

Abstract
Defects of the mitochondrial K+/H+ exchanger (KHE) result in increased matrix K+ content, swelling, and autophagic decay of the organelle. We have previously identified the yeast Mdm38 and its human homologue LETM1, the candidate gene for seizures in Wolf-Hirschhorn syndrome, as essential components of the KHE. In a genome-wide screen for multicopy suppressors of the pet− (reduced growth on nonfermentable substrate) phenotype of mdm38Δ mutants, we now characterized the mitochondrial carriers PIC2 and MRS3 as moderate suppressors and MRS7 and YDL183c as strong suppressors. Like Mdm38p, Mrs7p and Ydl183cp are mitochondrial inner membrane proteins and constituents of ∼500-kDa protein complexes. Triple mutant strains (mdm38Δ mrs7Δ ydl183cΔ) exhibit a remarkably stronger pet− phenotype than mdm38Δ and a general growth reduction. They totally lack KHE activity, show a dramatic drop of mitochondrial membrane potential, and heavy fragmentation of mitochondria and vacuoles. Nigericin, an ionophore with KHE activity, fully restores growth of the triple mutant, indicating that loss of KHE activity is the underlying cause of its phenotype. Mdm38p or overexpression of Mrs7p, Ydl183cp, or LETM1 in the triple mutant rescues growth and KHE activity. A LETM1 human homologue, HCCR-1/LETMD1, described as an oncogene, partially suppresses the yeast triple mutant phenotype. Based on these results, we propose that Ydl183p and the Mdm38p homologues Mrs7p, LETM1, and HCCR-1 are involved in the formation of an active KHE system.
Keywords: Mitochondria, Protein Purification, Protein-Protein Interactions, Subcellular Organelles, Yeast, K+/H+ Exchange, Morphology, Nigericin, Protein Complex, Volume Homeostasis
Introduction
The high, inside negative membrane potential (Δψ) of mitochondria favors uptake of cations through the inner mitochondrial membrane. Potassium is an osmotically active ion and the most abundant cation in the cytosol and in the mitochondrial matrix. The uncontrolled influx of K+ into mitochondria causes an increase of osmotic pressure of the organelles and their swelling. The presence of K+/H+ exchangers in mitochondria which, driven by the inside-directed pH gradient, extrude excess K+ from mitochondria was already postulated in the 1960s by Mitchell (1). Although the KHE4 has been studied extensively by physiological methods, its molecular identity remained obscure. Recently, our studies identified Mdm38/LETM1 as major players of this extrusion system (2–4).
Phenotypic analyses of mdm38Δ are consistent with the loss of KHE activity (4). These included increased matrix K+ content, swelling, and fragmentation of mitochondria, reduced mitochondrial Δψ, as well as reduced growth of cells on nonfermentable substrate. Further tests involving submitochondrial inner membrane particles (SMPs) confirmed the near total lack of KHE activity (2). Addition of the synthetic KHE nigericin to mdm38Δ cells restored all mitochondrial functions, including growth on nonfermentable substrates, Δψ, morphology, and KHE activity (4, 5). This result strongly supported the conclusion that Mdm38 acts as an essential regulator or subunit of the mitochondrial KHE, because it is unlikely that a protein with only one transmembrane domain like Mdm38 forms the KHE itself.
Mdm38p is conserved in all eukaryotic organisms. The human homologue, LETM1, has been implicated in the Wolf-Hirschhorn syndrome (6). The yeast Saccharomyces cerevisiae encodes a homologue, YPR125w. YPR125w had initially been identified as a multicopy suppressor of mutants lacking the mitochondrial Mg2+ transporter MRS2 and was named MRS7 (7). YPR125w/MRS7, also named YLH47 for yeast LETM1 homologue of 47 kDa (6), encodes a protein located in mitochondria (4, 8). Although disruption of MRS7 has a weak phenotype, its overexpression restores growth of mdm38Δ strains, showing a functional homology to Mdm38p (4). The human genome also encodes a second member of the Mdm38/LETM1 family, named HCCR-1 or LETMD1, which was found to be overexpressed in various human cancer cells (9).
Here, we characterize the role of four yeast multicopy suppressors of mdm38Δ as well as of LETM1 and HCCR-1 with respect to their potential to restore K+/H+ exchange activity. We find that like Mdm38p, Mrs7p and Ydl183cp are part of a large mitochondrial KHE protein complex. We discuss its putative composition and analyze the additive effects resulting from the triple deletion of MDM38, MRS7, and YDL183c.
EXPERIMENTAL PROCEDURES
Yeast Strains and Growth Media
The S. cerevisiae strains W303 (ATCC accession number 2012239) and DBY747 (ATCC accession number 204659) were used as wild type. W303 mdm38::HIS3 termed mdm38Δ was described previously (4). W303 cells were grown in YPD (yeast extract, bacto peptone, 2% dextrose), YPG (2% glycerol) or YPGal (yeast extract, bacto peptone, 2% galactose) media as indicated. YPG plates were supplemented with 2 μm nigericin when indicated. Synthetic minimal media (S-Gal, synthetic medium containing 2% galactose, or SD, synthetic medium with 2% dextrose, 2% glucose) were supplemented with amino acids and bases when appropriate.
Genomically tagged versions of MDM38 and MRS7 were constructed by homologous recombination. The TAP tag and the selection marker TRP1-KL were amplified by PCR from the vector pBS1479 (10). The following primers were used to create a C-terminally tagged version of MDM38 with His6 and the TAP tag consisting of two immunoglobulin binding domains of protein A and the calmodulin-binding peptide: MDM38HisTAPfw, 5′-TACCTCCCATTCCGGCCGATCAAGCTGCGAAGACTTTTGTCATTAAGAAAGATCATCACCATCACCATCACTCCATGGAAAAGAGAAG-3′; MDM38HisTAPrev, 5′-CCTGATGTACTCACATTTCCATCTGGTGAGGATGGAGGTGGAGACGTCGTAGACATGGAACCCTGTTTACGACTCACTATAGGG-3′. For tagging MRS7 with His6 and TAP tag, the following primers were used: MRS7HisTAPfw, 5′-AACCGCATGACACCAAGCCTATCGGAGAAGCCGCTGCCATCAAAGAGAAGCATCACCATCACCATCACTCCATGGAAAAGAGAAG-3′; MRS7HisTAPrev, 5′-TAGACACTCTATTCTTTGAGTAATTTTGAGGGAGAGCAGCAATGATTAACTACGACTCACTATAGGG-3′.
To create chromosomal, C-terminally His6-tagged versions of MDM38 and MRS7, the following forward primers were used: MDM38Hisfw, 5′-TTCCGGCCGATCAAGCTGCGAAGACTTTTGTCATTAAGAAAGATCATCACCATCACCATCACTGATCCATGGAAAAGAGAAG-3′; MRS7Hisfw, 5′-CGCATGACACCAAGCCTATCGGAGAAGCCGCTGCCATCAAAGAGAAGCATCACCATCACCATCACTAATCCATGGAAAAGAGAAG-3′. MDM38HisTAPrev and MRS7HisTAPrev served as reverse primers, respectively. To create chromosomal, C-terminally One-STrEP (11)-tagged versions of MDM38 and MRS7, the One-STrEP sequence (based on the plasmid pEXPR-IBA103, IBA BioTAGnology) was synthesized (Eurofins MWG GmbH) and cloned into the BamHI-linearized pBS1479 plasmid. Chromosomal integration was performed using the forward primers: MDM38OneSTrEPfw, 5′-CATTCCGGCCGATCAAGCTGCGAAGACTTTTGTCATTAAGAAAGATGAGAATTTGTATTTTCAGG-3′, and MRS7OneSTrEPfw 5′-GCATGACACCAAGCCTATCGGAGAAGCCGCTGCCATCAAAGAGAAGGAGAATTTGTATTTTCAGG-3′. MDM38HisTAPrev and MRS7HisTAPrev served as reverse primers, respectively.
Multicopy Suppressor Screen
The mdm38Δ mutant strain was transformed with 1 μg of genomic library (constructed in YEp181, a 2-μm plasmid marked with LEU2, gift of Juraj Gregan and Kim Nasmith, IMP, Vienna, Austria). Transformants growing on SD−leu plates were replica-plated on YPG plates and incubated at 37 °C. Three hundred ninety six positive putative candidates were selected and classified into strong or weak suppressors. To confirm that the suppression was plasmid-borne, the plasmids were recovered, amplified in Escherichia coli, used for retransformation of W303 mdm38Δ, and tested for growth on YPG plates at 37 °C. Confirmed plasmids were then analyzed by restriction digestion patterns to eliminate self-complementation, and the inserts of selected plasmids were sequenced by VBC-Biotech Services GmbH. The suppressor plasmids contained multiple ORFs. Individual ORFs were subcloned and tested for their ability to suppress the growth phenotype. The individual plasmids containing PIC2, MRS3, MRS7, or YDL183c were used in all experiments if not otherwise indicated.
Gene Deletions
Deletion of the genes was performed according to the one-step replacement protocol (12). The MRS7 ORF was disrupted from the start to the stop codon by replacement with the KANMX4 disruption cassette, which was amplified with the primers 5′-TAGGTTCGAGTAAAGAAAATTTCATAAAGAAATCAACAAGACACACGTACGCTGCAGCTCGAC-3′ and 5′-GCGGAGAGTGTATCGTGCGGTTTAATGGGCCAGGTGAAAACTGGGATCGATGAATTCGAGCTCG-3′. To delete YDL183c in W303, the whole ORF was replaced with a URA3 disruption cassette flanked by loxP sites, using the primers 5′-CATCGATAGAATCATTTTATCACAATACCAAAACTT-3′ and 5′-CTCAGGAATACCTGTTATGTATATTTACATGAGATA-3′. Following verification of the correct gene replacement using analytical PCR (12), the selection marker was removed with the CRE recombinase containing vector pSH63 (13). YDL183c deletion in DBY747 was performed by replacement with the LEU2 disruption cassette, using the primers 5′-TCACAATACCAAAACTTCATCCGGTGTATTTTAGATTAAAGCGTACGCTGCAGGTCGAC-3′ and 5′-ACCTGTTATGTATATTTACATGAGATAGTGGACAATCTACATAGGCCACTAGTGGATCTG-3′. Double and triple deletion strains were obtained in W303 by crossing and sporulation of the diploids or in DBY747 by stepwise disruptions.
