

Abstract
To fully understand how bacteria respond to their environment, it is essential to assess genome-wide transcriptional activity. New high-throughput sequencing technologies make it possible to query the transcriptome of an organism in an efficient unbiased manner. We applied a strand-specific method to sequence bacterial transcripts using Illumina's high-throughput sequencing technology. The resulting sequences were used to construct genome-wide transcriptional profiles. Novel bioinformatics analyses were developed and used in combination with proteomics data for the qualitative classification of transcriptional activity in defined regions. As expected, most transcriptional activity was consistent with predictions from the genome annotation. Importantly, we identified and confirmed transcriptional activity in areas of the genome inconsistent with the annotation and in unannotated regions. Further analyses revealed potential RpoN-dependent promoter sequences upstream of several noncoding RNAs (ncRNAs), suggesting a role for these ncRNAs in RpoN-dependent phenotypes. We were also able to validate a number of transcriptional start sites, many of which were consistent with predicted promoter motifs. Overall, our approach provides an efficient way to survey global transcriptional activity in bacteria and enables rapid discovery of specific areas in the genome that merit further investigation.
Pseudomonas syringae pathovar tomato DC3000 (DC3000), an agriculturally important bacterial plant pathogen, is a causal agent of bacterial speck on tomato and can infect the model plant Arabidopsis (18). It is also a well-known model organism for the study of plant-pathogen interactions (18). Completion of the genomic sequence several years ago provided an important resource for determining the molecular genetics of this bacterium, as well as of the other P. syringae pathovars (18). The annotation of the DC3000 reference genome is continually being updated and refined (http://pseudomonas-syringae.org/pst_home.html) to provide high-quality genome-associated information on which further P. syringae annotations will be based and also to provide more accurate insights into the function of many of the gene products to allow for more informed biological investigations.
Despite the availability of the genomic sequence for DC3000, very little is known about the expression of many predicted gene products and the complex regulatory mechanisms this bacterium uses to monitor and adapt to different environmental cues. We along with others have employed molecular and computational approaches to identify regulatory factors and genes used by DC3000 to adapt to specific environmental conditions (17, 28, 29, 45, 71). Although these studies have provided important information concerning the transcriptional regulation of predicted genes, more studies aimed at determining the location of promoters and regulatory sites, operon membership, and the identification of noncoding RNAs (ncRNAs) are needed to establish more complete and detailed regulatory gene networks for DC3000.
To better understand the full coding potential and determine the functional elements of the genome, it is important to obtain experimental confirmation of the predicted transcribed sequences in a comprehensive manner. Also, correctly assigning transcriptional activity to a particular strand is necessary since recent reports describing overlapping and antisense transcription suggest that more of the genome is transcribed than once thought (24, 40, 66, 80). High-throughput sequencing technologies, such as the 454 GS FLX (Roche), the Genome Analyzer (Illumina), and the ABI SOLiD (Life Technologies) have been used to analyze genome-wide RNA profiles of a number of organisms (20, 38, 50, 56, 69, 77, 79, 82, 83, 88). Because of the ease of working with eukaryotic mRNA, this methodology, termed RNA-Seq, was initially developed and used to investigate the transcriptomes of eukaryotic organisms (82). Not only has transcriptome sequencing been able to catalog transcripts expressed under a particular condition, it has also been used to confirm alternative splicing events and reveal sequence variations (82). In addition, the technologies have been shown to be reproducible for both technical and biological replicates (82). Most importantly, it has been shown that by analyzing a single RNA sample, RNA-Seq can be extremely informative (50, 77). Recently, with the advent of enrichment methods to deplete rRNAs, RNA-Seq has been extended to the study of a few microbes and has successfully confirmed transcriptional activity of annotated genes as well as revealing previously unannotated genes and identifying ncRNAs (2, 23, 55, 69).
