

Biological context
Interleukin-2 tyrosine kinase (Itk) is a non-receptor tyrosine kinase of the Tec family that is activated upon antigen binding to the T cell receptor (Schwartzberg et al. 2005; Berg 2007). Itk is comprised of four regulatory domains: PH (Pleckstrin homology), TH (Tec homology), SH3, SH2 and the catalytic kinase domain. SH3 domains share a common fold consisting of five anti-parallel β strands that form a β barrel and bind canonical proline rich ligands as well as a variety of noncanonical ligands (Agrawal and Kishan 2002).
In contrast to Src family kinases (Boggon and Eck 2004), Tec family kinases lack the C-terminal regulatory phosphorylation site and consequently must be regulated by a distinct mechanism. We previously reported that the Itk SH3 domain binds the Itk SH2 domain in a novel noncanonical fashion (Brazin et al. 2000; Breheny et al. 2003). The classical PXXP motif that is often the target of SH3 domains is not present in the Itk SH2 domain. Correspondingly, the classical phosphotyrosine motif recognized by SH2 domains is not present in the Itk SH3 domain. We have found that disruption of the non-canonical SH3/SH2 intermolecular interaction in the context of full length Itk leads to an increase in Itk activity (Min, Andreotti et al., submitted). Thus, the interaction between these domains plays a regulatory role in Itk mediated signaling and currently efforts are aimed at determining the structure of the SH3/SH2 complex. Chemical shift perturbation mapping of the interface residues of the SH3/SH2 complex has been done and indicates that binding contacts are concentrated in the canonical binding pocket of the SH3 domain and the CD and BG loops of the SH2 domain (Brazin et al. 2000).
A previously solved structure (pdb: 1awj) contained the Itk SH3 domain plus the adjacent N-terminal proline-rich region of Itk (Andreotti et al. 1997), henceforth called PrSH3. The proline stretch binds to the Itk SH3 domain in an intramolecular fashion (Andreotti et al. 1997; Laederach et al. 2003) and the corresponding structure is therefore not a suitable model for determining the structure of the SH3/SH2 complex. Conjoined rigid body/torsion angle-simulated annealing that is used to solve structures of protein complexes relies on the basic assumption that the unbound and bound structures are similar and do not undergo large conformational changes upon binding (McCoy and Wyss 2002). In order to utilize the PrSH3 structure, the N-terminal proline-rich region containing 16 residues would need to be removed leaving the remaining residues in a ligand bound conformation. With the differences in the nature of the two ligands, proline-rich peptide chain versus protein surface (SH2 domain), the basic assumption that the bound and unbound structures are similar may not be met. Thus, the high-resolution solution structure of the unbound Itk SH3 domain is presented here as the first step toward solving the structure of the Itk SH3/SH2 complex.
Methods and results
NMR sample preparation
Protein expression and purification were performed as described previously (Brazin et al. 2000). The NMR sample consisted of 3.4 mM 13C, 15N labeled Itk SH3 domain, 50 mM NaPO4, 75 mM NaCl, 2 mM dithiothreitol (DTT), 5% D2O and 0.02% (w/v) NaN3 at pH 7.4.
NMR spectroscopy
All NMR spectra were collected at 298 K on a Bruker AVII 700 spectrometer equipped with a 5 mm HCN z-gradient cryoprobe operating at 700.133 MHz 1H frequency. Chemical shift assignments were elucidated from double and triple resonance experiments: CBCA(CO)NH, HNCACB, HBHA(CO)NH, HBHANH, HNCO, (HB)CB (CGCDCE)HE, (HB)CB(CGCD)HD, and HCCH-TOCSY, along with a 3D 15N-edited TOCSY and 2D homonuclear TOCSY. NOE correlations were obtained from a 2D homonuclear NOESY, 3D 13C-edited aliphatic NOESY, 3D 13C-edited aromatic NOESY, and 3D 15N-edited NOESY spectra. All NOESY experiments were acquired with a mixing time of 100 ms.
IPAP 1H-15N correlation experiments were performed to measure residual dipolar coupling constants. DNH values were determined from the difference between splittings recorded in anisotropic (J + D) and isotropic (J) media. Weak anisotropic alignment was achieved through the addition of 22.5 mg ml−1 of Pf1 phage (Hansen et al. 1998; Zweckstetter and Bax 2001).