Plasmid Constructs
To provide MRS7, MDM38, and YDL183c with a C-terminal GFP tag, the entire respective ORFs were cloned into the centromeric vector pUG35 (14). MRS7 coding sequence was amplified by PCR from YEp351-MRS7 plasmid (7) with the 5′ primer 5′-ACAAGAATTCATGCTGAAATACAGGTC-3′ and the 3′ primer 5′-ACATGTCGACCTTCTCTTTGATGGC-3′ (EcoRI and SalI sites are underlined) and cloned into the EcoRI/SalI sites of the plasmid pUG35 carrying the methionine promoter. To clone YDL183c into the pUG35 plasmid, the entire ORF sequence was amplified by PCR from W303 genomic DNA by use of 5′ primer 5′-CGGGATCCATGATACGTTCAATATTTATACCGC-3′ and 3′ primer 5′-GCGTCGACAATTTTGTTTTTCTCTTGAGATTTCG-3′ introducing the underlined BamHI and SalI restriction sites. The amplified fragment was cloned into the BamHI- and SalI-linearized pUG35 plasmid. A C-terminally GFP-tagged version of the entire ORF MDM38 was obtained by cloning MDM38 in pUG35 by use of the forward primer 5′-TAATATGGATCCATGTTGAATTTCGCATCAAGAGCG-3′ and the reverse primer 5′-AATATCTATCGATCTTAATGACAAAAGTCTTCGC-3′ (BamHI and ClaI sites underlined).
To express YDL183c from its own promoter and in fusion with the triple HA epitope at the C-terminal end, the entire ORF and its flanking region, including 217 nucleotides upstream of the ATG, were amplified from W303 genomic DNA with the primers 5′-CTTGAGCTCGGATGGATGGACTTGACGGC-3′ and 5′-GCGTCGACCAATTTGTTTTTCTCTTGAGATTTCC-3′ and inserted in the SacI/SalI sites of the YCp33-HA vector. The vectors YCp33-MDM38-HA and Yep351-MDM38-HA expressing Mdm38p under the control of its native promoter and pVTU103-LETM1-HA expressing the human LETM1 from the ADH promoter were described previously (4). To express HCCR-1 in yeast, the entire ORF was amplified from MGC IRAT human (Invitrogen 6009854) with the forward primer 5′-AACGGGATCCCGGATGGCGCTCTCCAGGGTGTG-3′ and the reverse primer 5′-CATGCTCGAGTCAGTGGTGGTGGTGGTGGTGGCGCCTTGTCCCAAGGTAGT-3′ digested with BamHI and XhoI and inserted in the pVTU103 vector.
Isolation and Subfractionation of Mitochondria
Yeast mitochondria used for ion-flux measurements were isolated from cells growing overnight to stationary phase. For all other experiments, cells were grown to A600 = 1. Mitochondrial isolation and mitoplast preparation were done as described previously (15). Protein extraction with sodium carbonate was performed according to Ref. 16 followed by protein precipitation with trichloroacetic acid and Western blotting analysis. Proteinase K protection experiments were performed as described previously (17). Resuspended mitoplasts were incubated in the presence or absence of proteinase K as indicated for 20 min, and the proteinase K reaction was inactivated with 1 μm phenylmethylsulfonyl fluoride, and the proteins were trichloroacetic acid-precipitated. 50 μg of protein were loaded in each lane of a 12.5% SDS-PAGE, transferred onto polyvinylidene difluoride membrane, and immunoblotted in Tris-buffered saline/Tween plus 2.5% dry milk with the antibodies against the following: HA (laboratory stock; hexokinase-1 (Biotrend); F1β, Tim44, and Yme1 (generous gifts of Gottfried Schatz, Hans van der Spek, and Tom Fox, respectively). The proteins were visualized by use of the SuperSignalTM West Pico system (Pierce).
Blue Native PAGE
Proteins of isolated mitochondria were solubilized as indicated with 1.2% n-dodecyl-d-maltoside or Triton X-100, and after a clarifying spin, 200 μg of proteins (25 μl) per lane were separated by BN-PAGE according to Ref. 18 on 5–18% polyacrylamide gradient gels. Following electrophoresis, wet blotting to polyvinylidene difluoride membrane was performed for 1 h at 100 V. Protein complexes were detected by immune decoration. The calibration standards (Amersham Biosciences) used in the BN-PAGE were bovine thyroglobulin (669 kDa), horse spleen apoferritin (440 kDa), bovine liver catalase (232 kDa), bovine heart lactate dehydrogenase (140 kDa), and bovine serum albumin monomer (67 kDa).
Affinity Chromatography
180 mg of isolated mitochondria were used for standard protein purification. Isolated mitochondria were adjusted to a concentration of 20 mg/ml. For affinity chromatography using Ni-NTA Superflow resin (Qiagen), mitochondria were solubilized with 1.2% Triton X-100 on ice for 30 min in Hi 50 buffer (10 mm Tris-HCl, pH 7.8, 50 mm NaCl, 20 mm imidazole, protease inhibitor mixture (Complete Mini, Roche Applied Science)). After centrifugation at 43,000 × g for 30 min (4 °C) to remove nonsolubilized mitochondrial debris, the Triton concentration of the supernatant was reduced to 1% by addition of Hi 50 buffer. Ni-NTA Superflow resin (Qiagen) was washed two times with 10 ml of Hi 50 buffer containing 1% Triton X-100. The clarified supernatant was incubated with the resin for 30 min under gentle shaking and loaded on a Poly-Prep chromatography column (Bio-Rad). The column was washed twice with 15 ml of Hi 50 Wash buffer 1 (0.8% Triton X-100, 20 mm imidazole) and twice with 15 ml of Hi 50 Wash buffer 2 (0.6% Triton X-100, 30 mm imidazole). Finally, bound proteins were eluted with Hi 50 Elution buffer (10 mm Tris-HCl, pH 7.8, 50 mm NaCl, 200 mm imidazole, 0.6% Triton X-100 and, unless otherwise stated, complete protease inhibitor mixture).
For streptavidin affinity chromatography using Strep-Tactin Superflow (Qiagen), mitochondria were solubilized with 1.2% Triton X-100 in Strep 50 buffer (100 mm Tris-HCl, pH 7.8, 50 mm NaCl, protease inhibitor mixture). All other steps were performed as described for the Ni-NTA chromatography except that Strep-Tactin Superflow (Qiagen) was used as affinity resin, and bound proteins were eluted with Strep 50 buffer with a final concentration of 2.5 mm d-desthiobiotin.
All purification steps were performed at 4 °C. Control experiments were performed for each affinity chromatography with untagged or untransformed DBY747 wild-type or mdm38Δ strains. Two-dimensional gel electrophoresis was performed as described previously (19). Anti-His antibody was purchased from Qiagen, and anti-Mdm38 was generously provided by P. Rehling.
Coimmunoprecipitation (CoIP)
UltraLink immobilized protein A, covalently bound to HA antiserum with the cross-linker, was kindly provided by A. Pichler. Isolated mitochondria (2 mg of protein) expressing YEp-MDM38-HA (70 kDa) and either pUG-MDM38-GPF (92 kDa), YDL183-GFP (64 kDa), or AIF-GFP (68 kDa) were solubilized for 30 min in RIPA buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS containing 1.2% n-dodecyl-d-maltoside (Sigma)) and protease inhibitor mixture (complete Mini, Roche Applied Science) plus 1 mm phenylmethylsulfonyl fluoride and, after a clarifying spin, incubated under rotation for 1 h with 10 μl of the HA-coupled beads, washed four times in RIPA buffer, and eluted in Laemmli buffer. Proteins were separated on 12.5% SDS-PAGE, transferred, and analyzed by immunoblotting with GFP (Roche Applied Science) and HA (laboratory stock) antibodies.
Measurements of the Mitochondrial Membrane Potential
The membrane potential of isolated mitochondria was recorded in an LS 55 fluorescence spectrometer (PerkinElmer Life Sciences) by monitoring the fluorescence of 5,5′,6,6′-tetrachloro-1,1,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (Molecular Probes) according to Ref. 20. Measurements were carried out in breaking buffer (0.6 m sorbitol, 10 mm Tris-HCl, pH 7.4). For calibration, aliquots of the same preparation were hyperpolarized with 1 μm nigericin (Sigma) and depolarized with 1 μm carbonyl cyanide p-trifluoromethoxyphenylhydrazone (Sigma) in breaking buffer. The reading after addition of nigericin was set as 100% and that after carbonyl cyanide p-trifluoromethoxyphenylhydrazone as 0%. The values were linearized, and the relative membrane potential was calculated using y = kx + d.
K+/H+ Exchange Measurements in SMPs
Preparations of SMPs and loading with the K+- and H+-sensitive fluorescent dyes potassium-binding benzofuran isophthalate (PBFI) and BCECF (both Invitrogen) were made as described previously (2). SMPs were treated with 1 μm antimycin A and 1 μm oligomycin prior to measurements. To determine the kinetics of K+ and H+ transport across the membrane, 150 mm KCl was added to the SMPs. When indicated, incubation of SMPs with 10 μm nigericin (Sigma) was done at room temperature for 5 min before the measurements. All measurements were repeated at least three times with different preparations of SMPs.
Confocal Microscopy
The plasmids pHS72 (TOM72-YFP) (21) and pYX232-mtGFP (22) were gifts from H. Sesaki and B. Westermann, respectively, and served to label mitochondria. Alternatively, mitochondria were labeled with Mitotracker Red (100 nm). Vacuoles were stained with FM4-64 in a final concentration of 10 μm (Molecular Probes). Microscopy settings were used as described previously (5).
