

Abstract
Schizosaccharomyces pombe Dss1p and its homologs function in multiple cellular processes including recombinational repair of DNA and nuclear export of messenger RNA. We found that Tap-tagged Rad24p, a member of the 14-3-3 class of proteins, co-purified Dss1p along with mitotic activator Cdc25p, messenger RNA export/cell cycle factor Rae1p, 19 S proteasomal factors, and recombination protein Rhp51p (a Rad51p homolog). Using chromatin immunoprecipitation, we found that Dss1p recruited Rad24p and Rae1p to the double-strand break (DSB) sites. Furthermore, Cdc25p also recruited to the DSB site, and its recruitment was dependent on Dss1p, Rad24p, and the protein kinase Chk1p. Following DSB, all nuclear Cdc25p was found to be chromatin-associated. We found that Dss1p and Rae1p have a DNA damage checkpoint function, and upon treatment with UV light Δdss1 cells entered mitosis prematurely with indistinguishable timing from Δrad24 cells. Taken together, these results suggest that Dss1p plays a critical role in linking repair and checkpoint factors to damaged DNA sites by specifically recruiting Rad24p and Cdc25p to the DSBs. We suggest that the sequestration of Cdc25p to DNA damage sites could provide a mechanism for S. pombe cells to arrest at G2/M boundary in response to DNA damage.
Keywords: Cell Cycle, Checkpoint Control, DNA Damage, DNA Repair, Yeast, Double-strand Break, Dss1p, Rad24p, Rae1p, S. pombe
Introduction
Eukaryotic cells respond to double-strand breaks (DSBs)3 within DNA by activating DNA damage checkpoint proteins that send signals to the cells, ultimately resulting in cell cycle arrest. The arrest allows DNA damage to be repaired by the proteins of the DNA repair pathway before cells can enter mitosis (1, 2). In Schizosaccharomyces pombe, entry into mitosis at the G2/M boundary is regulated by the phosphorylation status of the mitotic regulator Cdc2p at the tyrosine 15 residue (3). The cells are maintained in G2 by phosphorylation of Cdc2p by Wee1p and Mik1p kinases, whereas their entry into mitosis is triggered by dephosphorylation of Cdc2p by the Cdc25p phosphatase (3).
Dss1p, or its Saccharomyces cerevisiae homolog Sem1p, is a small acidic protein that is required for efficient DNA repair and the nuclear export of messenger RNA (mRNA) (4–7). Dss1p is a co-factor for human breast cancer susceptibility protein BRCA2 (8). In Ustilago maydis, a Dss1p homolog is a co-factor for Brh2p, a homolog of the human BRCA2 (9). The association between Dss1p and BRCA2 regulates the function of recombination-repair protein Rad51p (a homolog of the bacterial RecA; Rhp51p in S. pombe) (10, 11). So far the homologs of BRCA2/Brh2p have not been reported in either S. cerevisiae or in S. pombe. S. cerevisiae Sem1p was shown to recruit to DSB sites following a pattern similar to Rad51p, with high enrichment around the break site and gradually decreasing away from the break in both directions (12). Both Dss1p and Sem1p were shown to associate with the 19 S subunit of the 26 S proteasomes (12–14). It was suggested that the function of Sem1p involves regulating the function of the proteasome complex in DNA repair (12). So far the corresponding role of S. pombe Dss1p in DNA recombination-repair has not been studied.
S. pombe Rad24p belongs to the 14-3-3 family of proteins that play a significant role as checkpoint factors in monitoring DNA damage in the G2 phase of the cell cycle (15–18). Their role in the cell cycle was first demonstrated in S. pombe, where the products of the rad24 and rad25 genes were shown to possess a DNA damage checkpoint function (19). Neither the rad24 nor the rad25 gene is essential for growth, but simultaneous loss of both genes is lethal (19). The loss of rad24, but not rad25, leads to premature entry of cells into mitosis, resulting in small, round cells. In addition, the loss of rad24 renders cells highly sensitive to DNA-damaging agents, whereas a rad25 null strain is only modestly sensitive (19). In response to DNA damage in the G2 stage, activated Chk1p kinase phosphorylates Cdc25p. Recently Mek1p was shown to phosphorylate Cdc25p independent of Chk1p (20). Rad24p binds phosphorylated Cdc25p and apparently blocks a nuclear localization signal within Cdc25p. The Rad24p-Cdc25p complex exits the nucleus by using a dedicated nuclear export pathway. It was originally suggested that the “nuclear exclusion” of Cdc25p prevents the dephosphorylation and activation of the target of Cdc25p, Cdc2p, at tyrosine 15 (21). More recently, it has been shown that the exclusion of Cdc25p from the nucleus is not essential for inhibiting cell cycle progression (22).
S. pombe Rae1p is essential for the nuclear export of mRNA and functions in cell cycle progression from G2 to M (23, 24). However, the role of Rae1p in the G2/M transition was shown to be independent of its role in mRNA export (25). Thus far, no role for Rae1p has been implicated in DNA damage. A human homolog of Rae1p was found to be required in metaphase in mitotic spindle assembly. Through its interaction with the nuclear mitotic apparatus protein, human Rae1p is thought to be involved in bipolar spindle pole body formation (26–28). Whether S. pombe Rae1p plays a similar role remains to be investigated.
In this study, by using the tandem affinity purification (TAP) method, we found that Rad24p-TAP co-purified Dss1p and Rae1p. Several DNA repair proteins (for example, 19 S proteasomal subunits and Rhp51p) known to interact with Dss1p homologs also co-purified with the Rad24p-TAP fusion. By using an S. pombe strain carrying the S. cerevisiae homothallic switching (HO)endonuclease cleavage site and a plasmid expressing inducible HO-endonuclease (29), we conducted chromatin immunoprecipitation (ChIP) experiments to develop evidence that upon DNA damage, Dss1p mediates recruitment of DNA recombination/repair, cell cycle, and checkpoint control proteins to the DNA DSB sites. Finally, we found that Dss1p and Rae1p have a DNA checkpoint function, with Δdss1 cells timing of entry into mitosis indistinguishable from Δrad24 cells. Taken together, these results suggest that Dss1p plays a critical role in linking DNA repair and checkpoint factors to damaged sites by recruiting Rad24p and Cdc25p to the DNA DSB sites. We suggest that the recruitment of Cdc25p to the DSB site could lead to cell cycle arrest at the G2/M boundary in S. pombe cells in response to DNA damage.
EXPERIMENTAL PROCEDURES
Strains and Culture
Basic genetic and cell culture techniques have been described previously (30, 31). The strains used in this study are described in Table 1. The rad24-tap strain was constructed by techniques described previously (32, 33), in which TAP is fused to the rad24 gene at the C terminus by homologous recombination using the kanamycin gene as a selectable marker. Strains carrying the HO-endonuclease site were kind gifts from the laboratory of Paul Russell (The Scripps Research Institute, La Jolla, CA).
TABLE 1.