In this paper, we describe a transcriptome analysis which couples strand-specific transcription sequence data with a qualitative computational analysis. We evaluated the transcriptome of P. syringae pathovar tomato DC3000 using Illumina technology and obtained 478 million bases of high-quality, strand-specific transcript data from a single biological sample. A qualitative analysis was developed that revealed the transcription of genes encoding hypothetical proteins and enabled identification of previously unannotated regions including putative coding sequences (CDSs) and ncRNAs. Interestingly, we identified transcriptionally active regions in the genome that contradict the annotation and represent antisense activity. Our efforts have provided further insights into the genomic sequence and transcriptional events in P. syringae DC3000.
MATERIALS AND METHODS
Bacteria and culture conditions.
P. syringae strain DC3000 was routinely cultured on King's B agar (42) medium at 30°C. For RNA isolation, bacteria were cultured as previously described (17). Briefly, bacteria were grown in iron-limited MG medium (10 g/liter of mannitol, 2 g/liter l-glutamic acid, 0.5 g/liter KH2PO4, 0.2 g/liter NaCl, 0.2 g/liter MgSO4; final pH of 7.0) (17) at 25°C in a bioreactor system (Sixfors). Bacterial cultures were collected at late exponential phase (optical density at 660 nm [OD660] of 0.6).
Isolation and enrichment of RNA.
RNA was isolated using Trizol (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Once isolated, RNA was treated with DNase (Ambion, Austin, TX) to remove residual DNA and then cleaned and concentrated using a MinElute kit (Qiagen, Valencia, CA). Removal of DNA was verified by quantitative real-time PCR with primers to the normalizing genes gap1 (PSPTO_1287) and gyrA (PSPTO_1745) (78). Integrity of the RNA was assessed using a Bioanalyzer (Cornell University Life Sciences Core Laboratory Center [CLC] Microarrays Facility, Cornell University). Total RNA was processed using a MicrobExpress kit (Ambion) with the Pseudomonas module to remove the 23S and 16S ribosomal RNAs (rRNAs). Removal of rRNAs was assessed using an Agilent Bioanalyzer.
RNA processing.
Approximately 100 ng of ribosomal depleted RNA was fragmented by the addition of fragmentation buffer (Ambion) and heating at 70°C for 10 min. Stop buffer was added, and tubes were placed on ice. Next, the fragmented RNA was treated with calf intestinal alkaline phosphatase (CIP), and fragments of ∼50 to 60 nucleotides (nt) were excised from the gel, eluted from the agarose into elution buffer (Small RNA Sample Prep Kit, version 1.5; Illumina) at room temperature for 4 h, and then precipitated with ethanol (EtOH). An adapter (/App/TCGTATGCCGTCTTCTGCTTGddC, where App is a preadenylated modification and ddC is dideoxycytosine) was ligated to the 3′ ends of the RNA fragments. The 3′ adapter is 5′ adenylated to allow for ATP-independent ligation and contains a 3′ dideoxycytosine to prevent adapter self-ligation. T4-truncated ligase 2 (New England Biolabs) was used for the ligation. Fragments were gel purified and then treated with polynucleotide kinase (PNK). Next, an adapter (GUUCAGAGUUCUACAGUCCGACGAUC) was ligated to the 5′ end of the fragments, and fragments were gel purified. Reverse transcription-PCR (RT-PCR) was performed with the purified ligated RNA with the primer CAAGCAGAAGACGGCATACGA. The products were then amplified in 15 cycles of PCR using the primers CAAGCAGAAGACGGCATACGA and AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA, and resultant products were subjected to gel electrophoresis. Constructs of ∼200 bp were gel purified and sequenced on an Illumina Genome Analyzer II. The reads correspond to the sequence conventionally designated the sense or coding strand, i.e., the sequence of the transcribed RNA.
5′ RACE, 3′ RACE, and RT-PCR.