Electron Microscopy
Cells were harvested at logarithmic growth phase (A600 = 1). Cryofixation, freeze substitution, thin sectioning, and image acquisition were performed as described previously (5).
RESULTS
PIC2, MRS3, MRS7, and a Novel Gene YDL183c Act as Multicopy Suppressors of mdm38Δ
Absence of Mdm38p in yeast cells (mdm38Δ mutants) results in reduced growth on nonfermentable substrate (pet− phenotype) (4). According to our previous data, the antibiotic nigericin, a KHE ionophore, acts as a multivalent suppressor of the mdm38Δ deletion phenotype. It restores cell growth on nonfermentable substrate, KHE activity, and Δψ and reverts matrix swelling and fragmentation of mitochondria (5).
To identify proteins substituting similarly for the function of MDM38, we have screened a yeast genomic library and selected suppressor genes that, being overexpressed, restored growth of mdm38Δ on nonfermentable substrate (Fig. 1). Among the suppressors, we found three previously described genes, PIC2, MRS3, and MRS7, encoding mitochondrial proteins. PIC2 and MRS3 encode mitochondrial carrier proteins involved in Pi transport and Fe2+ accumulation, respectively (23, 24). MRS7 encodes a functional homologue of Mdm38p, located in the inner membrane of mitochondria, with a weak deletion phenotype depending on the strain (7, 8). Additionally, we found one not yet characterized gene, YDL183c.
FIGURE 1.

Multicopy suppressors and their growth effects on mdm38Δ. Effects of YDL183c, PIC2, MRS3, and MRS7 on the nonfermentative growth of mdm38Δ cells are shown. W303 mdm38Δ mutant cells containing an empty vector or a vector overexpressing YDL183c, PIC2, MRS3, or MRS7 and wild-type (WT) cells were spotted onto YPD or YPG plates and grown at 28 °C for 3 or 5 days, respectively.
Up-regulation of Pic2, Mrs3, Mrs7, Ydl183c, or Human LETM1 Increases the Mitochondrial Membrane Potential of mdm38Δ
Δψ was found to be moderately reduced in ydl183cΔ or mrs7Δ and substantially impaired in mdm38Δ mitochondria. Having determined that overexpression of Pic2p, Mrs3p, Mrs7p, and Ydl183cp rescued the nonfermentative cellular growth of mdm38Δ, we asked whether this positive growth effect also correlated with a rise of the mitochondrial Δψ of the mdm38Δ mutant. Although Δψ was slightly increased upon overexpression of Pic2p and more significantly upon overexpression of Mrs3p in mdm38Δ, overexpression of Mrs7p and Ydl183cp in the mutant restored Δψ close to the wild-type levels (Table 1).
TABLE 1.
Relative Δψ of mdm38Δ (Δ) and mdm38Δ mrs7Δ ydl183cΔ (ΔΔΔ) mutants in function of the overexpressed suppressor genes
The relative Δψ of mitochondria are expressed in % relatively to hyperpolarization of the probe with nigericin. ND, no data.
| Strains | Vectors |
||||||
|---|---|---|---|---|---|---|---|
| Empty | PIC2 | MRS3 | MRS7 | YDL183 | MDM38 | LETM1 | |
| Δ | 48 | 62 ± 3.4 | 85 ± 2.7 | 98 ± 3 | 92 ± 6 | 95 ± 6 | ND |
| ΔΔΔ | 17 ± 2 | ND | ND | 89 ± 3.3 | 78.6 ± 3.3 | 62.3 ± 3.4 | 82.3 ± 5.2 |
Mitochondrial Morphology Is Restored upon Overexpression of the Suppressor Genes in mdm38Δ
Next, we investigated whether high copy expression of the suppressor genes reversed the fragmentation of mdm38Δ mitochondria. For this purpose, mdm38Δ cells expressing a GFP targeted to the mitochondrial matrix and a vector with or without the suppressor genes were observed under the confocal microscope. Mitochondria from mdm38Δ cells transformed with the empty vector appeared fragmented into large unconnected spheres (Fig. 2a). Compared with wild-type cells, mdm38Δ cells displayed wild-type-like elongated tubular mitochondria in only about 3% of the population. Mutant mdm38Δ cells overexpressing PIC2 exhibited a heterogeneous mixture of spherical and tubular mitochondria, indicating a partial reversion of the phenotype (Fig. 2c). Overexpression of MRS3, YDL183c, or MRS7 resulted in a tubular mitochondrial network (Fig. 2, d–f, respectively, and Table 2) similar to that displayed by wild-type cells (Fig. 2b). The percentage of elongated tubular mitochondria was shifted to almost 80% upon overexpression of Pic2 and to about 95% when Mrs3, Mrs7, or Ydl183c was overexpressed (Table 2).
FIGURE 2.
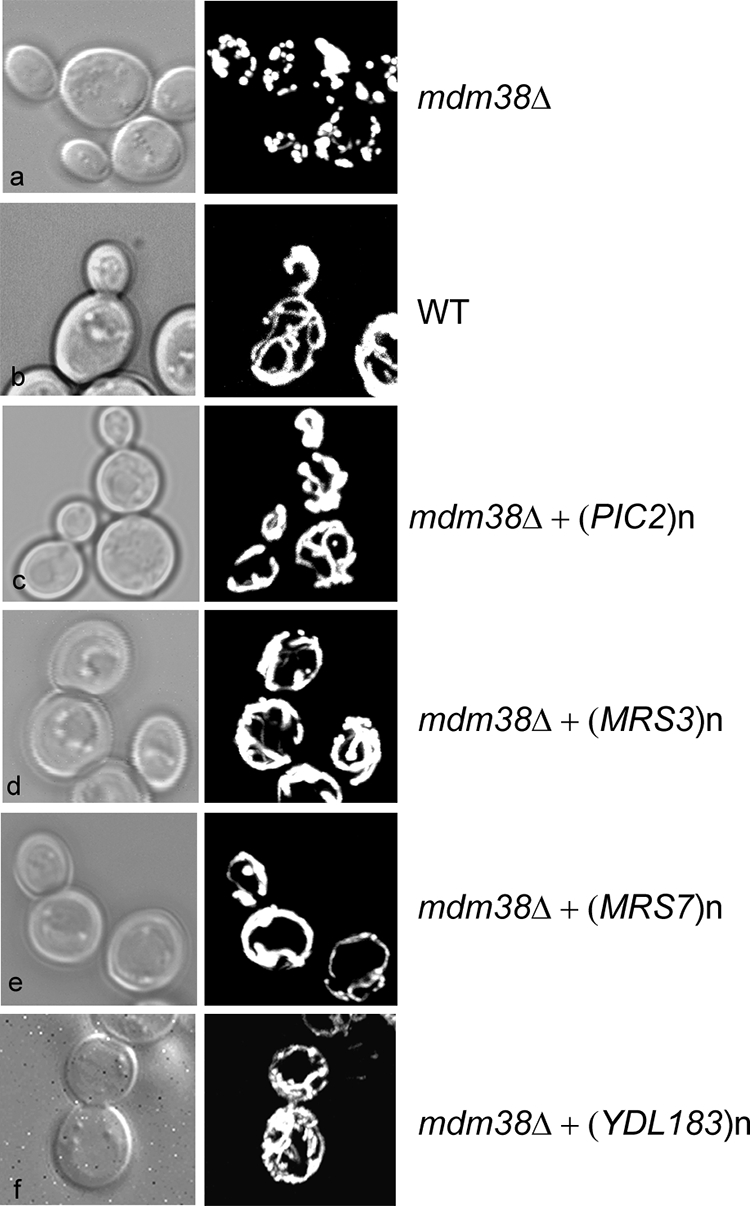
Mitochondrial morphology in function of overexpression of the proteins Pic2, Mrs3, Mrs7, or Ydl183c in W303 mdm38Δ mutant cells. Mitochondrial morphology of cells cotransformed with a mitochondrial matrix targeted GFP (pYX232-mtGFP) and the vector without (a) or with the following suppressor genes: PIC2 (c), MRS3 (d), YDL183c (f), and MRS7 (e) were compared with wild-type (WT) cells (b). Cells were grown in galactose-containing medium and analyzed by differential interference contrast (Nomarski) and confocal fluorescence microscopy.
TABLE 2.
Mitochondrial morphology of W303 mdm38D cells in function of the overexpressed suppressor gene
Strains were grown overnight, and mitochondrial morphology was visualized by detection of the expression of the mitochondrial targeted matrix GFP under fluorescence microscopy. Cells were counted with hidden identity.
| Strain | Total cells | % cells with fragmented mitochondria |
|---|---|---|
| Wild type | 602 | 2.6 ± 1.6 |
| mdm38Δ | 680 | 89.0 ± 5.8 |
| mdm38Δ+(MRS7)n | 1660 | 7.3 ± 2.6 |
| mdm38Δ+(PIC2)n | 870 | 20.3 ± 4 |
| mdm38Δ+(MRS3)n | 1126 | 10.6 ± 6.3 |
| mdm38Δ+(YDL183c)n | 1040 | 7.7 ± 3.8 |
YDL183c Is a Strong Suppressor for Mitochondrial KHE Activity in mdm38Δ
Because swelling, depolarization, and fragmentation of mdm38Δ mitochondria result from loss of mitochondrial KHE activity and mitochondrial K+ overload (5), we next asked whether overexpression of the suppressor genes restored the mitochondrial defects by modulating the KHE activity.
We have developed a method to measure the KHE activity across the mitochondrial inner membrane using SMPs with entrapped K+- and H+-sensitive fluorescent dyes PBFI and BCECF (2). This approach allows controlling internal and external ion milieus at will and recording of both proton and potassium fluxes. As shown previously and here in Fig. 3, SMPs prepared from wild-type mitochondria exhibited rapid, reciprocal translocation of K+ and H+ driven by concentration gradients of either. In contrast, SMPs from mdm38Δ failed to exhibit changes in [H+], and those in [K+] were drastically reduced. Nigericin restored K+ and H+ translocation in mutant SMPs to the wild-type level (Fig. 3B) (3).