S. pombe strains used in this study
| Genotypes | Source |
|---|---|
| h− leu1-32 ura4-D18 | Ref. 25 |
| h− leu1-32 ura4-D18 rae1-1 | Ref. 23 |
| h− leu1-32 ura4-D18 ade6-704 rae1-1 rad24::ura4+ | This study |
| h− leu1-32 ura4-D18 ade6-704 rad24::ura4+ | Ref. 19 |
| h− leu1-32 ura4-D18 rad24-TAP | This study |
| h+ leu+ ura4+ rad1-1 | Ref. 49 |
| h+ leu1-32 ura4-D18 ade− dss1::kan | Ref. 5 |
| h− leu1-32 ura4-D18 cdc25-GFPint cdc25::ura4+ | Ref. 50 |
| h− leu1-32::2xYFP-crb2+ -leu1+ ura4D18 crb2-D2::ura4+ his3-D1 arg3::HO site-kanMX4 | Ref. 29 |
| h− leu1-32::2xYFP-crb2+ -leu1+ ura4D18 crb2-D2::ura4+ his3-D1 arg3::HO site-kanMX4 dss1::kan | This study |
| h− leu1-32::2xYFP-crb2+ -leu1+ ura4D18 crb2-D2::ura4+ his3-D1 arg3::HO site-kanMX4 rad24::ura4+ | This study |
| h− leu1-32::2xYFP-crb2+ -leu1+ ura4D18 crb2-D2::ura4+ his3-D1 arg3::HO site-kanMX4 cdc25-GFPint cdc25::ura4+ | This study |
| h− leu1-32::2xYFP-crb2+ -leu1+ ura4D18 crb2-D2::ura4+ his3-D1 arg3::HO site-kanMX4 cdc25-GFPint cdc25::ura4+ rad24::ura4+ | This study |
| h− leu1-32::2xYFP-crb2+ -leu1+ ura4D18 crb2-D2::ura4+ his3-D1 arg3::HO site-kanMX4 cdc25-GFPint cdc25::ura4+ chk1::ura4+ | This study |
TAP Purification of Complexes
To identify mRNA export factors that may interact with Rad24p, an S. pombe strain expressing the Rad24p-TAP fusion protein from its chromosomal locus was used. As a control for these experiments, pNTAP41 was used. Purification of TAP-tagged proteins was performed by published methods using IgG-Sepharose beads (34). The complexes were cleaved from the beads by using TEV-protease. The cleaved fraction was next bound to calmodulin beads, and the bound proteins were eluted by calmodulin elution buffer and precipitated by trichloroacetic acid (34). Purified samples were separated on SDS-PAGE and visualized by Coomassie Blue staining, and associated proteins were identified using tandem mass spectrometry and/or Western blot analysis using specific antibodies (35, 36). The tandem mass spectrometry data were searched against the S. pombe data base from the Wellcome Trust Sanger Institute using BioWorks 3.3.1.
Western Blot Detection
Polyclonal antibodies to S. pombe mRNA export factors were made by expressing the proteins in Escherichia coli and injecting them into rabbits as previously described (5, 37, 38). Antibodies to S. pombe Rad24p and Rhp51p were made by expressing the full-length rad24 and rhp51 genes in pET14b. Polyclonal antibodies to S. pombe Mts2p were bought from Biomol Research La. Standard chemiluminescent methods (PerkinElmer Life Sciences) were used to detect the purified proteins.
ChIP Analysis
ChIP analysis was performed using published protocols (39, 40). Following cross-linking of proteins to DNA and precipitation by different antibodies, specific primer pairs were used to amplify segments of DNA fragments precipitated by the specific antibodies (Table 2). As a control for amplification, PCR primers (Table 2) that did not amplify the precipitated products were used with each experiment as an internal control. [α-32P]dCTP was added to the PCR. The amplified products were separated by electrophoresis on 6% Tris/borate/EDTA gel and quantified using phosphorimaging.
FIGURE 2.

Recruitment of S. pombe proteins to a genomic region containing a DSB site. A, a schematic of the DNA fragment (5.4 kb) surrounding an HO-endonuclease cleavable site within a KanMX4 gene integrated into the S. pombe genome (29). Location and corresponding number represent different nonoverlapping regions surrounding the HO site whose amplification was tested by PCR. Primers used for PCR reaction are shown in Table 2. B, a schematic diagram showing the calculation for relative fold enrichment of proteins based on the formula shown (36). IC represents PCR product generated by using a pair of primers (Table 2) as internal control of the PCR reaction. C, panels a–e, individual ChIP experiment data shown on the left. Antibodies used for immunoprecipitation are as indicated. WCE is whole cell extract; −IP, prebleed sera was used for immunoprecipitation; +IP, antibodies as indicated were used for immunoprecipitation. IC is the internal control used in every experiment. Graphical representations of the quantification of relative/fold enrichment at each of the nine fragments are shown on the right. The y axis shows fold enrichment and x-axis shows each of the nine fragments tested and the corresponding antibodies used for the immunoprecipitation. Each bar is an average of three independent experiments. The shaded bars represent control experiments done in the absence of HO-endonuclease, and the black bars represent experiments performed in presence of the HO-endonuclease.
TABLE 2.
Primers used in the ChIP experiments
| Primer | Sequence |
|---|---|
| ChIP 1 (forward) | 5′-TTTCATTGAGATAGACCACCT-3′ |
| ChIP 1 (reverse) | 5′-CTCTTTCTGTAGCTCGTAAAG-3′ |
| ChIP 2 (forward) | 5′-CCACCAACCTTGATAATAGCG-3′ |
| ChIP 2 (reverse) | 5′-GGCAATGCTTATTGAATTGCA-3′ |
| ChIP 3 (forward) | 5′-TGGCTGAAAATTACATCGGAAACC-3′ |
| ChIP 3 (reverse) | 5′-GAGAAGGGCTTCTTGCCAAAT-3′ |
| ChIP 4 (forward) | 5′-CATCCCTTTTCATTAGTTCTA-3′ |
| ChIP 4 (reverse) | 5′-GTCGACTACAAGGAACCCACA-3′ |
| ChIP 5 (forward) | 5′-CGCAGCTCAGGGGCATGATGT-3′ |
| ChIP 5 (reverse) | 5′-CTGATTGCCCGACATTATCGC-3′ |
| ChIP 6 (forward) | 5′-CTCTTCCGACCATCAAGCATT-3′ |
| ChIP 6 (reverse) | 5′-AATCACCATGAGTGACGACTG-3′ |
| ChIP 7 (forward) | 5′-AACTGCCTCGGTGAGTTTTCT-3′ |
| ChIP 7 (reverse) | 5′-CACATACGATTGACGCATGAT-3′ |
| ChIP 8 (forward) | 5′-CCTCTTAACTAGATATTTCAAGG-3′ |
| ChIP 8 (reverse) | 5′-AAGGAATTGGCGGAAGACAAG-3′ |
| ChIP 9 (forward) | 5′-GTAACCACAAAGGTAGTTTCT-3′ |
| ChIP 9 (reverse) | 5′-AGGGGCTCTAGAACTAATGAG-3′ |
| Internal control (forward) | 5′-CAACAGGAGCGCTATAATAA-3′ |
| Internal control (reverse) | 5′-CAGATAGCTTGGATAGATATG-3′ |
Chromatin Association Assay
The extracts were prepared from spheroplasts and nuclei according to published protocols (30, 41). The chromatin-associated fraction was separated from nuclear supernatant by layering the nuclear extract onto 30% sucrose cushion and centrifuged at 16,000 × g for 15 min (41). The chromatin-associated proteins were released by incubation with 277 units of DNase 1 for 10 min at 25 °C in a buffer containing 20 mm Hepes, pH 7.9, 1.5 mm magnesium acetate, 50 mm potassium acetate, 10% glycerol, 0.5 mm dithiothreitol, 150 mm Nacl, protease, and phosphatase inhibitors (42). Chromatin-associated protein was identified by Western blot analysis.