J-coupling data were collected from a quantitative J correlation HNHA experiment (Cavanagh 2007). The J-coupling constants were included as restraints in the Xplor-NIH structure calculation with the following Karplus coefficients and phase: A = 6.98, B = −1.38, C = 1.72, phase = −60.0.
Backbone torsion angle (φ, ψ and ω) data were obtained from 1HN, 15N, 1Hα, 13CO and 13Cβ chemical shifts using PREDITOR (Berjanskii et al. 2006). Only those angles with a confidence greater than or equal to 0.5 were included. J coupling phi restraints gathered from J-coupling data were given precedence and the corresponding phi angles obtained from PREDITOR were excluded.
Backbone hydrogen bonds were identified from a long range coupling HNCO hydrogen bond experiment (Cordier et al. 2003). Two structural restraints were added in Xplor-NIH (Schwieters et al. 2003) for each hydrogen bond: one restraint between the donor hydrogen and acceptor oxygen (1.5–2.3 Å) and one restraint between the donor nitrogen and acceptor oxygen (2.5–3.3 Å). All spectra were processed using TOPSPIN 1.3 and analyzed with CARA (Computer Aided Resonance Assignment) for chemical shift and NOE assignments (Keller 2005).
Resonance assignments for Itk SH3 domain
CARA provides very effective modular tools to facilitate the assignment process. The correlation of Hα, Hβ, Cα, Cβ, and C′ crosspeaks with the preceding residue’s crosspeaks was carried out using CBCA(CO)NH, HNCACB, HBHA (CO)NH, HBHANH, HNCO data in the polyscope module. The automatic strip matcher was used to predict polypeptide fragments that were then joined after intervening residues were assigned and fragments were confirmed by inspection of NOESY and TOCSY data. Side chain assignments were then completed using the systemscope module. Aromatic crosspeaks, Hδ and Hε were assigned using (HB)CB(CGCDCE)HE and (HB)CB(CGCD)HD spectra. The remaining aromatic peaks were assigned using 2D homonuclear TOCSY and 2D homonuclear NOESY data. The assigned 15N, 1H HSQC is shown in Fig. 1.
Fig. 1.

1H-15N HSQC spectrum of Itk SH3 at pH 7.4, 298K. Backbone and side chain NH assignments are indicated using the one letter amino acid code and full length Itk numbering. Side chain residues are labeled with Hε1 for tryptophans (W208 and W209), Hδ2 for asparagines (N232, N185, and N215) and Hε2 for glutamines (Q212, Q183, and Q188)
Structure of Itk SH3
For structure determination, NOE crosspeaks were picked in polyscope using the propose function of Cara. To obtain a preliminary structure, a subset of NOEs were used for simulated annealing from an extended conformation. This initial NOE subset consisted of NOEs for which resonance assignments were unambiguous and which were consistent with the general fold of the previously solved Itk SH3 structure (Andreotti et al. 1997). Additional NOEs were then added as restraints in subsequent structure refinement. NOEs that gave rise to violations during structure refinement were examined. Those that were determined to be affected either by resonance overlap or proximity to the solvent signal were loosened or eliminated.
Hydrogen bond, J-coupling, and torsion angle restraints were then added and a set of 200 structures were generated by simulated annealing from extended structures. To incorporate RDC restraints, starting alignment tensor termsDa (axial symmetry) and R (Rhombicity) were calculated via SVD (singular value decomposition) using the average minimized structure generated from the 20 lowest energy structures among the set of 200. Finally, 60 structures were generated with simulated annealing using the following restraints: NOEs, hydrogen bonds, J-couplings, torsion angles, and RDCs. The 20 lowest energy structures based on bond, angle, improper, RDC, NOE, and torsion restraint energies (Fig. 2b) were used to generate an average structure for the free Itk SH3 domain (Fig. 2a). During the minimization, Da and R were allowed to vary, the final values for the average structure were −5.68 and 0.609 respectively.
Fig. 2.