FIGURE 3.

KHE activity of mdm38Δ SMPs in the function of the suppressors Pic2, Mrs3, Mrs7, and Ydl183c. Submitochondrial inner membrane particles were prepared from wild-type and mdm38Δ mutant cells with entrapped K+-sensitive PBFI or H+-sensitive BCECF. Ratios of K+-bound or H+-bound to -unbound dyes were recorded at 25 °C at resting conditions and upon the addition of 150 mm KCl. A, shown are the effects on K+ and H+ fluxes in SMPs upon overexpression of the suppressor genes in W303 mdm38Δ. SMPs were prepared from mitochondria of wild type (WT) (black thin dashed line) or mutant mdm38Δ cells carrying the empty plasmid (gray solid) or the suppressor plasmid containing the genes PIC2 (black dotted line), MRS3 (black bold dashed line), MRS7 (gray square dotted line), or YDL183c (black solid line). B, increase of [K+]i and [H+]i observed in SMPs from DBY wild-type (black dashed line), single mutant mdm38 (gray dashed line), or triple mutant mdm38Δ mrs7Δ ydl183cΔ (black thin solid line) in the absence of nigericin or mdm38 (gray bold solid line) and mdm38Δ mrs7Δ ydl183cΔ (black bold solid line) in the presence of nigericin.
SMPs were then prepared from mdm38Δ mitochondria overexpressing the respective suppressors (Fig. 3A). Overexpression of the phosphate carrier Pic2p showed a mild increase in K+ fluxes and a stronger increase in H+ fluxes (Fig. 3A, round dotted line). Interestingly, when K2HPO4 was used as K+ salt instead of KCl, K+ fluxes were not significantly re-established, whereas H+ fluxes reached wild-type levels (data not shown), which are consistent with the role of Pic2 as PO4−/H+ transporter (23). Overexpression of Mrs3p poorly restored the K+ and H+ fluxes (Fig. 3A, black broken line). However, overexpression of Ydl183cp restored the K+ and H+ exchange activity to a wild-type level, as did overexpression of Mrs7p, the yeast Mdm38p homologue (Fig. 3A, black solid line and gray square dotted line, respectively) or addition of nigericin. These results confirmed that, in contrast to Pic2p or Mrs3p, Ydl183cp, like Mrs7p, can fully substitute for Mdm38p in providing mitochondria with KHE activity.
Ydl183cp Is an Integral Mitochondrial Protein
YDL183c encodes a protein of 320 amino acids with a molecular mass of about 37 kDa. The computer programs DAS and TMPRED (available on line) predict one transmembrane domain (196–212 amino acids) and a potential N-terminal mitochondrial targeting sequence. Homologues are found in fungi and in some green plants like Arabidopsis thaliana (At1g53760 accession number Q6NQN0). These proteins share one conserved putative transmembrane domain rich in proline residues (Fig. 4A). Except for the presence of a proline-rich putative transmembrane domain, there was no obvious sequence similarity between Ydl183cp and proteins of the Mdm38p/LETM1 family.
FIGURE 4.

Ydl183cp is a component of the mitochondrial inner membrane. A, Ydl183cp is a member of a novel protein family. Homologous proteins were identified by a BLAST search. A sequence alignment (ClustalW) of Ydl183cp and its homologues in A. thaliana (A.t.) and Neurospora crassa (N.c.) is shown here. Identical amino acids are highlighted in black and similar amino acids in gray. The putative potential N-terminal mitochondrial targeting sequence is marked with a dotted bar and the putative transmembrane domain with a solid bar. B, localization of the Ydl183c-GFP fusion protein analyzed under confocal microscopy. W303 cells expressing C-terminally GFP-tagged YDL183c gene were grown to log phase in galactose containing medium at 28 °C. Mitochondria are labeled with MitoTracker red chloromethyl-X-rosamine. C, subcellular and submitochondrial localization of Ydl183cp. Panel a, W303 cells expressing the Ydl183c-HA fusion protein (YCp-YDL183c-HA, 42 kDa) were grown to log phase in galactose-containing medium. Protoplasts were homogenized and separated into total cell (T), mitochondrial (M), and post-mitochondrial (C) fractions. Equal amounts of protein of subcellular fractions were subjected to SDS-PAGE, and immunodetection with antisera against the HA tag, Hxk1p and Por1p, was performed. Panel b, crude mitochondria (2 mg of protein) were treated with 0.1 m Na2CO3 and fractionated by centrifugation at 100,000 × g into pellet (P) and supernatant (SN). Both fractions (100 μg of protein/lane) were subjected to SDS-PAGE and immunoblotted with antisera against HA, Por1p, and F1β. Panel c, mitoplasts prepared by osmotic shock were separated into supernatant containing the inter-membrane space and pellet. Mitoplasts were aliquoted in equal amounts and incubated with or without proteinase K as indicated. Samples were analyzed by SDS-PAGE and immunoblotted with antisera against the HA tag and against mitochondrial proteins of the inner membrane Tim44p and Yme1p.
To determine the cellular localization of Ydl183c, cells expressing the fusion protein Ydl183c-GFP from the MET promoter encoded on the centromeric plasmid pUG35 were stained with Mito Tracker Red. Fluorescence confocal microscopy revealed the colocalization of GFP and red fluorescence, indicating the mitochondrial localization of Ydl183-GFP (Fig. 4B). To confirm these data, biochemical studies were performed with cells expressing the low copy vector encoding Ydl183cp from its own promoter and C-terminally tagged with the triple hemagglutinin (HA) epitope. Cell fractionation and immunoblotting showed Ydl183c-HA protein to cofractionate with a mitochondrial protein (Porin1, Por1p), whereas the cytosolic protein hexokinase 1 (Hxk1p) was detected in the post-mitochondrial fraction, excluding the possibility of cross-contamination of cytoplasmic and mitochondrial fractions (Fig. 4C, panel a). Fractionation of mitochondria into pellet and supernatant upon alkaline sodium carbonate treatment released the membrane-associated β subunit of the F1-ATPase (F1β) almost entirely into the supernatant, whereas the membrane protein Por1p was retained in the pellet fraction containing integral proteins. Ydl183c-HA was found in the membrane pellet (Fig. 4C, panel b, lane P). However, in contrast to Por1p, Ydl183c-HA was also partially found in the soluble fraction (Fig. 4C, panel b, lane SN). These results indicated that Ydl183cp is inserted into one of the mitochondrial membranes where it can be partly released by alkaline treatment.
For further determination of the topology of Ydl183cp, intact mitochondria were first treated with or without proteinase K. Ydl183cp was not degraded upon addition of proteinase K (data not shown). Mitoplasts were prepared by osmotic swelling and rupture of the mitochondria. Mitoplasts containing the inner membrane were treated with proteinase K (Fig. 4C, panel c). To control the intactness of mitoplasts, the topology of known proteins was also tested. Tim44p, a matrix-sided protein of the inner membrane, remained protected from proteinase K, indicating that the mitoplasts were intact. In contrast, Yme1p, an inner mitochondrial membrane protein partially exposed to the outside of mitoplasts, was accessible to proteinase K indicating that the outer membrane was disrupted. The C-terminally tagged Ydl183cp was resistant to 40 μg/ml proteinase K, whereas it became accessible to higher proteinase K concentrations. Proteinase K at 120 μg/ml degraded most of Ydl183-HA without generating proteolytic C-terminal fragments. When mitoplast were lysed with Triton X-100 and then treated with proteinase K, the protein was entirely degraded. Altogether, although alkaline extraction released some of the protein, these results qualify Ydl183p as an integral protein of the inner mitochondrial membrane, with a Cout (facing the intermembrane space) topology. Degradation of Ydl183c-HA occurred only in presence of high concentrations of proteinase K as compared with Yme1, either because it is shielded by other proteins or Ydl183cp is intrinsically more resistant to proteinase K.
Synthetic Growth Effect of Triple Disruptions of MDM38, MRS7, and YDL183c
The W303 and DBY ydl183cΔ disruptant strain showed reduced growth on nonfermentable carbon sources (YPG) at high temperature (37 °C) (data not shown). Reduced growth on nonfermentable substrate was also reported by Volckaert et al. (25) for a FY ydl183cΔ mutant at 30 °C and 37 °C. The double disruptants ydl183cΔ mrs7Δ exhibited a mild growth reduction on nonfermentable substrate at 16 and 28 °C (data not shown). Double disruptants mdm38Δ mrs7Δ in W303 or DBY747 essentially showed the same phenotype as the single mutant mdm38Δ at 28 or 35.5 °C, whereas a slight growth improvement was detected on YPG at 16 °C (Fig. 5A). Growth of mdm38Δ ydl183cΔ double mutants was reduced on YPG at 16 °C (Fig. 5A). Importantly, the triple deletion mutant mdm38Δ mrs7Δ ydl183cΔ resulted in synthetic phenotypes. Growth on nonfermentable substrates was virtually absent, and the DBY747 triple mutant strain also displayed a significant growth reduction on YPD plates at 28 and 35.5 °C (Fig. 5, A and B), consistent with a serious disturbance in mitochondrial function(s) essential for cell viability.
FIGURE 5.

Deletion growth phenotypes. A, serial dilutions of DBY. mdm38Δ, mdm38Δ mrs7Δ, mdm38Δ ydl183cΔ, and mdm38Δ mrs7Δ ydl183cΔ mutants were spotted onto YDP and YPG and incubated at the indicated temperatures. Growth on 28, 35.5, and 16 °C was observed after 3, 5, and 8 days, respectively. B, DBY wild-type (WT) and mdm38Δ mrs7Δ ydl183cΔ triple mutant cells expressing an empty control vector (pUG35) or YCp33-MDM38-HA, pUG35-MRS7-GFP, pUG35-YDL183c-GFP, or pVT-U-LETM1-HA. Serial dilutions were spotted onto YPD and YPG plates and incubated for 10 days at 16 °C or 3 or 5 days at 28 and 37 °C on YDP or YPG, respectively. C, effect of nigericin on the nonfermentative growth of DBY747 mdm38Δ single, mdm38Δmrs7Δ, mdm38Δydl183cΔ double, and mdm38Δ mrs7Δ ydl183cΔ triple mutant cells. Serial dilutions of the wild-type and mutant cells were spotted onto YPD and YPG plates containing (+) or not (−) 2 μm nigericin and incubated 10 days at 16 °C and 5 days at 28 °C.