Construction of Plasmids
The rad24 gene was isolated from a genomic library. For expression in bacteria, the rad24 gene was amplified and introduced into pGEX4T-3 (Amersham Biosciences) and pET14b (Novagen) to produce GST and His-tagged Rad24p, respectively. Similarly His-Rhp51p also made with pET14b. Construction of His-tagged fusions of Rae1p and Dss1p were described previously (5).
Sensitivity to DNA-damaging Agents
Sensitivity to UV light was determined by plating 750–1000 cells and irradiating with a Stratagene UV-Stratalinker 2400 at fixed energy levels. The cells were then incubated at 21 °C, and the number of colonies was scored after 5 days. Sensitivity of cells to other DNA-damaging agents was determined by incubating liquid cultures cells in 3 milliunits/ml bleomycin or 0.03% methyl methanesulfonate (MMS) or 15 mm hydroxyurea (HU) (43–45). In parallel, control cells were incubated similarly without the addition of any DNA-damaging agent. Following incubation of 500–700 cells on YES plates, the surviving colonies were counted after 5 days.
Checkpoint Analysis
The cells were synchronized by sedimentation through 7–30% lactose gradient and plated on YES medium (46). The G2 cells were irradiated with 0, 50, 100, and 150 J/m2 UV light in a Stratagene Stratalinker 2400. The control cells were plated on YES medium without exposure to UV radiation. The cells were then resuspended and incubated at 21 °C or at 27 °C. Samples were taken at the indicated times, fixed in methanol at −20 °C, mounted, stained with 4,6-diamidino-2-phenylindole and calcofluor, and visualized under a fluorescence microscope. The cells were judged to have passed mitosis if either nuclear division or septum formation occurred.
RESULTS
Dss1p and Interacting Components Co-purify with Rad24p-TAP Fusion Protein
An S. pombe strain was constructed in which rad24 gene was replaced by a rad24-tap gene fusion. As a negative control, TAP was expressed in wild type cells from a plasmid (pNTAP41) under the control of the nmt1 promoter. Fig. 1A shows a typical TAP-purification experiment using genomic Rad24p-Tap fusion. Compared with the Tap control (left lane), several distinct bands were visible when Rad24-TAP fusion was used to pull down interacting proteins whose identities were established by tandem mass spectrometric analysis. Elongation factors (eIF4G, eIF3a, and eIF3b), cell cycle factors (Cdc15p, Cdc22p, Cdc25p, and Rad25p), and 19 S proteasomal subunits (Mts4p, Rpn5p, Mts2p, and Rpn6p) were pulled down by Rad24p-Tap. DNA repair protein Rhp51p, Dss1p, and mRNA export factor Rae1p also co-purified with Rad24p-Tap fusion. Additional proteins identified using mass spectrometry (not shown in the gel in Fig. 1A) are presented in the supplemental Table 1S.
FIGURE 1.

Identification of Rad24p-TAP associated protein complex by tandem mass spectrometry and/or Western blot analysis. A, tandem mass spectrometry analysis of Rad24p-TAP and their identified proteins are indicated (lane 2). The asterisk indicates the 60 ribosomal protein L2 (rpl402). B, input extracts from strains expressing TAP control or Rad24p-TAP fusion as indicated (lanes 1 and 2). TAP control or Rad24-TAP associated proteins purified on IgG-Sepharose and calmodulin beads as indicated (lanes 3 and 4). Proteins identified by specific antibodies are indicated. WCE represents whole cell extract. See “Experimental Procedures” for details.
It is notable that only two known mRNA export factors (Rae1p and Dss1p) co-purified with Rad24p-Tap fusion protein. Western blot analysis was used to confirm these results. Fig. 1B shows the Western blot confirming that Rae1p and Dss1p co-purified with Rad24p-TAP but not with TAP alone (compare lane 4 versus lane 3). However, no other known mRNA export factor such as Uap56p, Mlo3p, Nup98p, Nup159p, and Sac3p co-purified with Rad24p-TAP. The absence of interaction with any other known mRNA export factor by Rad24p suggests that its interaction with Rae1p and Dss1p may represent a new complex, distinct from an mRNA export-related protein complex of these two proteins we previously described (5). By Western analysis we also confirmed interaction between Rad24p and Mts2p and Rhp51p. These results suggest that Rad24p physically interacts with at least some components of the proteasomes and the DNA recombination-repair pathway.
Dss1p, Rhp51p, and Mts2p Are Recruited to an HO-endonuclease-induced DSB Site in S. pombe Cells
We wanted to test whether in S. pombe Dss1p was recruited to DSB sites similar to Sem1p/human Dss1p. For this purpose, we used an S. pombe strain carrying the S. cerevisiae HO-endonuclease site integrated within the arg3 locus (obtained from Paul Russell) (29). A schematic of the 5.4-kb genomic region surrounding the HO-endonuclease cleavage site (∼2.1 kb upstream to ∼3.2 kb downstream) and specific regions/fragments 1–9 that were amplified by PCR are shown in Fig. 2A (for details see Fig. 2 legend and Table 2). The strain was transformed with a plasmid expressing HO-endonuclease in pJR1–41XH vector under the control of a thiamine repressible nmt1 promoter. By ChIP, DNA-bound proteins were precipitated using anti-Dss1p, anti-Rhp51p, or anti-Mts2p antibody from S. pombe cells in the presence or absence of HO-endonuclease expression. Quantitative PCR was performed to amplify specific fragments (bars 1–9 in Fig. 2A) corresponding to both sides of the HO-endonuclease site as well as from a nontranscribed internal control region (at a different chromosomal locus (24) from the immunoprecipitated DNA obtained by antibodies or prebleed sera. Relative fold enrichment of DNA was calculated by using established procedures following the formula shown in Fig. 2B. Based on this calculation, a protein was considered enriched at a region when the DNA fold increase value was above 1 (see details in Fig. 2B legend) (39).
We previously showed that Dss1p recruits to actively transcribed genes in wild type S. pombe cells (5). In the absence of HO-endonuclease (control), in the S. pombe strain carrying the HO site, Dss1p showed a relative fold enrichment above 1 throughout the tested region (that also carried a functional kanamycin gene) (Fig. 2C, panel a data and gray bars in graph). In the cells expressing HO-endonuclease, Dss1p showed a clear pattern of enrichment around the break site. Enrichment was highest near the break site (peak relative enrichment ∼5-fold proximal to the HO site; fragments 4–7) and gradually decreased away from the break site (fragments 1, 2, 3, 8, and 9).
Rad51p is known to recruit to DSB site in S. cerevisiae. We therefore tested whether Rhp51p similarly enriched at or around DNA break sites in S. pombe. Following a similar experimental procedure, we found that Rhp51p did not enrich within the test region in the control experiment (Fig. 2C, panel b, gray bars in graph). However, in the cells expressing the HO-endonuclease, there was significant increase in relative fold enrichment of the protein throughout the region (relative enrichment between 3–5-fold; fragments 1–9). Thus, in contrast to Dss1p, the pattern of enrichment shown by Rhp51p was different; it had an elevated level of enrichment both near and away from the break site, perhaps reflecting its nature of interaction with DNA.