Transcriptional start points were determined using Invitrogen's 5′ RACE system for rapid amplification of cDNA ends (version 2.0) as recommended by the manufacturer. RNA isolated from cells grown as described above was used as the template. Oligonucleotides used for reverse transcription and PCR are listed in Table S1 in the supplemental material. Products were separated by agarose gel electrophoresis to assess purity and product size. Products were excised, gel eluted (Zymogen), and sequenced. The sequencing results were interpreted by pairwise alignments of the 5′ RACE product sequence with the DC3000 genomic sequence. Sequencing data generated from 5′ RACE were performed by the CLC DNA Sequencing and Genotyping Facility.
3′ RACE was performed using a protocol adapted from Argaman et al. (10). Briefly, 1 μg of RNA was mixed with 100 pmol of RNA adapter (5′-phosphate-UUC ACU GUU CUU AGC GGC CGC AUG CUC-idT-3′), heat denatured at 95°C for 5 min, and then quick chilled on ice. The adapter was ligated at 17°C for 12 h with 40 units of T4 RNA ligase (New England Biolabs), 40 units of RNase OUT (Ambion) in a buffer containing 50 mM Tris-HCl (pH 7.9), 10 mM MgCl2, 4 mM dithiothreitol (DTT), 150 μM ATP, and 10% dimethyl sulfoxide (DMSO). Ligated RNA was purified using an RNA Clean and Concentrator-5 kit (Zymogen) and reverse transcribed using 20 pmol of a single primer complementary to the RNA adapter (5′-GAG CAT GCG GCC GCT AAG AAC AGT G-3′). Reverse transcription was performed using a Thermoscript reverse transcriptase system (Invitrogen) according to the manufacturer's protocol. Products were amplified using a 2-μl aliquot of the RT reaction mixture and 20 pmol of each gene-specific and adapter-specific primer. Bands of interest were excised, gel eluted, and cloned into pCR 2.1 TOPO vector (Invitrogen). Plasmids from three separate clones were sequenced.
RT-PCRs were performed using strand-specific primers and an Omniscript kit (Qiagen) according to the instructions provided by the manufacturer. RT-PCR negative controls were performed using non-reverse-transcribed RNA as a template.
Protein extraction.
Cell pellets were suspended in the extraction buffer at a ratio of 1 g of cells to 4 ml of buffer. The cells were disrupted in a French press (ThermoSpectronic) equipped with a 4-ml mini-cell. Streptomycin was added to the supernatant to give a final concentration of 4% (wt/vol), followed by centrifugation at 4,000 rpm (3,724 × g) for 30 min at 4°C. Protein was precipitated by the addition of 9 volumes of cold, acidified acetone. This suspension was incubated at −20°C for 1 h and then centrifuged at 4,000 rpm for 40 min (3,724 × g). The supernatant was decanted, and the pellet was resuspended twice with 5 ml of cold acetone and repelleted by centrifugation. After the final rinse the pellet was air dried for 30 min. The pellet was redissolved in iTRAQ dissolution buffer (0.5 M triethylammonium bicarbonate), and the protein concentration was determined using a Bradford assay (15). The material was stored at −80°C until used.
iTRAQ labeling.
Proteins (50 μg of each sample) were denatured by adding 1 μl of 2% SDS and reduced by addition of 2 μl of 50 mM Tris-(2-carboxyethyl) phosphine. The cysteine residues were blocked using 1 μl of 200 mM methyl methanethiosulfonate as described previously (62, 68). These samples were digested with trypsin (65), dried completely, and reconstituted in 50 μl of the dissolution buffer (500 mM triethylammonium bicarbonate). iTRAQ tags were added according to the manufacturer's instructions (8, 9) to enhance ionization and fragmentation. Four technical replicates were produced and analyzed.
2-D LC-MS/MS analysis.