Growth of the triple mutant on YPD and YPG was largely restored upon expression of MDM38 or overexpression of either MRS7, YDL183c, or human LETM1 (Fig. 5B). In comparison, overexpression of PIC2 in the triple mutant resulted in no growth improvement, and overexpression of MRS3 rescued the nonfermentable growth of the triple mutant only at 37 °C but not at 16 °C (data not shown).
Addition of nigericin, an electroneutral KHE ionophore, efficiently restored growth of the triple mutant mdm38Δ mrs7Δ ydl183cΔ on glycerol at 16 and 28 °C (Fig. 5C). This finding is important because it suggests that the growth defects on fermentable and nonfermentable substrates were essentially due to a lack of KHE activity.
Given the strong homology of the yeast proteins Mrs7 and Mdm38, we searched for human homologous proteins of LETM1. A BLAST search of the human protein data base revealed a protein containing a LETM1 domain and named LETMD1 or HCCR-1. The sequence alignment of Mrs7, Mdm38, LETM1, and HCCR-1 shows the conserved domains as highlighted (Fig. 6A). The recent work of Kim and co-workers (9, 26) showed that HCCR-1 was overexpressed in various human cancers and might function as a negative regulator of the p53 tumor suppressor. Having shown that overexpression of LETM1 from the ADH promoter restored growth of the triple mutant, we tested the suppression capacity of HCCR-1 expressed under the same promoter. Overexpression of HCCR-1 restored fermentative growth of the triple mutant to wild-type levels at 28 and 37 °C and nonfermentative growth on YPG at 28 °C (Fig. 6B). However, growth was only poorly increased on YPG at 16 °C and not at all at 37 °C (data not shown). As described previously, HCCR-1 was characterized as a mitochondrial protein (26). We verified its subcellular localization when heterologously expressed in yeast. Cell fractionation of wild-type (data not shown) and mdm38Δ mrs7Δ ydl183Δ triple mutant cells expressing HCCR-1-His and Western blotting analysis revealed that HCCR-1 was detected as a protein of 35 kDa in the total and mitochondrial fractions (Fig. 6C). Por1p was also recovered in total and mitochondrial fractions and Hxk1p in total and cytoplasmic fractions. Accordingly, in yeast HCCR-1-His was exclusively found in mitochondria, although in significantly less abundant amounts than Por1p (Fig. 6C).
FIGURE 6.

Suppression effect of human HCCR-1. A, sequence alignments of Mdm38, Mrs7, Letm1, and HCCR-1. ClustalW alignments of the amino acid sequences over the homologous regions are shown. Identities are highlighted in black and similarities in gray. Amino acid residues identical over all four sequences are in boldface and boxed. Bar is over the transmembrane domain. B, growth effect of HCCR-1 expression in yeast triple mdm38Δ mrs7Δ ydl183Δ mutants (ΔΔΔ). Wild-type (WT) and triple mutant cells expressing pVTU103 with or without HCCR-1 were spotted onto SD−ura, YPD, and YPG plates and grown at the indicated temperatures for 6, 3, and 6 days, respectively. C, subcellular localization of HCCR-1 in yeast. Yeast triple mdm38Δ mrs7Δ ydl183Δ mutants (ΔΔΔ) expressing HCCR-1 were fractionated into total (T), mitochondrial (M), and post-mitochondrial (C) fractions, and Western blotting was performed.
Severe Loss of the Mitochondrial Membrane Potential in the Absence of Mrs7p, Ydl183cp, and Mdm38p
Most importantly, the mitochondrial Δψ was dramatically reduced in the triple mutant mdm38Δ mrs7Δ ydl183cΔ (Table 1). We tested if overexpression of the individual suppressors also restored the mitochondrial Δψ in the triple mutant mdm38Δ mrs7Δ ydl183cΔ. We found that expression of Mdm38p and overexpression of Mrs7p or Ydl183cp restored the reduced Δψ of the triple mutant to a reasonable level (Table 1), a result comparable with that observed after overexpression of human LETM1. These findings suggest that cellular growth and increase of mitochondrial Δψ are mechanistically linked.
Dramatic Changes of Organelle Morphology in mdm38Δ mrs7Δ ydl183cΔ Cells
In addition to growth impairment and profound depolarization, the triple deletion mutant mdm38Δ mrs7Δ ydl183cΔ differed most strikingly from the mdm38Δ single deletion mutant in its organellar morphology. Confocal microscopic analysis of triple mutant cells expressing the mitochondrial matrix-targeted GFP showed that mitochondria appeared fragmented in spherical units, were less numerous than in the single mdm38Δ mutant, and were somewhat clumped together. Furthermore, costaining of cells with the specific vacuole dye FM4-64 consistently showed a multiple lobed morphology of the vacuoles (Fig. 7A). To look into the structure of the organelles at higher resolution, electron microscopy was performed. Remarkably, numerous vesicles were visible in each section, all looking almost alike in size and electron density. The recognition of single or double vesicle-surrounding membranes was the only morphological criterion to discriminate between mitochondrial and vacuolar vesicles. Yet a distinction of the organelles was not always possible (Fig. 7B, panels a and b, right panels). Surprisingly, a large number of cells showed vesicular mitochondria containing undefined material suggesting either internalized membranes or paracrystalline structures (Fig. 7B, panel b, right panel). Most importantly, wild-type-like morphology of the cells was restored upon addition of nigericin (Fig. 7B, panel c) with reversion of mitochondria from swollen, fragmented, and electron-transparent to condensed, elongated, and electron-dense organelles. This key finding links the morphological phenotype of the triple mutant to a defect of K+ homeostasis, which can be compensated by nigericin.
FIGURE 7.

Mitochondrial and vacuolar morphology in absence of Mdm38p, Mrs7p, and Ydl183cp. Cells were grown to logarithmic phase in galactose (A and B)- or galactose- and raffinose (C)-containing medium. Shown are representative fluorescent and electron microscopy images. A, confocal microscopy analysis of W303 wild-type cells (a), isogenic mdm38Δ (b) and isogenic mdm38Δ mrs7Δ ydl183cΔ triple mutant cells (c) expressing the mitochondrial matrix targeted GFP. Vacuoles were stained with FM4-64. B, electron micrographs of mdm38Δ mrs7Δ ydl183cΔ cells. Panels a and b show the organellar ultrastructure of the triple mutant grown as described above. Whole cells are shown in right panels. The cells display mitochondria with aberrant morphologies (details showing mitochondria are in the left panels). Panel c shows the organellar ultrastucture of cells from the same culture to which nigericin (2 nm) has been added for the last growth generation. Right panel, whole cells; left panel, mitochondrion after nigericin treatment. Bar, 200 nm (left panels) and 1 μm (right panels). C, confocal microscopy analysis of wild-type cells (panels a–d) and mdm38Δ mrs7Δ ydl183cΔ cells (panels e–h) expressing the mitochondrial targeted YFP (panels a and e) to the outer membrane (pHS72). Vacuoles are indicated by FM4-64 (panels b and f). Merged fluorescence is shown in panels c and g. The yellow fluorescence detected indicates the colocalization of mitochondria and vacuoles. Differential interference contrast microscopy of wild-type (panel d) and mdm38Δ mrs7Δ ydl183cΔ (panel h) cells is shown.
For better discrimination of the origin of the visualized organelles, we used a mitochondrial YFP targeted to the outer membrane (pHS72) and the vacuolar stain FM4-64. Confocal microscopy showed stained wild-type mitochondria and vacuoles as clearly distinct organelles (Fig. 7C, panels a–d). In contrast, triple mutant cells exhibited widely overlapping fluorescence of FM4-64 (vacuoles) and YFP directed to the outer mitochondrial membrane (Fig. 7C, panels e–h), indicative of the colocalization of both organelle markers that occurs in mitophagy (27).
Mitochondrial KHE Is Totally Absent in the Triple Deletion Strain mdm38Δ mrs7Δ ydl183cΔ
Remarkably, SMPs from the triple mutant mdm38Δ mrs7Δ ydl183cΔ failed to exhibit H+ fluxes, and most importantly, residual K+ fluxes observed in the mdm38Δ single mutant SMPs were fully eliminated in the triple mutant (Fig. 3B). However, KHE was fully active in ydl183cΔ and moderately reduced in mrs7Δ (data not shown). Consistent with data reported above on cell growth and Δψ, disruption of all three genes had additive effects on the KHE activity. However, preincubation of the mdm38Δ mrs7Δ ydl183cΔ SMPs with nigericin led to the activation of the K+ and H+ transport across the SMPs membrane, although not to full wild-type levels (Fig. 3B).
K+ and H+ flux measurements carried out in triple mutant SMPs revealed efficient restoration of activities by Mdm38p expressed from a single copy vector, whereas its homologue Mrs7p required expression from a multicopy vector (Fig. 8, B, black solid line, and A, black square dotted line, respectively). Overexpression of Ydl183cp also restored some of the K+/H+ fluxes in the triple mutant but not fully (Fig. 8A, black broken line). Finally, overexpression of the human homologue of Mdm38p, LETM1 in the triple mutant strains, restored most of the KHE activity (Fig. 8B, gray solid line).
FIGURE 8.