Proteasomal components are known to recruit to DSB sites. We tested for the recruitment of Mts2p, a component of the proteasomes, to the DSB site. Mts2p was not significantly enriched in cells that did not express the HO-endonuclease (Fig. 2C, panel c, gray bars in graph). Relative fold enrichment of Mts2p was observed in the vicinity of the DSB site (highest enrichment, ∼4.5-fold; Fig. 2C, panel c, black bars in graph, fragments 3–7) similar to the recruitment pattern observed for Dss1p in the strain expressing the HO-endonuclease. These results demonstrate that Dss1p, Rhp51p, and Mts2p are recruited preferentially surrounding the region of a DSB site introduced into the S. pombe cells, consistent with observations made with their homologs (12).
Rad24p and Rae1p Are Enriched at DSB Sites in S. pombe Cells
We previously showed that Rae1p was not recruited to active genes (5, 24). Also, it is not known whether Rad24 or any of its homologs recruit to genes. We wanted to test whether Rad24p and Rae1p were recruited to the DSB site in S. pombe cells. Following a similar experimental plan as above, we tested relative fold enrichment of Rad24p and Rae1p around the DSB site. Rad24p did not recruit efficiently within the 5.4-kb region in the cells not expressing the HO-endonuclease (Fig. 2C, panel d, control experiment). However, in presence of the HO-endonuclease, there was a significant relative fold enrichment of Rad24p with a pattern similar to Dss1p; there was maximum enrichment proximal to the break site (∼5-fold) and a gradual decrease away from the break site in either direction (Fig. 2C, panel d, black bars in graph).
Finally, we tested for relative enrichment of Rae1p to the same 5.4-kb region in the presence or absence of the HO-endonuclease. Rae1p did not show preferential recruitment to any portion of the 5.4-kb region in cells not expressing the HO-endonuclease. However, in the cells expressing the HO-endonuclease, there was a significant relative fold enrichment of Rae1p around the DSB site (highest relative enrichment, ∼4.5-fold; Fig. 2C, panel e, black bars in graph).
In a previous work, we showed that in S. pombe cells, Rae1p predominantly localizes at the nuclear pore (25). The current results are the first indication that some fraction of Rae1p could be physically (and functionally) associated with the genome. In addition, these results suggest that although under the normal cellular conditions Rad24p did not interact with the DNA region around the HO site, following DNA damage it was recruited to the DSB site similar to Dss1p, Rhp51p, Mts2p, and Rae1p. By considering the co-purification of proteins with Rad24p-TAP and the pattern of enrichment at the DSB site described above, it is likely that some of these proteins may function as a multiprotein complex(s) on DNA around the DSB site.
Dss1p Is Required for Recruitment of Rad24p, Rae1p, Rhp51p, and Mts2p to DSB Sites
We wanted to explore whether Dss1p-interacting proteins depended on Dss1p for their recruitment onto the DSB sites. In a Δdss1 strain, the recruitment of Rad24p, Rae1p, Rhp51p, and Mts2p was not detectable when HO-endonuclease was expressed (Fig. 3, A–D). We tested the amount of proteins present in the extract used for the ChIP experiment (shown in Fig. 3E) to rule out the possibility that lack of recruitment was not due to loss of proteins in the Δdss1 strain. As can be seen, the level of each protein in Δdss1 strain was comparable with the level observed in wild type cells (Fig. 3E). The lack of recruitment of all four proteins in the Δdss1 strain is strikingly different from the enrichment pattern observed in wild type cells (see Fig. 2C for comparison). These results strongly suggest that at the DSB site and the surrounding region, the recruitment of all four proteins depends on the presence of Dss1p.
FIGURE 3.

Dss1p-dependent recruitment to the DSB site of proteins that co-purified with Rad24p-TAP. A–D and F–H, as in Fig. 2C, graphical representations of the relative fold enrichment at each of the nine fragments are shown. Strain backgrounds and specific antibodies used in each experiment are indicated. See Fig. 2C for nomenclature and details of the experiments. The experimental data are provided as supplemental Fig. 1S. E and I, protein levels tested in wild type, Δdss1, and Δrad24 strains. Lane 1, HO-endonuclease uninduced; lane 2, HO-endonuclease induced. Uap56p was used as an experimental control.
In a Δrad24 strain, we found that the recruitment of Dss1p was unaffected, suggesting that Dss1p recruitment was independent of Rad24p (Fig. 3F, compare with Fig. 2C, panel a). Similarly, Rhp51p was also enriched in Δrad24 strain at the DSB site similar to the pattern observed in the wild type cells (Fig. 3G, compare with Fig. 2C, panel b). Thus, Rhp51p was recruited independent of Rad24p. However, we found that Rae1p was not recruited significantly at the break site in a Δrad24 strain (Fig. 3H; see also supplemental Fig. 1S). Rae1p levels were tested in the Δrad24 strain, and no significant drop in the expression level of Rae1p in the Δrad24 strain was detected (Fig. 3I). These results indicate that (i) Dss1p is required for the recruitment of Rad24p, Rae1p, Rhp51p, and Mts2p to DSB sites and (ii) Rad24p is required for the recruitment of Rae1p.
Cdc25p Is Recruited to DSB Sites Dependent on Rad24p and Dss1p
The recruitment of Rad24p raised the interesting possibility that its interacting partner Cdc25p may associate with it and be recruited to DSB site upon DNA damage in S. pombe cells. To test this hypothesis, we constructed a wild type test strain carrying the HO-endonuclease cleavage site and a genomic copy of cdc25-gfp gene. Following the experimental plan as before, we tested the recruitment of Cdc25p-GFP in the presence or absence of HO-endonuclease in the strain. In the absence of the HO-endonuclease, there was no detectable enrichment of Cdc25p-GFP around the HO site (see control, gray bars in the histogram shown at the right of Fig. 4A, panel a). In contrast, there was a significant relative enrichment of Cdc25p-GFP around the DSB site when the HO-endonuclease expression was induced. These results clearly show that Cdc25p-GFP recruited to the DSB site in S. pombe cells during DNA damage.
FIGURE 4.

Recruitment of Cdc25p-GFP to a DSB-containing region. A, panels a–d, extracts from cells expressing the integrated Cdc25p-GFP and HO-endonuclease site were used for immunoprecipitation using an anti-GFP polyclonal antibody. ChIP experiments were performed using wild type, Δrad24, Δchk1, and Δdss1 cells carrying integrated Cdc25p-GFP and an HO-endonuclease site. Experimental details and figure descriptions are as in Fig. 2. B, protein levels of Cdc25p-GFP in wild type, Δrad24, Δchk1, and Δdss1 cells. Equal amounts of proteins from various cell extracts were first immunoprecipitated by monoclonal antibodies to GFP, and following gel electrophoresis and Western blotting Cdc25p-GFP was detected by a polyclonal anti-GFP antibody (Clontech). Uap56p was used as an experimental control. C, chromatin association of Cdc25p-GFP was analyzed in the absence of HO-endonuclease (control) and in the presence of HO-endonuclease (HO) as shown. Total cell lysates (T), total nuclei (TN), nuclear supernatant (NS), and chromatin-associated fractions (C) were analyzed by Western blotting using anti-GFP polyclonal antibody (Clontech). The nuclear protein Uap56p was used as an experimental control. D, asynchronized wild type cells expressing the integrated Cdc25p-GFP were treated without (−IR) or 300-gray γ ionizing radiation (+IR). The samples were taken at indicated time intervals (h). Total cell lysate and chromatin-associated fractions were analyzed by Western blot using anti-GFP polyclonal antibody. As a loading control, anti-α-tubulin was used against total cell lysates, and Coomassie staining for chromatin-associated fractions was performed to show the presence of proteins.