The dried iTRAQ-labeled peptide mixtures were reconstituted in 0.5 ml of solvent A (10 mM potassium phosphate, pH 2.75, 20% acetonitrile) and fractionated on a PolySulfoethyl A column (5 μm, 200 Å, 2.1 mm by 100 mm; PolyLC Inc. Columbia, MD) using an AKTA high-performance liquid chromatography (HPLC) system (GE Healthcare Bio-Sciences, Piscataway, NJ). Peptides were eluted with a segmented linear gradient of 0 to 10% solvent B (10 mM potassium phosphate, pH 2.75, and 20% acetonitrile with 1 M KCl) for 2 min, 10 to 18% solvent B for 30 min, and 18 to 50% solvent B for 5 min at a flow rate of 0.2 ml/min. These fractions were desalted by solid-phase extraction using Waters C18 cartridges, dried, and reconstituted with 30 μl of 2% acetonitrile-0.5% formic acid for analysis by nano-liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI MS/MS) as described previously (89). iTRAQ labeling and two-dimensional (2-D) LC-MS/MS processing was done by the CLC Proteomics and Mass Spectrometry Facility.
Protein identification.
Spectra collected during nano-LC ESI MS/MS runs were submitted to Mascot, version 2.2, for a search against a database of all maximal open reading frames (ORFs) represented in the P. syringae DC3000 genome using a mass tolerance of 1.5 Da for precursor mass and 0.6 Da for fragment mass. Search parameters allowed for one missed cleavage of semitrypsin, oxidation (M)/iTRAQ4plex (Y) as variable modifications, and iTRAQ4plex for K side chains and N-terminal residues and Methylthio C as fixed modifications. The protein identifications were obtained by requiring the detection of at least one peptide with an E-value less than or equal to 0.005.
Alignment and filtering of Illumina sequences.
A total of 29,622,520 reads were generated. These sequences were aligned to the DC3000 genome using ELAND, and all reads that aligned with the genome ambiguously or with mismatches were discarded. This resulted in a filtered set of 14,951,312 unique perfect matches (UPMs). We observed that the ELAND alignment tool did not account for the circular topology of the DC3000 replicons. Using string matching, we realigned the nonaligned reads with the sequences that spanned the origins of replication and found that an additional 20 UPMs could be uniquely and perfectly aligned to DC3000. The final data set used for our analysis contained 14,951,332 UPMs.
Sequence analysis.
Let L be the formal language defined as the set of all sequences that could be produced by the high-throughput sequencing platform used in the experiment, and let s ∈ L represent an individual sequence or word in this language. For this study, L is the set of all 32-mers composed of the letters A, C, G, and T.
A high-throughput sequencing experiment is represented as a function, E, that maps individual sequences to a count of the number of times that it was observed during the experiment. In other words, E : L → N, where N is the set of nonnegative integers.
Let G be the set of positions within a genome and p ∈ G represent an individual position. The protocol is strand specific, so positions can be represented by triples of the following form: p = 〈replicon,strand,index〉.
The result of aligning the sequences produced by an experiment, E, against a genome, G, will be represented by two functions: POSE,G maps a sequence to a set of positions on the genome, and 5′E,G maps a sequence to its 5′ end positions, represented as POSE,G : L → 2G and 5′E,G : L → 2G, respectively.
Every sequence that is aligned is assigned both a position and a 5′ end. 5′ ends must lie within the set of aligned positions. Thus, we take the following as axioms:
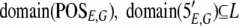 |
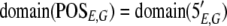 |
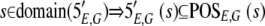 |
The POS function was constructed only for the sequences that aligned to precisely one region of the genome and without errors. Thus, the POS function is not defined for sequences that did not match exactly or uniquely align with G. Also, because each UPM is aligned with only one set of positions on the genome, 5′E,G(s) will always be a singleton set.
The “overlap” of a position, p, is the set of sequences from any experiment, E, whose alignment can contain p: OverlapG(p) = ∪E {s : s ∈ L | p ∈ POSE,G (s)}.
