigations reveal more and more complexity. The interactions between a protein and its environment lead to divergence from free diffusion, so ligand binding-dependent modulation of receptor dynamics are expected (34).
Binding of the antagonist CTAP to T7-EGFP-hMOP receptor in SH-SY5Y cells does not induce any signalization and has presumably no associated conformational changes; consistent with these observations, there was no change of the diffusional behavior of T7-EGFP-hMOP receptor. This is also compatible with our previous observation that CTAP does not induce internalization of the receptors (21). These findings, however, are not applicable to all cases because internalization of delta opioid receptor, belonging to the same opioid receptor family, has been convincingly demonstrated in NG108–15 cells after antagonist binding (35). Moreover, SPT analysis of hMOP receptor in NRK cells revealed that the antagonist CTAP induced modifications of the receptor diffusion behavior similar to those following binding of the agonist DAMGO (36). These findings provide additional evidence of variability according to the receptor and/or cell type.
Conversely, and consistent with most data for GPCR, the binding of the agonists DAMGO and morphine induced a decrease of the domain size and/or the diffusion coefficient. Note, however, that the domain sizes we measured were significantly larger that those reported elsewhere (12). Again, variability between cell types may be an explanation for these differences; they may also be due to the different experimental techniques and devices or data analysis methods used.
It is likely that lipid-protein or protein-protein interactions are responsible for the confinement of the receptors. Agonist-induced dynamic changes may reflect either the strengthening of these pre-existing interactions or the appearance of new interactions through the involvement of new partners.
The involvement of relocalization of the receptors into rafts appears to be unlikely as the proportion of receptors found in detergent-insoluble membrane fractions in the absence of ligand is negligible as reported by Mouledous et al. (37) and remains unchanged after addition of agonists in agreement with previous findings of Gaibelet et al. (38). We therefore did not investigate any putative raft localization of the receptor by studying the effects of cholesterol depletion on receptor diffusion. We focused on investigating of the relationship between a diffusional behavior and receptor function.
The receptor is rapidly internalized following exposure to DAMGO (21). It is therefore likely that receptors undergoing slow diffusion and confined to small domains (Fig. 6) in the presence of DAMGO enter the clathrin-coated pit pathway of internalization. This was supported by the inhibition of internalization by the presence of sucrose. Note that the proportion of long-range free diffusing receptors was similar to that of receptors remaining at the cell surface, i.e. non-internalized, as estimated from the fluorescence microscopy images of transfected SH-SY5Y cells in the presence of DAMGO (21).
FIGURE 6.

Schematic model for the dynamic organization of T7-EGFP-hMOP receptors in the SH-SY5Y membrane in the basal state (A), and in the presence of morphine (B) or DAMGO (C). A, before activation by the agonist, receptors diffuse in joined semi-permeable domains allowing long-range diffusion. B, after morphine binding the receptors diffuse in smaller joined semi-permeable domains. This modification is also observed in the presence of hypertonic sucrose but inhibited by a pretreatment with PTX. C, DAMGO binding induces the slow diffusion confined to small isolated domains for half of T7-EGFP-hMOP receptors; the other half has a more rapid free diffusion. This change is also observed in the case of a pretreatment with PTX but inhibited in the presence of hypertonic sucrose.
The changes of receptor dynamics following morphine binding, which maintained the receptors confined to joined permeable domains, but of smaller size (Fig. 6), may appear surprising. This effect of morphine persisted in the presence of sucrose and was abolished by inhibition of the G-protein activation. Thus, the early events of the signaling cascade, which consist of the association of various proteins with the receptors, modulate their diffusion behavior. We believe that this reveals protein-protein interactions in the membrane plane to be the major source of receptor confinement, consistent with theoretical analysis (39). Indeed, the association between signaling partners and the receptor may change the interaction between the receptor and its membrane environment and consequently modulate the size of the confinement domains. Interestingly, this idea is supported by the change of the diffusion coefficient, twice as large as the change of the domain size, in agreement with the relationship D ∼ L2 predicted by the interacting protein membrane model (39).
We thus report, for the first time, and using a single cell system, two distinct dynamic organizations for hMOP receptor depending on the agonist bound. Furthermore, we describe the phenomena underlying these organizations (Fig. 6): receptors bound to DAMGO entering into endocytosis displayed slow diffusion confined to isolated small domains; and morphine-bound receptors involved in signal transduction displayed diffusion restricted to joined permeable domains, smaller than those for free receptors.