To further explore how Cdc25p could be recruited to the DSB site, Cdc25p-GFP recruitment was tested in a Δrad24 strain. There was no detectable recruitment of Cdc25p-GFP in a Δrad24 strain with or without HO-endonuclease induction, demonstrating that there was no significant enrichment of Cdc25p-GFP to the DSB site (Fig. 4A, panel b). Thus, Cdc25p recruitment depended on Rad24p.
The activation of Chk1p is known to be required for Cdc25p-Rad24p interaction. To test whether Cdc25p-GFP recruitment was dependent on Chk1p, a Δchk1 strain carrying the HO site along with a genomic copy of Cdc25p-GFP was constructed. As can be seen in Fig. 4A (panel c), in the Δchk1 strain, Cdc25p-GFP did not recruit to the DSB site.
Cdc25p protein levels can vary within the cell at different time points in the cell cycle. So we wanted to exclude the possibility that loss of recruitment of Cdc25p-GFP was due to low levels of protein in the cell. Thus, we tested the relative amount of Cdc25p-GFP present in the wild type strain (carrying the HO site and genomic tagged Cdc25p-GFP) and in isogenic Δrad24 and Δchk1 strains before and after HO-endonuclease induction (Fig. 4B). Compared with the uninduced condition (first, third, and fifth lanes), under the induced condition (second, fourth, and sixth lanes) for the three strains, there was a significant increase in Cdc25p-GFP level as measured by detection of immunoprecipitated protein. The increase in the amount of Cdc25p-GFP in Δrad24 and Δchk1 cells (fourth and sixth lanes) is expected and is consistent with data from other observations (21) and suggests that the lack of recruitment of Cdc25p-GFP was not due to a low amount of the protein in the tested strains. These results are consistent with the suggested functional role of Chk1p kinase in Cdc25p-Rad24p interaction.
We finally tested whether the recruitment of Cdc25p-GFP to the DSB site depended on the presence of Dss1p. There was no detectable increase in the recruitment of Cdc25p-GFP to the HO site in the presence of HO-endonuclease in Δdss1 cells (Fig. 4A, panel d). We compared the relative amounts of Cdc25p-GFP protein under the HO-endonuclease induced and uninduced conditions. As expected there was a slight increase in the level of detectable Cdc25p-GFP in the HO-endonuclease-induced cells (Fig. 4B, compare seventh and eighth lanes). These results suggest that the lack of increase in the recruitment of Cdc25p-GFP to the DSB site in Δdss1 cells under the conditions of DNA damage was not due to the lack of available protein. Taken together, the recruitment of Cdc25p-GFP to the DSB site was dependent on the presence of Dss1p.
Following DSB, Cdc25p in the Nucleus Is Found Chromatin-associated
We next wanted to determine what fraction of nuclear Cdc25p-GFP is chromatin-associated following the formation of DSBs. We used the wild type strain carrying the HO-endonuclease cleavage site expressing cdc25-gfp to test the relative amounts of Cdc25p-GFP in total lysate (in Fig. 4C, lanes T; see figure legend for experimental details), total nuclear lysate (TN), nuclear supernatant (NS), and chromatin-associated fraction (C) in the presence and absence of the HO-endonuclease (Fig. 4C). In the absence of the HO-endonuclease, the nuclear Cdc25p-GFP was found to be present in the nuclear supernatant but not associated with the chromatin (compare NS and C lanes in the Control panel). In contrast, in cells expressing the HO-endonuclease, Cdc25p-GFP was detected in the chromatin-associated fraction but not in the nuclear supernatant (see NS and C in the HO panel). We used Uap56p, a nuclear protein, as a control to exclude the possibility that the lack of Cdc25p-GFP in the nuclear supernatant was not due to the absence of proteins in the extract. Fig. 4C shows that a comparable amount of nuclear supernatant protein was used in control and HO-endonuclease induced cells. These results indicate that in cells with DSB, all of the detectable nuclear Cdc25p is chromatin-associated.
We next investigated whether Cdc25p was also chromatin-associated in DNA damaged cells using ionizing radiation. The association of Cdc25p-GFP with chromatin was analyzed in asynchronous wild type cells expressing Cdc25p-GFP with and without γ radiation (300 grays) treatment (Fig. 4D). Following γ radiation chromatin was purified at 0, 1, 2, 3, and 4 h for determining its association with Cdc25p-GFP. Cdc25p-GFP was found to be associated with chromatin by 1, 2, and 3 h with maximum association at 2 h after treatment (Fig. 4D). α-Tubulin was used as an internal control for the total amount of protein loaded, and Coomassie staining of the gel was performed to show that proteins were present in samples that were chromatin-purified (Fig. 4D). These results indicate that Cdc25p is chromatin-associated even when cells are damaged using ionizing radiation.
Rad24p Directly Binds to Rae1p and Dss1p
Based on the ChIP experiments described above, one way that Rad24p could be recruited onto the DSB site was by direct physical interaction with Dss1p. Using proteins expressed and purified from E. coli extracts, we tested direct interaction between Dss1p and Rad24p. Dss1p was able to bind GST-Rad24p fusion protein on beads but not GST alone (Fig. 5A, lane 4 versus lane 5).
FIGURE 5.

In vitro binding of GST-Rad24p to His-Dss1p and His-Rae1p. Proteins as indicated were made and purified from E. coli extracts. The presence or absence of a protein in the binding reaction is indicated by + or − on top of the respective lanes. Individual protein bands are shown. GST or GST-tagged proteins were immobilized on glutathione beads. A, binding of His-Dss1p by GST-Rad24p. Lanes 1–3 are input. Lane 4 is binding of His-Dss1p to GST-Rad24p on beads, and lane 5 is binding of His-Dss1p to GST alone. B, binding of His-Rae1p by GST-Rad24p. Lanes 1–3 are input. Lane 4 is binding of His-Rae1p with GST-Rad24p, and lane 5 is binding of His-Rae1p with GST. C, binding of His-Rhp51p by GST-Dss1p. Lanes 1–3 are input. Lane 4 is binding of His-Rhp51p with GST-Dss1p, and lane 5 is binding of His-Rhp51p with GST alone. 5-fold more His-Dss1p, His-Rae1p, and His-Rhp51p over input was used in the binding reactions in A, B, and C, respectively. The bound proteins were analyzed by SDS-PAGE and Coomassie staining.
The ChIP experiment also suggested that Rae1p recruitment was dependent on Rad24p. We therefore tested their interaction in vitro. GST-Rad24p, but not GST alone, was able to bind His-Rae1p (Fig. 5B, lane 4 versus lane 5). Because Rae1p recruitment onto DSBs depended on Rad24p (see previous section), these results clearly support the view that Rae1p-Rad24p interaction is essential for recruitment of Rae1p onto the DSB site, and Rad24p mediates Rae1p recruitment to DSB site. We previously showed direct physical interaction between Dss1p and Rae1p in a proposed messenger ribonucleoprotein complex critical for mRNA export (5). In contrast, here our results indicate that direct physical interaction with Dss1p does not determine recruitment of Rae1p.
Others have shown that homologs of Rhp51p associate with a Dss1p homolog indirectly within the recombination-repair complex (8, 9). Consistent with those studies, we detected no direct association between GST-Dss1p and His-Rhp51p (Fig. 5C, lane 4). Finally, direct physical interaction between Dss1p and Mts2p was not tested because their homologs are components of the 19 S proteasomal subcomplex and are known to interact with each other (12).