We define the measure of local uniqueness (MLU) at position p as the cardinality of OverlapG(p). That is, MLUG(p) = |OverlapG(p)|.
Because only 32 UPMs of length 32 can overlap a position p, MLUG(p) will be 32 in regions of the genome consisting of unique strings and 0 in regions that are highly repetitive. Note that OverlapG(p) and MLUG(p) are properties inherent to the genome G and not any particular high-throughput experiment E.
The “sinister,” or S, function of a position, p, is the sum of the counts of the sequences whose 5′ end aligns with p. Because of the alignment strategies, there can be exactly 0 or 1 sequence, s, whose 5′ end is p; so S can be defined as follows: SE,G(p) = E(s) where {p} = 5′E,G(s); otherwise, SE,G(p) = 0.
The “in media res,” or IMR, function of a position, p, is the geometric mean of the count of the sequences that overlap p:
 |
For the subsequent analysis based on geometric means, we used the logarithmic transformation of two closely related functions that include an offset to avoid undefined values:
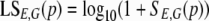 |
 |
Profile generation.
The positions on the chromosome are represented as pairs, p = 〈s,i〉, where s refers to strand, which will be either plus or minus, and i refers to the index. We did not consider the two plasmids of DC3000.
Many of the functions defined here are represented as numerical vectors and arrays and therefore are amenable to visualization using Artemis and manipulation using linear algebra toolkits, such as Matlab. To represent these functions numerically, we must account for the fact that many are defined only for a subset of the positions of the genome. When a function is represented as a numerical array, the value of each array element will represent the value of the function evaluated at a single genomic position. Thus, using standard array notation, F[s,i] will represent the value of F(〈s,i〉), if F(〈s,i〉) is defined. Otherwise, F[s,i] = 0. Note: square brackets, “F[...],” will be used instead of parentheses, “F(...),” to distinguish matrices from their corresponding functions. F[s,i] = 0.
Thus, the functions MLUG(p), LSE,G(p), and LIMRE,G(p) can all be represented as the arrays MLU[s,i], LS[s,i], and LIMR[s,i]. The terms “arrays” and “profiles” are used interchangeably. It can be shown that MLU[P,i] = MLU[M,i], so we use MLU[i].
Evaluating disjointedness and technical reproducibility.
First, the sets of results were reexpressed as matrices. To evaluate the disjointedness of the two strands, two vectors were constructed from the two strands of the LIMR profile: ai = LIMR[Plus,i] and bi = LIMR[Minus,i]. Disjointedness was measured using Pearson's uncentered correlation, which is the cosine of the angle between these two vectors: r = cos θ = (aTb)/(||a|| ||b||), where T indicates the matrix transpose operator. For the DC3000 chromosome under these conditions, the Pearson's uncentered correlation is 0.067 (theta = 86.24°). This indicates that the two strands are uncorrelated, and therefore expression is essentially disjoint with little overlap.
Similarity between technical replicates was measured by first embedding results of the four technical replicates into a single matrix, A, as follows: Aij = LIMRj[Plus,i] if i ≤ n, and Aij = LIMRj[Minus,i − n] if i > n, where n is the length of the genome and LIMRj is the LIMR profile computed using the reads from the jth replicate.
Next, the singular value decomposition (SVD) of A was computed as follows: A = U Σ VT. Since the singular values of matrix Σ appear in decreasing order and U is a unitary matrix, the first column of matrix V gives the most contribution to all of columns of A. Thus, if the columns of A are similar, then σ1 will be much larger than the other σi's. Our similarity index is defined as follows: similarity = σ12/(Σi σi2). The similarity index was 98.41%, suggesting that variation is very small when sequences are determined from the same library preparation. More information concerning other properties of the SVD and how it can be computed efficiently can be found in reference 31. (Other applications to biological problems can be found in references 3-6, 16, 36, and 44.)
Computing region-based transcriptional activity scores.