Itk SH3 domain structure. (a) Stereo view of the Itk SH3 average minimized structure. The five β strands that form the β barrel are colored in yellow while the loops are green. Aromatic residues located in the binding pocket: Y180, Y225, W208, Y182, and Y220, are colored in cyan. (b) Ensemble generated from the superposition of the 20 lowest energy structures on the backbone atoms (Cα, C′, N) for residues spanning 173–229. Color scheme and side chains shown are as in (a). (c) Overlay of the Itk SH3 and PrSH3 average minimized structures colored in black and gray, respectively. Structural differences between Itk SH3 and PrSH3 are evident in the β1, β2 strands, and RT loop. The side chains of the proline-rich region (KPLPPTP) are shown for the PrSH3 structure. The different N- and C-termini for the two structures are labeled and the linker within PrSH3 is indicated
The quality of the final murine Itk SH3 structure was evaluated using PROCHECK_NMR (Laskowski et al. 1996) and WHATCHECK (Hooft et al. 1996). Ramachandran plot statistics in the core, allowed, generously allowed, and disallowed regions for residues in the average structure are 76.9%, 17.0%, 4.6%, and 1.5% respectively. The average backbone RMSD (N, Cα, C′) is 0.55 Å and average heavy atom RMSD is 1.27 Å (Table 1). All figures were created using PYMOL (DeLano 2002). The Itk SH3 structure has the same secondary structural elements found in other SH3 domains including five β strands that form a β barrel and a short 310 helix at the C terminus (Fig. 2).
Table 1.
Structural statistics
| Experimental restraints | Number of restraints | ||
|---|---|---|---|
| Total NOEs | 1,260 | ||
| Intra-residuea | |i − j| = 0 | 657 | |
| Sequential | |i − j| = 1 | 261 | |
| Medium | 1<|i − j|<5 | 77 | |
| Long range | |i − j|>5 | 265 | |
| Backbone hydrogen bondb | 30 | ||
| cdih total | 88 | ||
| φ | 8 | ||
| ψ | 24 | ||
| ω | 55 | ||
| J coupling | 23 | ||
| Residual dipolar coupling | NH | 27 | |
| Total restraints | 1,428 | ||
| Restraint statistics | Deviation | ||
| RMSD from standard geometry | |||
| Bonds (Å) | 0.004 | 0.0005 | |
| Angles (deg) | 0.508 | 0.017 | |
| Improper (deg) | 0.401 | 0.023 | |
| RMSD from experimental restraints | |||
| Distance (Å) | 0.041 | 0.002 | |
| cdih (deg) | 0.44 | 0.102 | |
| Ramachandran plot | |||
| Core | 76.9 | 3.7 | |
| Allowed | 17.0 | 3.6 | |
| Generously allowed | 4.6 | 2.5 | |
| Disallowed | 1.5 | 1.4 | |
| Average RMSD | Backbone | N,Cα,C′ | 0.55 Å |
| ItkSH3 | Heavy atoms | 1.27 Å | |
The variables i and j refer to residue numbers
There are two restraints for each of 15 hydrogen bonds
Coordinates
The coordinates have been deposited in the Protein Data Bank (accession codes: 2rn8, 2rna) Complete resonance assignments have been deposited into BioMagResBank (accession code 11018).
Discussion and conclusion
The Itk SH3 domain was solved to high resolution using NMR spectroscopy as a first step in determining the structure of the binary complex between Itk SH3 and Itk SH2. This Itk SH3 domain structure (Fig. 2a, b) is a better model than the previous Itk PrSH3 structure for use in conjoined rigid body/torsion angle-simulated annealing (McCoy and Wyss 2002). As expected, the overall folds of the free Itk SH3 domain and PrSH3 are similar yet the RMSD for backbone atoms (N, Cα, C′) between SH3 and PrSH3 for residues 173 through 229 is 2.52 Å. This can be compared to the corresponding backbone RMSD of 1.44 Å between the free Itk SH3 domain solved here and the Btk SH3 domain (pdb: 1awx) (Hansson et al. 1998). The larger RMSD between the free Itk SH3 domain structure and PrSH3 might therefore be significant. The largest differences are localized to the N-terminal half of the β2 strand and the β1 strand of the SH3 domain (Fig. 2c). Indeed, such differences were previously noted between the structure of the free Btk SH3 domain and Itk PrSH3 (Hansson et al. 1998).
The presence of the extended N-terminal proline-rich region of PrSH3 is the likely explanation for the differences between the two Itk SH3 structures (and for differences observed in the comparison between Btk SH3 and Itk PrSH3 (Hansson et al. 1998)). Intramolecular interaction between the proline-rich region and the aromatic binding cleft of PrSH3 (Andreotti et al. 1997; Laederach et al. 2003) appears to alter the conformation of the N-terminal region of the SH3 domain (including the β1 strand and the RT loop) and the β2 strand. The location of the observed structural differences are consistent with the fact that these elements are all close in space to the linker region that connects the proline-rich sequence and the N-terminus of the SH3 domain (Fig. 2c) suggesting that the intramolecular tether between SH3 domain and proline-rich ligand perturbs part of the SH3 structure.