Dss1p Is Required for DNA Damage Checkpoint Function in S. pombe
We and others have shown that dss1 mutants are sensitive to DNA-damaging agents such as UV light (5, 12). We wanted to test whether Δdss1 mutant cells were capable of activating the DNA damage checkpoint in response to UV light. As a control we used Δrad24 and wild type cells. Using a lactose gradient, we synchronized wild type, Δrad24, and Δdss1 cells at G2 stage. The G2 synchronized cells were then exposed to varying doses of UV light, and the percentage of cells passing through mitosis over time at 27 °C was monitored and counted (Fig. 6A). Both Δdss1 and Δrad24 cells entered mitosis prematurely compared with the wild type cells. Strikingly, the timing of entry to mitosis by Δdss1 and Δrad24 cells was almost indistinguishable. One consequence of the Δdss1 cells entering mitosis prematurely was that the cells had abnormal morphologies (cut, anucleated, and multiseptated phenotypes) (Fig. 6B), similar to that seen for Δrad24 cells. These results indicate that Dss1p is required for monitoring DNA damage in the G2 stage of the cell cycle.
FIGURE 6.

Sensitivity to DNA-damaging agents and analysis of the checkpoint control of S. pombe wild type and mutant strains. A, analysis of DNA damage checkpoint control in wild type, Δrad24, and Δdss1 cells. G2 synchronized cells were treated with different doses of UV light at 27 °C, and the cells were scored as having passed through mitosis if the nuclei had divided or septum had formed (see “Experimental Procedures” for details). B, morphology of cells treated with 4,6-diamidino-2-phenylindole (DAPI) and calcofluor following treatment with UV light with 100 J/m2 when 20–25% of mutant cells had passed through mitosis. C, survival analysis at 21 °C of wild type, rae1-1, Δrad24, and rae1-1 Δrad24 treated with UV light, MMS, bleomycin, and HU as indicated (see “Experimental Procedures” for experimental details). D, analysis of checkpoint control in wild type, rae1-1, and rad1-1 cells. See “Experimental Procedures” for details. The cells were scored as having passed through mitosis if the nucleus had divided or a septum had formed. E, morphology of cells following irradiation with UV light. When 20–25% of cells had passed through mitosis following irradiation with 100 J/m2, the cells were fixed and stained with 4,6-diamidino-2-phenylindole and calcofluor. The morphology of wild type, rae1-1, and rad1-1 cells is shown.
rae1-1 Allele Is Sensitive to DNA-damaging Agents
Because Rae1p requires both Dss1p and Rad24p for recruitment to DSB site, we wanted to determine whether the rae1 gene has any role in DNA damage in S. pombe. Wild type, rae1-1, Δrad24, and rae1-1 Δrad24 double mutant cells were treated with DNA-damaging agents such as UV light, bleomycin, MMS, and HU. Fig. 6C shows that wild type cells were not sensitive to any of the DNA-damaging agents. In contrast and as expected, Δrad24 cells were sensitive to UV, bleomycin, and MMS but not to HU. Similarly, rae1-1 cells were sensitive to UV, MMS, and bleomycin and not to HU (Fig. 6C). The sensitivity of rae1-1 cells was intermediate between wild type and Δrad24 mutant cells. The sensitivity of the rae1-1 Δrad24 double mutant was similar to that of the more sensitive Δrad24 mutant cells, suggesting that in DNA damage, rae1-1 mutation is epistatic to Δrad24. Similar to Δrad24 mutant cells, rae1-1 and the rae1-1 Δrad24 double mutant also did not exhibit any sensitivity to HU, thus suggesting that Rae1p is unlikely to have a role in monitoring DNA damage during replication (Fig. 6C).
rae1-1 Mutation Shows Partial Loss of DNA Damage Checkpoint Function
Because the rad24 gene is known to be involved in monitoring DNA damage in G2, we tested whether rae1-1 cells exposed to DNA-damaging agents such as UV light could fully activate the DNA damage checkpoint. Wild type, rae1-1, and rad1-1 cells (rad1 is a known DNA damage checkpoint gene) were synchronized in G2 using a lactose gradient. These strains were exposed to varying doses of UV light, and the percentage of cells for each culture passing through mitosis over time was determined (Fig. 6D). In contrast to rad1-1 cells, which showed essentially no arrest following treatment with UV light, rae1-1 cells showed an initial checkpoint arrest but entered mitosis prematurely when compared with wild type cells. For example, following treatment with 150 J/m2 of UV light, at 21 °C rae1-1 mutant cells (these cells are temperature-sensitive at 25 °C) began to enter mitosis after a delay of 5 h as compared with 9 h for wild type cells (Fig. 6D).
The consequence of premature entry of rae1-1 cells into mitosis was the generation of cells with abnormal morphologies. At 6.5 h following exposure to 100 J/m2 of UV light, approximately 22% of the rae1-1 cells had passed through mitosis, whereas less than 1% of wild type had done so. Of these, ∼25% of rae1-1 cells and 40% of rad1-1 cells displayed either a cut or anucleated phenotype (Fig. 6E). These results suggest that rae1-1 cells pass through mitosis prior to completely repairing their DNA and that Rae1p has a role in monitoring DNA damage in G2.
DISCUSSION
In this work, we show that Dss1p has a DNA damage checkpoint function in S. pombe at the G2/M boundary and functions by recruiting the checkpoint protein Rad24p, cell cycle protein Cdc25p, and mRNA export/cell cycle factor Rae1p to DSB sites. To our knowledge, this is the first evidence of recruitment of these proteins to the damaged DNA sites, linking their functions in DNA damage and checkpoint via Dss1p. The similarity of timing and premature entry into mitosis by Δdss1 cells and Δrad24 cells suggests an epistatic relationship. This relationship is further revealed by direct physical interaction between Rad24p and Dss1p, placing them functionally together in DNA damage response. We further show that following DNA damage the nuclear Cdc25p is chromatin-associated. Finally, we propose that the recruitment of Cdc25p onto DSB site causes its intranuclear sequestration that could delay the timing of mitosis by arresting the cells at the G2/M boundary in response to DNA damage.
Dss1p Is Critical for Linking DNA Repair and Checkpoint Functions in S. pombe Cells
ChIP experiments shed new light into how Dss1p functions in S. pombe. In this view, Dss1p is a key physical link between two functional complexes: one relating to DNA repair-recombination, and the other relating to the DNA damage checkpoint.
First, our results confirm observations made in other eukaryotic organisms about the physical association of Dss1p to DSB sites (8, 12). In addition, our data physically connect the recruitment of Rhp51p and Mts2p to DSB site through Dss1p.
Second, Dss1p is also essential for the recruitment of two additional proteins Rad24p and Rae1p. By examining the patterns of recruitment of Rad24p and Rae1p in wild type and Δdss1 backgrounds, a scheme of their recruitment on the DSB emerged. A simple sequential recruitment scheme (Dss1p < Rad24p < Rae1p) could explain the observed pattern. Proteins purified in E. coli allowed us to confirm direct physical interactions among Dss1p, Rad24p, and Rae1p. Consistently, we found evidence of direct physical interaction between Rad24p and Dss1p and between Rad24p and Rae1p. We could not directly investigate the formation of a ternary complex of Dss1p-Rad24p-Rae1p, because Dss1p and Rae1p also interact in vitro directly (5).