KHE activity of mdm38Δ mrs7Δ ydl183c Δ SMPs. [K+]-driven changes of [K+]i and [H+]i in submitochondrial inner-membrane particles prepared from wild-type and mdm38Δ mrs7Δ ydl183cΔ mutant cells with entrapped K+-sensitive PBFI or H+-sensitive BCECF were recorded as described in Fig. 3. A, effect of overexpression of Mrs7p (black square dotted line) or Ydl183cp (black thin dashed line) on [K+]-driven changes of [K+]i and [H+]i in DBY triple mutant mdm38Δ mrs7Δ ydl183cΔ SMPs (black solid line) in comparison with wild-type SMPs (black dotted line). B, effect of Mdm38p (expressed from YCp33, bold black solid line) or LETM1 (expressed from pVTU-(bold gray solid line) on [K+]-driven changes of [K+]i and [H+]i in DBY mdm38Δ mrs7Δ ydl183cΔ SMPs (black thin solid line) in comparison with wild-type SMPs (bold square dotted line).
Ydl183cp, Mrs7p, and Mdm38p Form High Molecular Weight Complexes
The genetic data presented here support the notion that Mdm38, Mrs7, and Ydl183c proteins are functionally equivalent in contributing to the formation of an active KHE. All three proteins are single-pass transmembrane proteins and thus are unlikely to form an exchanger without self-association or without association with other yet unidentified proteins (homo- or hetero-oligomerization, respectively). To address the question of whether the proteins were part of high molecular weight complexes, we first performed chemical cross-linking. We used crude mitochondria expressing Mdm38p or Ydl183cp in cross-linking experiments using disuccinimidyl suberate at increasing concentrations. Based on the electrophoretic mobility of the cross-linked products, our data confirmed that Mdm38p and Ydl183cp were part of large protein complexes (supplemental material). To improve the molecular weight size resolution of the complexes, we performed BN-gel electrophoresis followed by immunodetection. Mitochondria were isolated from DBY wild-type strains expressing chromosomally tagged Mdm38-His or Mrs7-His or extra-chromosomal Ydl183-GFP from the pUG vector. Isolated mitochondria were solubilized with mild detergents and separated on nondenaturing gels prior to Western blotting and immunodetection. Probing with the anti-His antibody revealed that Mdm38-His (∼67 kDa) migrated at ∼500, <232, and <140 kDa (Fig. 9A, left panel). Of note, Mdm38-His was detected in protein complexes of the same molecular weights irrespective of the presence of Mrs7p or Ydl183cp (data not shown). Furthermore, Mrs7-His (53 kDa) appeared in three bands around ∼500 kDa and an additional band of >232 kDa (Fig. 9B, lane 1). BN-PAGE analysis of solubilized mitochondria expressing Ydl183-GFP yielded a product of an apparent molecular mass of about 67 kDa, which corresponds to its molecular weight as GFP-tagged monomer and to additional bands of <232 kDa, representing YDL183GFP-containing complexes. However, in the background of a mutant mdm38Δ strain, Ydl183-GFP partly shifted to a major band of ∼500 kDa (Fig. 9C).
FIGURE 9.

Mdm38p, Mrs7p, and Ydl183cp are part of a high molecular weight complex. A, DBY chromosomally Mdm38-His-tagged mitochondria were solubilized with 1.2% Triton X-100. Left panel, one part of the preparation was immediately separated on BN-PAGE. The anti-His antibody recognized three protein complexes of ∼500, <232, and < 40 kDa. Middle panel, other part of the same preparation was used for a further step involving nickel-affinity chromatography. Mdm38-His was recovered as part of a complex of <232 kDa. Right panel shows Mdm38-STrEP after STrEP-affinity chromatography elution separated on BN-PAGE. The anti-STrEP antibody recognized the complexes of <232 and >440 kDa. M, marker. B, DBY mitochondria expressing the chromosomally His-tagged Mrs7 were solubilized as in A and analyzed by BN-PAGE (left panel, lane 1), and parallel fractions were used for further isolation of a Mrs7-His complex by affinity chromatography (left panel, lane 2). Solubilized mitochondrial proteins and elution fractions from the affinity purification were separated on the same BN gel, transferred to a common membrane for Western blotting, and probed with an antiserum against His. DBY mitochondria expressing the chromosomal Mrs7-STrEP were solubilized, affinity-purified, and recovered in complexes of <140, >232, and >440 kDa. C, mitochondria expressing pUG-YDL183c-GFP in different backgrounds as follows: wild-type (lane 1) or mdm38Δ (lane 2) were solubilized with 1.2% n-dodecyl β-d-maltoside. Equal amounts of proteins were separated on BN-PAGE and immunoblotted with an antibody against GFP.
Next, we affinity-purified the chromosomally His-tagged Mdm38p and Mrs7p. The proteins were bound to the resin and eluted from the column prior to analysis on BN-PAGE. Surprisingly, despite changing the experimental conditions such as the incubation times with the Ni-NTA beads or using different detergents or NaCl concentrations, the eluted Mdm38-His exclusively appeared as a single band of a molecular mass slightly smaller than 232 kDa (Fig. 9A, middle panel). This was an unexpected result. To find out if the protein complex of ∼500 kDa containing the His-tagged Mdm38p as detected on BN prior affinity purification had become inaccessible to the column possibly because it was hidden by additional proteins of the larger complex, we decided to affinity-purify Mdm38 fused to the One-STrEP tag. This tag containing a linker region makes the tagged component of a protein complex more accessible to the column. In fact, using the chromosomal One-STtrEP-tagged version of MDM38, the purified Mdm38p was recovered within high molecular complexes ranging between ∼500 and < 600 kDa in addition to the complex of <232 kDa (Fig. 9A, right panel).
Affinity chromatography of solubilized mitochondria chromosomally expressing Mrs7-His followed by BN-PAGE recovered Mrs7-His within three complexes of <140, >232, and between 440 and 669 kDa as seen in Fig. 9B, left panel, lane 2. Similar results were obtained using Mrs7OneStrep instead of Mrs7His (Fig. 9, right panel). In the next step, we solubilized mitochondria from chromosomally Mrs7-His-tagged cells coexpressing either YCp-Mdm38-HA or YCp-Ydl183-HA. Mitochondrial expression of Mdm38-HA and Ydl183c-HA was confirmed by Western blotting (data not shown). Affinity purification followed by BN-PAGE and Western blotting analyses, including immunodetection with anti-His and HA antibodies, was performed. Although the anti-His antibody recognized Mrs7-His, neither Mdm38-HA nor Ydl183-HA was detectable when the eluted fractions were probed with the anti-HA antibody, excluding a direct interaction of Mrs7-His and Mdm38-HA or Mrs7-His and Ydl183-HA (Fig. 10, A and C, respectively). Second dimension SDS-PAGE confirmed that Mdm38-HA was not part of the Mrs7-His complex (Fig. 10B, right panel). Taken together, our experiments did not suggest any direct interaction between Mdm38-HA and Mrs7-His.
FIGURE 10.

Interaction of Mrs7-His with Mdm38-HA and YDL183c-HA. A, affinity chromatography and preparative BN-PAGE of solubilized mitochondria coexpressing chromosomally His-tagged Mrs7 and extra-chromosomal YCp-Mdm38-HA in different backgrounds as follows: wild-type (WT) (lanes 1 and 2) and mdm38Δ (lanes 3–5). 120 μl (lanes 1, 3, and 5) and 60 μl (lanes 2 and 4) of the eluted fractions were applied to the same gel. Lanes 1–4 were probed with an antibody against His. Lane 5 served for the additional immunodetection with an antibody against HA. M, marker. B, second dimension SDS-PAGE of lane 3. Left panel, the antibody against His recognizes a product of ∼55 kDa corresponding to Mrs7-His. The signal is in perfect agreement with the signals of the first dimension (BN-PAGE). Right panel, immunodetection with anti-HA antibody of the same blot after mild stripping. C, affinity chromatography and BN-PAGE of solubilized mitochondria coexpressing chromosomally His-tagged Mrs7 (lanes 1 and 3) or His-TAP-tagged Mrs7 (lanes 2 and 4) and extra-chromosomal Ydl183-HA (lanes 1–4). Lanes 1 and 2 and lanes 3 and 4 were probed with antibodies against HA and His, respectively.
These results are in contrast to data reported previously by Frazier et al. (8), indicating a direct interaction of a protein A-tagged Mdm38 with numerous other mitochondrial proteins, including Mrs7p. In fact, when we used a strain expressing the Mrs7 protein C-terminally tagged with a His fused to protein A (Mrs7-His-TAP), we found that Mdm38 coeluted with Mrs7-His-TAP in the ∼232-kDa complex (Fig. 11, B and C). However, a direct interaction between Mrs7-His-TAP and Ydl183c was not detectable (Fig. 10C). We asked whether the tags affected the suppression of the mutant phenotype. Mrs7-His or Mrs7-His-TAP was introduced into the mdm38Δ mutant, and nonfermentative growth was tested. We found that mdm38Δ cells expressing Mrs7-His-TAP did not grow as well as Mrs7-His or wild-type cells (Fig. 11D). Thus, these data altogether suggest that Mrs7p and Mdm38p are not interacting directly.
FIGURE 11.

Interaction of Mdm38p with Mrs7-His and Mrs7-His-TAP. A, affinity chromatography and BN-PAGE of solubilized DBY mitochondria expressing either chromosomally His-tagged or His-TAP-tagged Mrs7. Eluted fractions 1–2 containing Mrs7-His were applied on lanes 1 and 2 and 5 and 6 and eluted fractions 1–2 containing Mrs7-His-TAP on lanes 3 and 4 and 7 and 8. BN-PAGE was performed and followed by immunostaining with an antibody against His (lanes 1–4) and Mdm38p (lanes 5–8). M, marker. B, preparative affinity chromatography and BN-PAGE of DBY mitochondria expressing chromosomally Mrs7-His-TAP prior to second dimension SDS-PAGE. The membrane was first incubated with an anti-His primary antibody (lane 1). Thereafter, the blot was mildly stripped and reincubated with an antibody against Mdm38p (lane 2). C, second dimension SDS-PAGE. Left panel, the blot was probed with the anti His antibody. Right panel, same blot probed with the anti Mdm38p antibody after mild stripping of the membrane. D, suppression effect of Mrs7-His and Mrs7-His-TAP in mdm38Δ. DBY wild type (WT) with YEp112 empty and mdm38Δ with YEp112 empty, MRS7-His, or MRS7-His-TAP were grown overnight. Serial dilutions were spotted onto YPD and YPG plates and incubated at the indicated temperatures.