We next examined side chain conformations in the conserved SH3 domain binding cleft for both free Itk SH3 domain and PrSH3. Three aromatic residues make direct contact to canonical proline ligands; Y180, Y225 and W208 (Itk numbering). There are discernable differences in the relative orientation of Y180 and Y225 between the free (structure solved here) and ligand bound (PrSH3) Itk SH3 structures. The angle between these two tyrosines appears to widen to accommodate the proline motif (Fig. 3a). Inspection of a number of other SH3 domains for which both free and ligand bound structures are available reveals similar changes in these two conserved tyrosines. As shown in Fig. 3b–d, the SH3 domains of Crk, Grb2, and Hck all show a similar trend in the position of Tyr 180 and 225 (Itk numbering) between free and ligand bound structures. In all of these cases, the angle between the two tyrosine side chains is smaller for the free SH3 domain and widens slightly upon ligand binding. For the Crk SH3 domain (Fig. 3b), it is notable that even for several different ligand bound structures the angle between the two tyrosine residues widens to a nearly identical extent upon ligand binding. It should be noted however, that the small changes evident between these free and bound SH3 domain structures are not universally observed as demonstrated by the SH3 structures of p40phox; ligand bound and unbound structures in that case overlay extremely well (Fig. 3e). Moreover, differences between structures that have been determined by NMR versus X-ray crystallography could contribute in part to the putative conformational changes and further systematic studies are needed to fully describe subtle SH3 structural changes that might occur on binding proline-rich ligands.
Fig. 3.
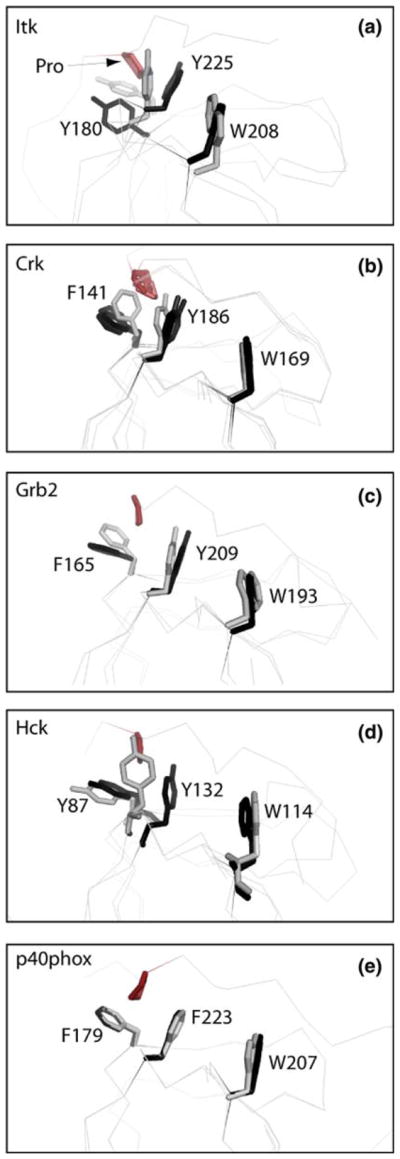
Superpositions of SH3 domain structures either bound to a proline-rich peptide ligand (colored black) or unbound (colored gray). The bound and unbound structures for each SH3 domain were superimposed using all SH3 domain atoms. Three aromatic residues within the SH3 binding pocket are depicted (Y180, Y225 and W208 for Itk) and labeled for each structure. Residue labels correspond to the full length numbering for each protein. For each bound structure the proline from the ligand that contacts Y180 and Y225 (Itk numbering) is indicated in red. The pdb code for each free and bound structure is: (a) Itk: this study (free), 1awj (bound); (b) c-Crk: 1m30 (free), 1bo7 (bound), 1cka (bound), 1ckb (bound); (c) Grb2: 1gfc (free), 1io6 (bound); (d) Hck: 4hck (free), 2oj2 (bound); (e) p40phox: 1w6x (free), 1w70 (bound)
With the high-resolution Itk SH3 domain structure now solved, NMR data acquired for the Itk SH3/SH2 complex can be more effectively analyzed to determine the molecular details of the non-classical interaction between the Itk SH2 and Itk SH3 domains. Additionally, this structure is more appropriate for comparisons with other SH3 structures than the previously solved PrSH3 structure.