Interestingly, our findings can be well accounted for by the model of agonist-selective mechanisms of GPCR desensitization proposed by Kelly and co-workers (40, 41). In this model, based on pharmacological analyses on heterologous cells and neurons, DAMGO binding leads to G-protein activation together with competition with receptor phosphorylation by GRK, mediating a rapid internalization/desensitization, whereas morphine activates also signal transduction but together with a much slower PKC-dependent phosphorylation and subsequent internalization of the receptor. Indeed, as expected for two independent pathways, PTX pretreatment did not inhibit receptor entry into the internalization pathway after DAMGO binding. Also, in the presence of sucrose, receptors bound to DAMGO, most probably phosphorylated, behaved differently from receptors bound to morphine, presumably not yet phosphorylated in the time interval of our experiment.
So, according to the model of Kelly et al. (41), it is likely that the slow phosphorylation of the receptors after morphine binding allows visualization of their dynamic changes accompanying G-protein activation. These changes are not detected after DAMGO binding because GRK phosphorylation induces rapid internalization of the receptors. It seems likely that the difference in the kinetics of the events following the binding of the two agonists investigated allowed us to reveal the different dynamic behaviors. Our investigation of hMOP receptor diffusion has shed new light on these mechanisms. Substantial progress in the elucidation of these complex signaling processes can be expected from further studies coupling dynamics and biochemistry of agonist-activated receptors.
Supplementary Material
This work was supported by the CNRS and MENESR joint DRAB program.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- GPCR
- G-protein-coupled receptor
- hMOP
- human mu opioid
- DAMGO
- [d-Ala2, N-Me-Phe4, Gly-ol5]enkephalin
- CTAP
- d-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2
- DRM
- detergent-resistant membrane
- PTX
- pertussis toxin
- PBS
- phosphate-buffered saline
- FRAP
- fluorescence recovery after photobleaching.
REFERENCES
- 1.Bockaert J., Pin J. P. (1999) EMBO J. 18, 1723–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luttrell L. M. (2008) Mol. Biotechnol. 39, 239–264 [DOI] [PubMed] [Google Scholar]
- 3.Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 4.Bünemann M., Hosey M. M. (1999) J. Physiol. 517, 5–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolfe B. L., Trejo J. (2007) Traffic 8, 462–470 [DOI] [PubMed] [Google Scholar]
- 6.Chidiac P. (1998) Biochem. Pharmacol. 55, 549–556 [DOI] [PubMed] [Google Scholar]
- 7.Hur E. M., Kim K. T. (2002) Cell Signal. 14, 397–405 [DOI] [PubMed] [Google Scholar]
- 8.Neubig R. R. (1994) Faseb J. 8, 939–946 [DOI] [PubMed] [Google Scholar]
- 9.Engelman D. M. (2005) Nature 438, 578–580 [DOI] [PubMed] [Google Scholar]
- 10.Marguet D., Lenne P. F., Rigneault H., He H. T. (2006) EMBO J. 25, 3446–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vereb G., Szöllosi J., Matkó J., Nagy P., Farkas T., Vigh L., Mátyus L., Waldmann T. A., Damjanovich S. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 8053–8058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker A., Saulière A., Dumas F., Millot C., Mazères S., Lopez A., Salomé L. (2007) Eur. Biophys. J. 36, 849–860 [DOI] [PubMed] [Google Scholar]
- 13.Cézanne L., Lecat S., Lagane B., Millot C., Vollmer J. Y., Matthes H., Galzi J. L., Lopez A. (2004) J. Biol. Chem. 279, 45057–45067 [DOI] [PubMed] [Google Scholar]