We used CoIP to ask whether Mdm38 homo- or hetero-oligomerizes with Ydl183cp. Mdm38 self-oligomerization was confirmed by CoIP experiments performed on mdm38Δ cells coexpressing Mdm38-HA (72 kDa) and Mdm38-GFP (92 kDa). Although Mdm38-HA was successfully bound to HA-coated protein A beads, only Mdm38-GFP was pulled down, and Ydl183c-GFP did not copurify in the protein A-bound fractions like Aif-GFP serving as negative control (Fig. 12).
FIGURE 12.

CoIP of isolated mdm38Δ mitochondria coexpressing YEp-MDM38-HA (72 kDa) and pUG-MDM38-GFP (92 kDa) (A), YDL183-GFP (65 kDa) (B), or AIF-GFP (68 kDa) (C). F, flow-through fraction; B, HA-coated protein A-bound fraction.
DISCUSSION
We previously characterized Mdm38p as a mitochondrial protein essential for KHE activity (2, 4). Because this protein has only one transmembrane domain, it appears unlikely to be solely responsible for the KHE process. To explore the possible existence of additional proteins involved in KHE, we carried out a genome-wide suppressor screen.
We identified the mitochondrial carriers Pic2p and Mrs3p as weak suppressors. Their overexpression rescued the growth defect of mdm38Δ. We showed that mitochondrial morphology of mdm38Δ was restored to wild-type upon overexpression of PIC2 and MRS3. However, in mdm38Δ SMPs, KHE was not seen after overexpression of Pic2p and was only marginally restored by Mrs3p. Because overexpression of Pic2p, a Pi carrier, had no effect on mitochondrial K+ fluxes in mdm38Δ mitochondria and resulted in a marginal increase of the mitochondrial Δψ, we hypothesize that a contribution to Δψ above a threshold is sufficient to heal the growth and morphology phenotype. Alternatively, Pic2p might act indirectly by modulating proton fluxes or mitochondrial pH. Overexpression of Mrs3p, a Fe2+ carrier, increased the mitochondrial Δψ and moderately the KHE. Deletion of MRS3 had no effect on KHE, and mrs3Δ mdm38Δ mutants remained without synthetic phenotype,5 excluding a role of Mrs3p as the KHE. These findings suggest an indirect role of Mrs3p on the KHE activity. In fact, several ions have been stated to play a direct or indirect role in mitochondrial K+ homeostasis (28–31).
The suppressor screen also identified two additional genes, MRS7 and the novel gene YDL183c encoding an unknown protein, which were found to restore both growth and KHE activity of mdm38Δ mutant cells. Although Mdm38p and Mrs7p are phylogenetically related, Ydl183cp is likely not related to them. Each of these proteins contains a single transmembrane domain and appears to be part of a high molecular weight protein complex. They are functionally redundant in establishing a functional KHE in mitochondria.
In contrast to Mdm38p, absence of either Mrs7p or Ydl183cp alone or in combination did not seriously affect the growth of yeast cells. Yet the triple mutant mdm38Δ mrs7Δ ydl183cΔ had a dramatically stronger negative growth phenotype than the single mdm38Δ mutant or double mutants. This mutant completely failed to grow on respiratory substrates and exhibited a strain- and temperature-dependent reduced growth on fermentable substrates. This synthetic phenotype of the triple mutant indicates the following: (i) all three proteins are functionally expressed in yeast; (ii) loss of all three proteins dramatically impairs mitochondrial volume homeostasis through a disturbance that can be rescued by nigericin; and (iii) impaired volume homeostasis causes mitochondrial dysfunction affecting cell vitality as indicated by the reduction in growth on fermentable substrates.
Overexpression of Mrs7p or Ydl183cp fully compensated the growth defects of the mdm38Δ single and the mdm38Δ mrs7Δ ydl183cΔ triple disruptant. Accordingly, either protein could fully substitute for Mdm38p when expressed at high abundance. Addition of the exogenous KHE nigericin equally compensated for the growth defect of mdm38Δ single as well as for the even stronger growth defects of the triple mutant. This finding supports the conclusion that the triple mutant growth phenotype is essentially due to a lack of KHE activity.
This important point was proved by a direct test for KHE activity on inner membrane SMPs, a system that entirely avoids any interference of osmotically swollen mutant mitochondria. A comparison between single and triple mutants clearly revealed that the single mdm38Δ mutant retained a minor KHE activity, whereas the triple mutant totally lacked this activity. Thus, single and triple mutants most likely differ only in the degree to which they have lost KHE activity. These data correlate well with the reduction in growth of the single and triple mutants. However, mild reduction in KHE activity detected in single mutants mrs7Δ and ydl183cΔ indicates that expression of Mrs7p and/or Ydl183p is necessary for full KHE activity in wild-type cells. Yet the physiological effects of mrs7Δ or ydl183cΔ mutations are too weak to result in reduced growth of mutant cells. In assays performed on the single mdm38Δ mutant, overexpression of Ydl183cp or Mrs7 could equally restore transmitochondrial K+/H+ fluxes, like Mdm38p or LETM1. However, in the triple mutant mdm38Δ mrs7Δ ydl183c, Ydl183cp restored the mitochondrial KHE to a lesser extent than Mdm38p Mrs7p, or LETM1, most likely resulting from a less abundant expression of Ydl183c as compared with Mdm38 or Mrs7 (shown in supplemental Fig. S4). Taken together, these results indicate that Mdm38p, Mrs7p, and Ydl183cp are functionally redundant, but only Mdm38p is essential.
Mitochondrial depolarization in the mutants and its rescue by expression of either Mdm38p or Mrs7p or Ydl183cp correlated with loss and recovery of KHE activity, respectively, and with cell growth that was mildly affected on YPG when Δψ fell below 60% of wild-type values and increasingly more with lower values. The lowest Δψ values observed in the triple mutant also affected growth on fermentable substrate, indicating that essential functions of mitochondria, possibly protein import, were affected. The loss of Δψ in mitochondria of the disruptants may be a direct consequence of the absence of H+ fluxes into the mitochondria in exchange for the efflux of K+ rather than an additional effect resulting from the K+ accumulation and swelling of mitochondria.
Total loss of KHE activity of the triple mutant was accompanied by more dramatic changes in organelle morphology than in the single mdm38Δ mutant. Both mitochondria and vacuoles appeared to be heavily fragmented and were shown to frequently colocalize, suggesting intense mitophagy. Notably, hyperosmotic stress has been reported to result in significant changes of the vacuole morphology of wild-type cells. In fact, the one to three large vacuoles usually present in wild-type cells underwent fragmentation to numerous smaller multilobe vacuoles (32). Interestingly, treatment of triple mutant cells with the K+/H+ ionophore nigericin efficiently reversed swelling and restored a near normal mitochondrial network. As this involves the mitochondrial fusion (33, 34), we assume that proteins regulating the fusion activity are not affected by the absence of Mdm38, Mrs7, and Ydl183c. Vacuolar fragmentation was efficiently reverted together with re-establishing mitochondrial KHE activity by nigericin (data not shown). This raises the question of how the loss of KHE and swelling of mitochondria cause fragmentation of the vacuole. In sum, this study provides strong evidence for a role of all three proteins in contributing to an active mitochondrial KHE.
A genome-wide screen in Drosophila S2 cells recently identified LETM1 as strongly affecting mitochondrial Ca2+ and H+ homoeostasis. Absence of LETM1 resulted in reduced mitochondrial Ca2+ uptake in situ, a finding that led the authors to conclude that Letm1 is the mitochondrial Ca2+/H+ antiporter (35). This conclusion is puzzling, because down-regulation of the mitochondrial Ca2+/H+ exchanger would rather have been expected to result in decreased Ca2+ efflux and therefore in increased mitochondrial Ca2+ accumulation, because mitochondrial cation/H+ antiporters protect cells from mitochondrial cation overload by mediating cation efflux from energized mitochondria (1). Jiang et al. (35) also found that reconstitution of LETM1 in liposomes catalyzed Ruthenium red-sensitive Ca2+/H+ exchange, which raises further questions since decades of work on mitochondria indicate that Ca2+/H+ exchange is insensitive to Ruthenium red (36). Previous evidence that LETM1 is essential for mitochondrial K+/H+ exchange is compelling (4, 5, 37), and the present study demonstrated that LETM1 fully restores mitochondrial KHE activity of the yeast triple mutant mdm38Δ mrs7Δ ydl183c like the exogenous bona fide KHE nigericin. We believe that further studies will be needed to clarify whether the highly conserved LETM1 proteins exert different cation-specific functions in different eukaryotic organisms or rather disturbances of mitochondrial K+ homeostasis can secondarily affect mitochondrial cation transport. Relevant to this discussion, we have shown that yeast mitochondria depleted of Mdm38p display a considerably reduced influx of Mg2+ and Ca2+ resulting from decreased mitochondrial Δψ (4).
Human LETM1 has previously been shown to be part of a complex of about 550 kDa by CoIP of GFP- and HA-tagged isomers (38). Rehling and co-workers (8) reported an interaction of Mdm38-protein A with various proteins, including numerous mitochondrial ribosomal proteins and Mrs7p. In our hands, hetero-oligomerization of Mrs7p with Mdm38p was not detected unless Mrs7p was fused to a tag, including protein A and calmodulin-binding protein.