Transcriptional activity score (TAS) over a region was defined as follows. Let Region[s,b,e,c] be the set of positions, p, between positions b and e on strand s, where MLU[p] is greater than or equal to cutoff, c. Further, let |Region[s,b,e,c]| be the cardinality of Region[s,b,e,c]. Then,
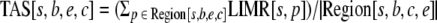 |
if |Region[s,b,e,c]| > 0. If |Region[s,b,e,c]| = 0, then TAS[s,b,e,c] = 0. The value c is the MLU cutoff. For the classification analysis, an additional length cutoff, l, was used on |Region[s,b,e,c]|. When we impose an MLU cutoff of c and a length cutoff of l, we compute TAS[s,b,e,c] only when |Region[s,b,e,c]| ≥ l.
Because the LIMR profile values are the logarithm of a geometric mean of the counts of UPMs overlapping a position, the TAS can be interpreted as the logarithm of a geometric mean of the pseudocounts of UPMs overlapping the region.
Computing the threshold for classifying transcriptional activity.
A receiver operating characteristic (ROC)-like analysis was used for setting the threshold, t, for classifying transcriptional activity. First, the TAS was computed for both annotated and opposite strands of all CDSs that overlapped and had the same orientation as any of the peptides obtained from the proteomics data. When computing these scores, we used the cutoffs c = 31 and l = 50. Next, the threshold, t, was chosen to maximize
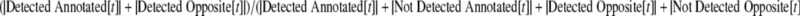 |
where |Detected Annotated[t]| is the number of CDSs whose annotated strand TAS value is ≥ t, and the other variables are defined similarly.
The threshold was 0.0754, which corresponds to an annotated strand detection rate of 99.36% and an opposite strand detection rate of 2.97%.
Classification.
Using the classification threshold t, all annotated CDS regions on the chromosome were classified using an MLU cutoff of c = 31 and a length cutoff of l = 50. For the regions defined by the Rfam predictions and other putative ncRNAs, the same method for computing TAS values of the annotated and opposite strands was used, but an MLU cutoff of zero was used because these regions are very short. We do not report results for tRNAs and rRNAs because of their low MLU values.
RESULTS
Sequencing, assembly, and sequence analyses.
To characterize the transcriptome of P. syringae DC3000, total RNA was isolated from bacteria grown under iron-limited conditions in a defined medium that allowed for virulence factor expression (17). We then used a commercially available kit to capture and deplete the 23S and 16S rRNAs. Approximately 95% of rRNA was removed by this procedure, and the remaining sample contained ∼50% rRNA and ∼50% mRNA by mass (see Fig. S1 in the supplemental material). The RNA was converted to cDNA in a manner that retained strand specificity and was subjected to sequencing on the Illumina Genome Analyzer II (GAII) sequencing system.
To maximize coverage and enable the analysis of technical reproducibility (see Materials and Methods), the cDNA library was analyzed on four independent lanes and two different flow cells (two independent runs). In total, 29,622,520 reads were generated, each with a length of 32 nucleotides (Table 1). Since technical replicates were in close agreement with one another (see Materials and Methods), subsequent analyses were performed using the merged data.
TABLE 1.
Summary of sequencing statistics
| Read type | No. of reads for the indicated replicate |
|||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Reads with unique alignments | 4,053,237 | 4,332,106 | 4,444,145 | 4,207,913 |
| Reads with unique and perfect alignments | 3,502,766 | 3,815,662 | 3,978,342 | 3,654,542 |
| Total | 7,072,779 | 7,523,131 | 7,678,259 | 7,348,351 |
Alignments to the DC3000 genome were generated, and reads that did not align with the genome uniquely and perfectly were discarded, including those that corresponded to the rRNAs. This resulted in a filtered set of 14,951,312 reads with unique perfect matches (UPMs) to the genome, which represents ∼50% of the original data set and is consistent with ∼50% of the sample being rRNA.