Recruitment of Rad24p to the DSB site raised the intriguing possibility of whether Cdc25p was also recruited by its interaction with Rad24p under the conditions of DNA damage. Indeed, a clear enrichment of Cdc25p following the pattern of Rad24p was discernible in the ChIP experiments. Cdc25p recruitment was also dependent on Dss1p. These results are consistent with Dss1p-mediated recruitment of Cdc25p via Rad24p into a single complex analogous to the Dss1p-Rad24p-Rae1p complex suggested above. Indeed, it is not unreasonable to speculate that a four protein complex of Dss1p, Rad24p, Rae1p, and Cdc25p may exist in vivo on or near DNA DSB sites.
Mechanism of Checkpoint Control in S. pombe Cells
In S. pombe, the Rad24p-Cdc25p interaction is well characterized in mediating G2/M checkpoint control. The function of Rad24p is thought to be mediated by its binding to phosphorylated Cdc25p (47). Also, the binding of Rad24p to Cdc25p is preceded by activation and phosphorylation of Cdc25p by Chk1p kinase or independently by Mek1p (20, 47). Cdc25p dephosphorylates Cdc2p (47). It is thought that to establish G2/M checkpoint control, Rad24p-Cdc25p complex exits the nucleus (nuclear exclusion), thereby physically removing the enzyme (Cdc25p) from its substrate (Cdc2p). However, recently it has also been suggested that nuclear exclusion of Cdc25p may not be essential for cell cycle arrest in response to DNA damage (22).
Our results here support a new mechanistic scenario. In this scenario, DNA DSB formation signals activation of Chk1p kinase. At the DSB site, DNA repair factors including Dss1p are recruited (Fig. 7). Rad24p and Cdc25p (phosphorylated by activated Chk1p kinase) are recruited concurrently or sequentially to the break site via Dss1p. Rae1p is also likely recruited to the same site, dependent on Rad24p and Dss1p. The recruitment of Cdc25p physically separates it from its target Cdc2p and thus prevents it from activating Cdc2p. In the absence of active Cdc2p, the cells are prevented from entering mitosis. Once the DNA is repaired, the protein complexes are likely disassembled, releasing Cdc25p into the nucleoplasm to interact with and activate Cdc2p. This in turn allows the cells to enter mitosis.
FIGURE 7.
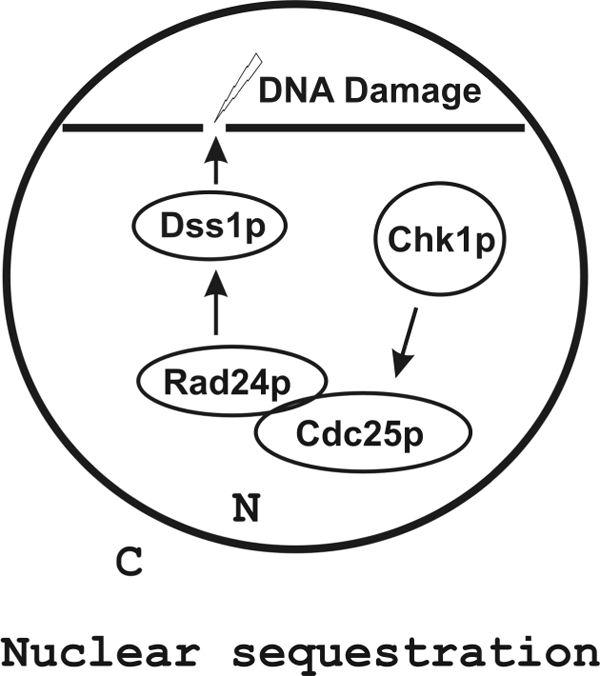
Nuclear sequestration model of Cdc25p in response to DNA damage. DNA damage leads to activation of Chk1p kinase that phosphorylates Cdc25p. Phosphorylated Cdc25p binds to Rad24p within the nucleoplasm (as shown in the schematic diagram) or after prebinding to DNA via Dss1p. In either case, Cdc25p is sequestered from its target Cdc2p.
What Is the Role of Rae1p in Checkpoint Control in S. pombe?
Several experiments now link Rae1p to the S. pombe cell cycle progression. First, rae1-1 temperature sensitive mutant at the restrictive temperature causes cells to arrest at G2/M boundary (25, 37). Sensitivity of rae1-1 cells to DNA-damaging agents shown here is consistent with a functional role in monitoring DNA damage and/or cell cycle. Interestingly, another mutant of rae1 (rae1-167) was not sensitive to any DNA-damaging agent (data not shown), suggesting allele specificity of the function of Rae1p in DNA damage. Second, we have previously shown the role of Rae1p in mRNA export within a protein complex comprised of mRNA export factors Uap56p, Dss1p, and Mlo3p. Most of the S. pombe Rae1p is localized to the nuclear pore in association with nucleoporin Nup98 (25). Our hypothesis is that a Rae1p-containing mRNA export complex functions at the nuclear pore. In contrast, the recruitment of Rae1p to the DSB site described in this paper is reflective of a separate protein complex with components that are not known to be linked to mRNA export. In particular, neither Nup98 nor Nup159 associated with the Rad24p-purified complex that pulled down Rae1p. (Rae1p is stably bound to Nup98 in mRNA export complexes, and these proteins co-purify in vivo/in vitro (48)). Third, the direct physical interaction between Rae1p and Rad24p in vitro and Rad24p-dependent recruitment of Rae1p in vivo suggest a molecular basis for Rae1p function through Rad24p in checkpoint control. Finally, for establishing cell cycle delay, Cdc25p function must be inhibited so that it does not activate Cdc2p. The timing of the function of Rae1p in the cell cycle also places it prior to Cdc2p activation (23, 25). Based on the work described here, the key future question regarding the role of Rae1p in checkpoint control is whether its function affects Cdc2p directly or indirectly (i.e. via Cdc25p) or via another target protein.
Supplementary Material
Acknowledgments
We thank Dr. Paul Russell, Dr. Paul G. Young, and Dr. Kathleen L. Gould for providing strains and plasmids, lab members for helpful discussions, and Dr. Joseph Landry and National Institutes of Health Fellows Editorial Board members for reading and correcting the manuscript.
This work was supported, in whole or in part, by National Institutes of Health NCI intramural program.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1S and Table 1S.
- DSB
- double-strand break
- TAP
- tandem affinity purification
- GST
- glutathione S-transferase
- MMS
- methyl methanesulfonate
- HU
- hydroxyurea
- GFP
- green fluorescent protein
- ChIP
- chromatin immunoprecipitation
- mRNA
- messenger RNA
- HO
- homothallic switching.