We have shown that the Mdm38p, Mrs7p, and Ydl183cp form oligomers. Data provided here confirmed that Mdm38p self-dimerizes in mitochondria. However, it may hetero-oligomerize also with a yet unknown protein as suggested by BN and affinity chromatography data. Mdm38p appeared as part of high molecular complexes of ∼140, 232, and 500 kDa. Ydl183cp appeared as part of protein complexes with variable molecular sizes depending on the presence or absence of Mdm38p. Remarkably, in the absence of Mdm38p, Ydl183-GFP appeared as part of a complex of ∼500 kDa. In any case, a direct interaction between Ydl183cp and Mdm38p was not found according to the CoIP experiments performed in this work. Furthermore, Mrs7 was shown to be part of several protein complexes, including a complex of ∼500 kDa, independently of the presence or absence of Mdm38p. Thus, data presented here exclude a direct interaction between Mrs7p and Mdm38p or Ydl183cp. Altogether, the three proteins may act as cofactors interacting with a so far unidentified KHE, which in their absence would be completely inactive. This would be reminiscent of what has been observed for plasma membrane cation exchangers, where the exchanger is regulated by essential cofactors (39, 40). We propose that either Mdm38p or Mrs7p or Ydl183cp bind to a yet unknown protein or protein complex to activate the KHE activity. Although the composition of the 500-kDa complex and the molecular mechanisms through which Mdm38p, Mrs7p, and Ydl183cp modulate KHE activity remain to be elucidated, this study has identified intriguing new players that are amenable to further genetic analysis.
Finally, we included here for the first time a human gene, HCCR-1, which has been shown to play a role in cancer development (34). Furthermore, HCCR-1 shows sequence homologies to LETM1. We suggest a related role of HCCR1 to LETM1. Discrepancies in the quality as suppressor might result from its weak expression in yeast. Further studies will be required to test the direct role of HCCR-1 in the KHE.
Supplementary Material
Acknowledgments
We thank Kristina Djinovic for constructive discussions; Andrea Pichler for experimental advice; Paolo Bernardi for critical reading of the manuscript and supportive discussions; and Mirjana Iliev for expert technical assistance. We are indebted to Juraj Gregan for generously providing the yeast genomic library; Benedikt Westermann and Hiromi Sesaki for sharing the YFP and GFP vectors; and to Peter Rehling, Gottfried Schatz, Tom D. Fox, and Hans van der Spek for the antisera against Mdm38, F1β, Yme1, and Tim44, respectively.
This work was supported by the Austrian Science Fund (to R. J. S.) and SYSMO (to R. J. S. and K. N.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4 and an additional reference.
This paper is dedicated to the memory of Rudolf Schweyen, who tragically passed away during the writing of this manuscript.
G. Wiesenberger and K. Nowikovsky, unpublished data.
- KHE
- K+/H+ exchanger
- SMP
- submitochondrial particle
- FM4-64
- N-(3-triethylammoniumpropyl)-4-(6-(4-(diethylamino)phenyl)hexatrienyl)pyridinium dibromide
- GFP
- green fluorescent protein
- HA
- hemagglutinin
- CoIP
- coimmunoprecipitation
- ORF
- open reading frame
- Ni-NTA
- nickel-nitrilotriacetic acid
- GFP
- green fluorescent protein
- BCECF
- 2′,7′-bis(carboxyethyl)-5,6-carboxyfluorescein
- BN
- Blue Native
- YFP
- yellow fluorescent protein
- PBFI
- potassium-binding benzofuran isophthalate.
REFERENCES
- 1.Mitchell P. (1966) Biol. Rev. Camb. Philos. Soc. 41, 445–502 [DOI] [PubMed] [Google Scholar]
- 2.Froschauer E., Nowikovsky K., Schweyen R. J. (2005) Biochim. Biophys. Acta 1711, 41–48 [DOI] [PubMed] [Google Scholar]
- 3.McQuibban A. G., Joza N., Megighian A., Scorzeto M., Zanini D., Reipert S., Richter C., Schweyen R. J., Nowikovsky K. (2010) Hum. Mol. Genet. 19, 987–1000 [DOI] [PubMed] [Google Scholar]
- 4.Nowikovsky K., Froschauer E. M., Zsurka G., Samaj J., Reipert S., Kolisek M., Wiesenberger G., Schweyen R. J. (2004) J. Biol. Chem. 279, 30307–30315 [DOI] [PubMed] [Google Scholar]
- 5.Nowikovsky K., Reipert S., Devenish R. J., Schweyen R. J. (2007) Cell Death Differ. 14, 1647–1656 [DOI] [PubMed] [Google Scholar]
- 6.Zollino M., Lecce R., Fischetto R., Murdolo M., Faravelli F., Selicorni A., Buttè C., Memo L., Capovilla G., Neri G. (2003) Am. J. Hum. Genet. 72, 590–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waldherr M., Ragnini A., Jank B., Teply R., Wiesenberger G., Schweyen R. J. (1993) Curr. Genet. 24, 301–306 [DOI] [PubMed] [Google Scholar]
- 8.Frazier A. E., Taylor R. D., Mick D. U., Warscheid B., Stoepel N., Meyer H. E., Ryan M. T., Guiard B., Rehling P. (2006) J. Cell Biol. 172, 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho G. W., Shin S. M., Namkoong H., Kim H. K., Ha S. A., Hur S. Y., Kim T. E., Chai Y. G., Kim J. W. (2006) Gene 384, 18–26 [DOI] [PubMed] [Google Scholar]
- 10.Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. (1999) Nat. Biotechnol. 17, 1030–1032 [DOI] [PubMed] [Google Scholar]
- 11.Junttila M. R., Saarinen S., Schmidt T., Kast J., Westermarck J. (2005) Proteomics 5, 1199–1203 [DOI] [PubMed] [Google Scholar]
- 12.Wach A., Brachat A., Pöhlmann R., Philippsen P. (1994) Yeast 10, 1793–1808 [DOI] [PubMed] [Google Scholar]
- 13.Thorpe H. M., Smith M. C. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 5505–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cormack B. P., Bertram G., Egerton M., Gow N. A., Falkow S., Brown A. J. (1997) Microbiology 143, 303–311 [DOI] [PubMed] [Google Scholar]
- 15.Zinser E., Daum G. (1995) Yeast 11, 493–536 [DOI] [PubMed] [Google Scholar]
- 16.Fujiki Y., Hubbard A. L., Fowler S., Lazarow P. B. (1982) J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daum G., Gasser S. M., Schatz G. (1982) J. Biol. Chem. 257, 13075–13080 [PubMed] [Google Scholar]
- 18.Schägger H., von Jagow G. (1991) Anal. Biochem. 199, 223–231 [DOI] [PubMed] [Google Scholar]
- 19.Schamel W. W. (2008) Curr. Protoc. Cell Biol. 6, Unit 6.10.1–6.10.21 [DOI] [PubMed] [Google Scholar]
- 20.Nowikovsky K., Devenish R. J., Froschauer E., Schweyen R. J. (2009) Methods Enzymol. 457, 305–317 [DOI] [PubMed] [Google Scholar]
- 21.Sesaki H., Southard S. M., Yaffe M. P., Jensen R. E. (2003) Mol. Biol. Cell 14, 2342–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westermann B., Neupert W. (2000) Yeast 16, 1421–1427 [DOI] [PubMed] [Google Scholar]
- 23.Hamel P., Saint-Georges Y., de Pinto B., Lachacinski N., Altamura N., Dujardin G. (2004) Mol. Microbiol. 51, 307–317 [DOI] [PubMed] [Google Scholar]
- 24.Mühlenhoff U., Stadler J. A., Richhardt N., Seubert A., Eickhorst T., Schweyen R. J., Lill R., Wiesenberger G. (2003) J. Biol. Chem. 278, 40612–40620 [DOI] [PubMed] [Google Scholar]
- 25.Volckaert G., Voet M., Van der Schueren J., Robben J., Vanstreels E., Vander Stappen J. (2003) Yeast 20, 79–88 [DOI] [PubMed] [Google Scholar]
- 26.Ha S. A., Shin S. M., Lee Y. J., Kim S., Kim H. K., Namkoong H., Lee H., Lee Y. S., Cho Y. S., Park Y. G., Jeon H. M., Oh C., Kim J. W. (2008) Int. J. Cancer 122, 501–508 [DOI] [PubMed] [Google Scholar]
- 27.Kissová I., Salin B., Schaeffer J., Bhatia S., Manon S., Camougrand N. (2007) Autophagy 3, 329–336 [DOI] [PubMed] [Google Scholar]
- 28.Cortés P., Castrejón V., Sampedro J. G., Uribe S. (2000) Biochim. Biophys. Acta 1456, 67–76 [DOI] [PubMed] [Google Scholar]
- 29.Lee W. K., Spielmann M., Bork U., Thévenod F. (2005) Am. J. Physiol. Cell Physiol. 289, C656–C664 [DOI] [PubMed] [Google Scholar]
- 30.Rasheed B. K., Diwan J. J., Sanadi D. R. (1984) Eur. J. Biochem. 144, 643–647 [DOI] [PubMed] [Google Scholar]
- 31.Wojtczak L., Nikitina E. R., Czyz A., Skulskii I. A. (1996) Biochem. Biophys. Res. Commun. 223, 468–473 [DOI] [PubMed] [Google Scholar]
- 32.Bonangelino C. J., Nau J. J., Duex J. E., Brinkman M., Wurmser A. E., Gary J. D., Emr S. D., Weisman L. S. (2002) J. Cell Biol. 156, 1015–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamoto K., Shaw J. M. (2005) Annu. Rev. Genet 39, 503–536 [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y., Chan D. C. (2007) FEBS Lett. 581, 2168–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang D., Zhao L., Clapham D. E. (2009) Science 326, 144–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernardi P. (1999) Physiol. Rev. 79, 1127–1155 [DOI] [PubMed] [Google Scholar]
- 37.Dimmer K. S., Navoni F., Casarin A., Trevisson E., Endele S., Winterpacht A., Salviati L., Scorrano L. (2008) Hum. Mol. Genet. 17, 201–214 [DOI] [PubMed] [Google Scholar]
- 38.Hasegawa A., van der Bliek A. M. (2007) Hum. Mol. Genet. 16, 2061–2071 [DOI] [PubMed] [Google Scholar]
- 39.Pang T., Su X., Wakabayashi S., Shigekawa M. (2001) J. Biol. Chem. 276, 17367–17372 [DOI] [PubMed] [Google Scholar]
- 40.Weinman E. J., Cunningham R., Wade J. B., Shenolikar S. (2005) J. Physiol. 567, 27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.