- 14.Jacquier V., Prummer M., Segura J. M., Pick H., Vogel H. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 14325–14330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lill Y., Martinez K. L., Lill M. A., Meyer B. H., Vogel H., Hecht B. (2005) Chem. Phys. Chem. 6, 1633–1640 [DOI] [PubMed] [Google Scholar]
- 16.Ostrom R. S. (2002) Mol. Pharmacol. 61, 473–476 [DOI] [PubMed] [Google Scholar]
- 17.Ostrom R. S., Insel P. A. (2004) Br. J. Pharmacol. 143, 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chini B., Parenti M. (2004) J. Mol. Endocrinol. 32, 325–338 [DOI] [PubMed] [Google Scholar]
- 19.Pike L. J. (2003) J. Lipid Res. 44, 655–667 [DOI] [PubMed] [Google Scholar]
- 20.Pucadyil T. J., Chattopadhyay A. (2006) Prog. Lipid Res. 45, 295–333 [DOI] [PubMed] [Google Scholar]
- 21.Saulière A., Gaibelet G., Millot C., Mazères S., Lopez A., Salomé L. (2006) FEBS Lett. 580, 5227–5231 [DOI] [PubMed] [Google Scholar]
- 22.Lopez A., Dupou L., Altibelli A., Trotard J., Tocanne J. F. (1988) Biophys. J. 53, 963–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker A. M., Saulière A., Gaibelet G., Lagane B., Mazères S., Fourage M., Bachelerie F., Salomé L., Lopez A., Dumas F. (2007) J. Biol. Chem. 282, 35163–35168 [DOI] [PubMed] [Google Scholar]
- 24.Salomé L., Cazeils J. L., Lopez A., Tocanne J. F. (1998) Eur. Biophys. J. 27, 391–402 [DOI] [PubMed] [Google Scholar]
- 25.André A., Gaibelet G., Le Guyader L., Welby M., Lopez A., Lebrun C. (2008) Biochim. Biophys. Acta 1778, 1483–1492 [DOI] [PubMed] [Google Scholar]
- 26.Cao T. T., Mays R. W., von Zastrow M. (1998) J. Biol. Chem. 273, 24592–24602 [DOI] [PubMed] [Google Scholar]
- 27.Smith S. M., Lei Y., Liu J., Cahill M. E., Hagen G. M., Barisas B. G., Roess D. A. (2006) Endocrinology 147, 1789–1795 [DOI] [PubMed] [Google Scholar]
- 28.Minnis J. G., Patierno S., Kohlmeier S. E., Brecha N. C., Tonini M., Sternini C. (2003) Neuroscience 119, 33–42 [DOI] [PubMed] [Google Scholar]
- 29.Hegener O., Prenner L., Runkel F., Baader S. L., Kappler J., Häberlein H. (2004) Biochemistry 43, 6190–6199 [DOI] [PubMed] [Google Scholar]
- 30.Heuser J. E., Anderson R. G. (1989) J. Cell Biol. 108, 389–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov A. I. (2008) Methods Mol. Biol. 440, 15–33 [DOI] [PubMed] [Google Scholar]
- 32.Krueger K. M., Barbieri J. T. (1995) Clin. Microbiol. Rev. 8, 34–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prather P. L., Tsai A. W., Law P. Y. (1994) J. Pharmacol. Exp. Ther. 270, 177–184 [PubMed] [Google Scholar]
- 34.Lopez A., Salome L. (2009) Cell Mol. Life Sci. 66, 2093–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belcheva M. M., Barg J., Gloeckner C., Gao X. M., Chuang D. M., Coscia C. J. (1992) Mol. Pharmacol. 42, 445–452 [PubMed] [Google Scholar]
- 36.Daumas F. (2002) PhD. Thesis, Diffusion latérale du récepteur mu aux opioïdes analysée par suivi de particule unique à la surface de cellules vivantes: relation organisation dynamique - fonction. In., Université Paul Sabatier, Toulouse [Google Scholar]
- 37.Moulédous L., Merker S., Neasta J., Roux B., Zajac J. M., Mollereau C. (2008) Biochem. Biophys. Res. Commun. 373, 80–84 [DOI] [PubMed] [Google Scholar]
- 38.Gaibelet G., Millot C., Lebrun C., Ravault S., Sauliere A., Andre A., Lagane B., Lopez A. (2008) Mol. Membr. Biol. 25, 423–435 [DOI] [PubMed] [Google Scholar]
- 39.Destainville N., Dumas F., Salomé L. (2008) J. Chem. Biol. 1, 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson E. A., Oldfield S., Braksator E., Gonzalez-Cuello A., Couch D., Hall K. J., Mundell S. J., Bailey C. P., Kelly E., Henderson G. (2006) Mol. Pharmacol. 70, 676–685 [DOI] [PubMed] [Google Scholar]
- 41.Kelly E., Bailey C. P., Henderson G. (2008) Br. J. Pharmacol. 153, S379–S388 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.