Acknowledgments
This work is supported by grants from the National Institutes of Health (NIAID, AI43957) and a Roy J. Carver Charitable Trust graduate fellowship to A.S.
Abbreviations
- SH3
Src-homology 3 domain
- SH2
Src-homology 2 domain
- NMR
Nuclear magnetic resonance
- NOE
Nuclear overhauser effect
- NOESY
Nuclear overhauser enhancement spectroscopy
- TOCSY
Total correlation spectroscopy
- Itk
Interleukin-2 tyrosine kinase
- Btk
Bruton’s tyrosine kinase
- RDC
Residual dipolar coupling
References
- Agrawal V, Kishan KV. Promiscuous binding nature of SH3 domains to their target proteins. Protein Pept Lett. 2002;9:185–193. doi: 10.2174/0929866023408760. [DOI] [PubMed] [Google Scholar]
- Andreotti AH, Bunnell SC, Feng S, Berg LJ, Schreiber SL. Regulatory intramolecular association in a tyrosine kinase of the Tec family. Nature. 1997;385:93–97. doi: 10.1038/385093a0. [DOI] [PubMed] [Google Scholar]
- Berg LJ. Signalling through TEC kinases regulates conventional versus innate CD8(+) T-cell development. Nat Rev Immunol. 2007;7:479–485. doi: 10.1038/nri2091. [DOI] [PubMed] [Google Scholar]
- Berjanskii MV, Neal S, Wishart DS. PREDITOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 2006;34:W63–W69. doi: 10.1093/nar/gkl341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggon TJ, Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004;23:7918–7927. doi: 10.1038/sj.onc.1208081. [DOI] [PubMed] [Google Scholar]
- Brazin KN, Fulton DB, Andreotti AH. A specific intermolecular association between the regulatory domains of a Tec family kinase. J Mol Biol. 2000;302:607–623. doi: 10.1006/jmbi.2000.4091. [DOI] [PubMed] [Google Scholar]
- Breheny PJ, Laederach A, Fulton DB, Andreotti AH. Ligand specificity modulated by prolyl imide bond cis/trans isomerization in the Itk SH2 domain: a quantitative NMR study. J Am Chem Soc. 2003;125:15706–15707. doi: 10.1021/ja0375380. [DOI] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ. Protein NMR principles and practice. Elsevier; Burlington: 2007. [Google Scholar]
- Cordier F, Barfield M, Grzesiek S. Direct observation of Calpha-Halpha.O=C hydrogen bonds in proteins by interresidue h3JCalphaC′ scalar couplings. J Am Chem Soc. 2003;125:15750–15751. doi: 10.1021/ja038616m. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. DeLano Scientific; San Carlos: 2002. [Google Scholar]
- Hansen MR, Mueller L, Pardi A. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat Struct Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- Hansson H, Mattsson PT, Allard P, Haapaniemi P, Vihinen M, Smith CI, Hard T. Solution structure of the SH3 domain from Bruton’s tyrosine kinase. Biochemistry. 1998;37:2912–2924. doi: 10.1021/bi972409f. [DOI] [PubMed] [Google Scholar]
- Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- Keller R. Biology Diss. ETH Zurich NO. 15947; Zurich: 2005. Optimizing the process of nuclear magnetic resonance spectrum analysis and computer aided resonance assignment. [Google Scholar]
- Laederach A, Cradic KW, Fulton DB, Andreotti AH. Determinants of intra versus intermolecular self-association within the regulatory domains of Rlk and Itk. J Mol Biol. 2003;329:1011–1020. doi: 10.1016/s0022-2836(03)00531-x. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- McCoy MA, Wyss DF. Structures of protein-protein complexes are docked using only NMR restraints from residual dipolar coupling and chemical shift perturbations. J Am Chem Soc. 2002;124:2104–2105. doi: 10.1021/ja017242z. [DOI] [PubMed] [Google Scholar]
- Schwartzberg PL, Finkelstein LD, Readinger JA. TEC-family kinases: regulators of T-helper-cell differentiation. Nat Rev Immunol. 2005;5:284–295. doi: 10.1038/nri1591. [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Zweckstetter M, Bax A. Characterization of molecular alignment in aqueous suspensions of Pf1 bacteriophage. 2001. [DOI] [PubMed] [Google Scholar]