In order to enable visualization and data analysis, we generated several profiles. The first profile constructed was the measure of local uniqueness, or MLU profile, which is unstranded (assigning a single value per base pair). The MLU of a position is defined as the number of unique 32-mers in the genome that overlap that position (51). MLU values range from 32, when all 32 overlapping 32-mers are unique, to 0, when none of the 32-mers is unique. A total of 94.50% of the positions in the main chromosome have MLU values of 31 or greater. The remainder consists largely of rRNA genes, highly represented families of transposable elements (18), and repetitive extragenic palindromic (REP) sequences (73). Since the two plasmids carried by DC3000 contain many duplicate sequences and have low MLU values, they were not analyzed further. The second profile constructed was the sinister, or S, profile which is stranded (assigning a value to the positive and negative strand for each base pair). In this profile, the number of UPMs whose 5′ ends (hence, sinister, or left) align with each position on the positive and negative strands of the genome were counted. A logarithmic transformation was applied to each value of the S profile to produce the logarithmic sinister, or LS, profile. The final profile, also stranded, is called the logarithmic in medias res (meaning in the middle), or LIMR profile, whose values are the logarithm of the geometric mean of the number of UPMs that overlap a position weighted by the number of unique 32-mers that overlap the same position (i.e., MLU). All of the profiles are available in Files S1, S2, and S3 in the supplemental material in a format suitable for viewing in Artemis.
Figure 1 (see also Files S2 and S3 in the supplemental material) shows the LS and LIMR profiles layered onto the DC3000 genome as visualized using the Artemis genome browser (http://www.sanger.ac.uk/Software/Artemis/). These figures show a position-by-position snapshot of the transcriptional activity. The resulting profiles have continuous stretches of transcriptional activity and sharp transitions. We should note that the profiles are also nonuniform, as has been previously reported (21, 52, 53, 57, 86), due to a number of factors such as stochastic artifacts from PCR amplification, fragmentation, and other steps during sample preparation.
FIG. 1.

Profiles displayed using Artemis depicting the disjointedness of the transcriptional activity (A) and overlapping transcriptional activity (B). The transcriptional profile (orange and purple) is shown above the annotated genome. An orange trace indicates transcription occurring from left to right (on the positive strand), and a purple trace represents transcription occurring from right to left (on the negative strand). The green regions represent the CDSs that demonstrate transcriptional activity consistent with the annotation (described in “A global qualitative classification of transcriptional activity” in the Results section,). The yellow regions represent the CDSs that demonstrate transcriptional activity on both strands. The regions that represent promoter motifs are depicted in cyan. Hrp box, the motif recognized by the sigma factor HrpL; RpoN box, the motif recognized by the sigma factor RpoN.
Identification of transcriptional start sites.
The profiles generated from the RNA sequencing provided the opportunity to investigate the transcription start sites of transcripts or stable 5′ ends. Visual inspection of the LS profiles revealed many cases in which transcriptional activity rises abruptly just upstream of annotated genes. To confirm if these abrupt changes corresponded to transcriptional start sites, we determined the 5′ ends of a number of transcripts and compared the results with the RNA-Seq transcript profiles. The transcripts were chosen because (i) we had either previously determined the transcriptional start site/stable 5′ end by 5′ RACE, (ii) they contained putative promoter predictions of interest, or (iii) they represented unusual cases of expression (i.e., possible misannotation and ncRNAs). The transcripts evaluated included annotated ORFs, unannotated ORFs, and putative ncRNAs (see Table S1 in the supplemental material). In nearly all cases (58/66), the primary transcriptional start site mapped by 5′ RACE was consistent with the apparent transcriptional start site determined by global transcript sequencing. Eight transcripts were not considered to be consistent with the RNA-Seq data either because there was no clear abrupt spike in transcriptional activity indicating a transcriptional start site, two 5′ ends were identified using 5′ RACE (one which was consistent with the profile and one which was not), or the 5′ end mapped by 5′ RACE was further upstream than the start site indicated by the RNA-Seq data.