REFERENCES
- 1.Bucher N., Britten C. D. (2008) Br. J. Cancer 98, 523–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., Linn S. (2004) Annu. Rev. Biochem. 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 3.Berry L. D., Gould K. L. (1996) Prog. Cell Cycle Res. 2, 99–105 [DOI] [PubMed] [Google Scholar]
- 4.Mannen T., Andoh T., Tani T. (2008) Biochem. Biophys. Res. Commun. 365, 664–671 [DOI] [PubMed] [Google Scholar]
- 5.Thakurta A. G., Gopal G., Yoon J. H., Kozak L., Dhar R. (2005) EMBO J. 24, 2512–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H., Jeffrey P. D., Miller J., Kinnucan E., Sun Y., Thoma N. H., Zheng N., Chen P. L., Lee W. H., Pavletich N. P. (2002) Science 297, 1837–1848 [DOI] [PubMed] [Google Scholar]
- 7.Zhou Q., Kojic M., Cao Z., Lisby M., Mazloum N. A., Holloman W. K. (2007) Mol. Cell. Biol. 27, 2512–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudmundsdottir K., Lord C. J., Witt E., Tutt A. N., Ashworth A. (2004) EMBO Rep. 5, 989–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kojic M., Yang H., Kostrub C. F., Pavletich N. P., Holloman W. K. (2003) Mol. Cell 12, 1043–1049 [DOI] [PubMed] [Google Scholar]
- 10.Jang Y. K., Jin Y. H., Shim Y. S., Kim M. J., Yoo E. J., Choi I. S., Lee J. S., Seong R. H., Hong S. H., Park S. D. (1996) Mol. Gen. Genet. 251, 167–175 [DOI] [PubMed] [Google Scholar]
- 11.Muris D. F., Vreeken K., Carr A. M., Broughton B. C., Lehmann A. R., Lohman P. H., Pastink A. (1993) Nucleic Acids Res. 21, 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krogan N. J., Lam M. H., Fillingham J., Keogh M. C., Gebbia M., Li J., Datta N., Cagney G., Buratowski S., Emili A., Greenblatt J. F. (2004) Mol. Cell 16, 1027–1034 [DOI] [PubMed] [Google Scholar]
- 13.Funakoshi M., Li X., Velichutina I., Hochstrasser M., Kobayashi H. (2004) J. Cell Sci. 117, 6447–6454 [DOI] [PubMed] [Google Scholar]
- 14.Jossé L., Harley M. E., Pires I. M., Hughes D. A. (2006) Biochem. J. 393, 303–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruckmann A., Steensma H. Y., Teixeira De Mattos M. J., Van Heusden G. P. (2004) Biochem. J. 382, 867–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hermeking H., Benzinger A. (2006) Semin. Cancer Biol. 16, 183–192 [DOI] [PubMed] [Google Scholar]
- 17.van Heusden G. P. (2005) IUBMB Life 57, 623–629 [DOI] [PubMed] [Google Scholar]
- 18.van Heusden G. P., Steensma H. Y. (2006) Yeast 23, 159–171 [DOI] [PubMed] [Google Scholar]
- 19.Ford J. C., al-Khodairy F., Fotou E., Sheldrick K. S., Griffiths D. J., Carr A. M. (1994) Science 265, 533–535 [DOI] [PubMed] [Google Scholar]
- 20.Pérez-Hidalgo L., Moreno S., San-Segundo P. A. (2008) Cell Cycle 7, 3720–3730 [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Girona A., Furnari B., Mondesert O., Russell P. (1999) Nature 397, 172–175 [DOI] [PubMed] [Google Scholar]
- 22.Lopez-Girona A., Kanoh J., Russell P. (2001) Curr. Biol. 11, 50–54 [DOI] [PubMed] [Google Scholar]
- 23.Brown J. A., Bharathi A., Ghosh A., Whalen W., Fitzgerald E., Dhar R. (1995) J. Biol. Chem. 270, 7411–7419 [DOI] [PubMed] [Google Scholar]
- 24.Thakurta A. G., Selvanathan S. P., Patterson A. D., Gopal G., Dhar R. (2007) J. Biol. Chem. 282, 17507–17516 [DOI] [PubMed] [Google Scholar]
- 25.Whalen W. A., Bharathi A., Danielewicz D., Dhar R. (1997) Yeast 13, 1167–1179 [DOI] [PubMed] [Google Scholar]
- 26.Blower M. D., Nachury M., Heald R., Weis K. (2005) Cell 121, 223–234 [DOI] [PubMed] [Google Scholar]
- 27.Jeganathan K. B., Malureanu L., van Deursen J. M. (2005) Nature 438, 1036–1039 [DOI] [PubMed] [Google Scholar]
- 28.Wong R. W., Blobel G., Coutavas E. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 19783–19787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du L. L., Nakamura T. M., Moser B. A., Russell P. (2003) Mol. Cell. Biol. 23, 6150–6158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alfa C., Fantes P., Hyams J., Mcleod M., Warbrick E. (1993) Experiments with Fission Yeast, Cold Spring Harbor Press, Cold Spring Harbor, NY [Google Scholar]
- 31.Moreno S., Klar A., Nurse P. (1991) Methods Enzymol. 194, 795–823 [DOI] [PubMed] [Google Scholar]
- 32.Bähler J., Wu J. Q., Longtine M. S., Shah N. G., McKenzie A., 3rd, Steever A. B., Wach A., Philippsen P., Pringle J. R. (1998) Yeast 14, 943–951 [DOI] [PubMed] [Google Scholar]
- 33.Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. (1999) Nat. Biotechnol. 17, 1030–1032 [DOI] [PubMed] [Google Scholar]
- 34.Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B. (2001) Methods 24, 218–229 [DOI] [PubMed] [Google Scholar]
- 35.Ilani T., Khanna C., Zhou M., Veenstra T. D., Bretscher A. (2007) J. Cell Biol. 179, 733–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simkus C., Bhattacharyya A., Zhou M., Veenstra T. D., Jones J. M. (2009) Immunology 128, 206–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bharathi A., Ghosh A., Whalen W. A., Yoon J. H., Pu R., Dasso M., Dhar R. (1997) Gene 198, 251–258 [DOI] [PubMed] [Google Scholar]
- 38.Yoon J. H., Love D. C., Guhathakurta A., Hanover J. A., Dhar R. (2000) Mol. Cell. Biol. 20, 8767–8782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noma K., Allis C. D., Grewal S. I. (2001) Science 293, 1150–1155 [DOI] [PubMed] [Google Scholar]
- 40.Partridge J. F., Borgstrøm B., Allshire R. C. (2000) Genes Dev. 14, 783–791 [PMC free article] [PubMed] [Google Scholar]
- 41.Lygerou Z., Nurse P. (1999) J. Cell Sci. 112, 3703–3712 [DOI] [PubMed] [Google Scholar]
- 42.Nishitani H., Lygerou Z., Nishimoto T., Nurse P. (2000) Nature 404, 625–628 [DOI] [PubMed] [Google Scholar]
- 43.Enoch T., Carr A. M., Nurse P. (1992) Genes Dev. 6, 2035–2046 [DOI] [PubMed] [Google Scholar]
- 44.Morishita T., Tsutsui Y., Iwasaki H., Shinagawa H. (2002) Mol. Cell. Biol. 22, 3537–3548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhind N., Russell P. (2001) Mol. Cell. Biol. 21, 1499–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barbet N. C., Carr A. M. (1993) Nature 364, 824–827 [DOI] [PubMed] [Google Scholar]
- 47.Russell P. (1998) Trends Biochem. Sci. 23, 399–402 [DOI] [PubMed] [Google Scholar]
- 48.Blevins M. B., Smith A. M., Phillips E. M., Powers M. A. (2003) J. Biol. Chem. 278, 20979–20988 [DOI] [PubMed] [Google Scholar]
- 49.Rowley R., Subramani S., Young P. G. (1992) EMBO J. 11, 1335–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chua G., Lingner C., Frazer C., Young P. G. (2002) Genetics 162, 689–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roguev A., Shevchenko A., Schaft D., Thomas H., Stewart A. F., Shevchenko A. (2004) Mol. Cell Proteomics 3, 125–132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.