Interestingly, a number of transcriptional start sites were verified to lie within an annotated ORF or at the translational start codon (e.g., PSPTO_2030, PSPTO_3157, PSPTO_3836, PSPTO_3841, PSPTO3976, and PSPTO_5516). For PSPTO_2030, the predicted protein appears to be conserved in P. syringae pv. phaseolicola 1448A and in P. syringae pv. tabaci ATCC 11528 and is predicted to be considerably shorter (311 amino acids) than the protein in DC3000 (333 amino acids). This is also the case for PSPTO_3157, which is predicted to be 210 amino acids in DC3000 and 177 and 183 amino acids in P. syringae pv. tabaci and P. syringae pv. syringae B728a, respectively. For PSPTO_3836, PSPTO_3841, PSPTO3976, and PSPTO_5516, conserved proteins are found in a number of the other pseudomonads, and the predicted lengths are equal. These results suggest that the translation initiation sites may be misannotated or that there are multiple overlapping transcripts of the same region that cannot be resolved by these methods.
Next, we globally determined if the predicted motifs for the transcriptional repressor Fur and the sigma factor HrpL correlated with predicted transcriptional start sites indicated by the transcript profiles. We previously reported 12 operons in DC3000 that are iron regulated and are associated with a predicted Fur motif (17). Of those 12 operons, 9 have obvious transcriptional start sites within 20 bp of the predicted Fur motif. For the remaining three cases, the predicted Fur motif lies either in a region where there is divergent transcription or in a region of overlapping transcriptional activity, so we did not make a determination as to which gene(s) to assign the putative Fur binding site. For HrpL, transcriptional start sites could be identified for 34 of the 50 genes which have a predicted HrpL promoter motif (28). Sixteen could not be resolved because the transcripts were not expressed or the predicted promoter motif was located in the middle of a CDS. Overall these findings indicate that visual inspection of the LS profile in combination with computational predictions for promoter motifs facilitates the identification of candidate transcriptional start sites.
A global qualitative classification of transcriptional activity.
A global assessment indicated that expression is essentially disjoint, occurring on separate strands (see Materials and Methods and below). However, through visual inspection, we observed several regions of the genome with significant expression on both strands at the same base pair. An example of such coincident expression is shown in Fig. 1B. Because identification of these areas by visually scanning the entire genome is time-consuming and subjective, we wanted to develop a method to quickly identify regions that have transcriptional activity above background and also determine whether transcriptional activity within regions of the genome is inconsistent with annotation or displays antisense activity. Therefore, we devised a system to make a qualitative assessment of gene expression.
To do this we incorporated additional biological information derived from the qualitative analysis of protein expression in the same sample. A key biological insight we exploit is that if a specific protein is detected in our sample (which was taken during exponential growth phase), then the mRNA encoding that protein must be present as well, at least in a subpopulation of cells (11). We also make two assumptions that make it possible to exploit the qualitative analysis of mRNA expression inferred from the existence of encoded proteins. First, for a given CDS, the level of mRNA expression, and thus the frequency of strand-specific Illumina sequence reads, is expected to be higher on the sense strand than on the antisense strand. Second, we assume that in cases where proteomics data are consistent with the annotation, then transcription will overwhelmingly occur on the sense strand. Therefore, the set of opposite strands relative to protein coding genes with confirmed protein expression can be used to characterize background levels of RNA transcription.
To obtain the proteomics data, we conducted an MS/MS analysis using proteins taken from the same sample used to generate the sequence data. A total of 5,320 peptides were aligned with the chromosome (see File S4 in the supplemental material). Of these, 5,310 overlapped and had the same orientation with 471 CDSs in the current annotation. The analysis revealed protein expression from a wide variety of genes, including those that code for core functions such as ATP biosynthesis, ribosomal proteins, and biosynthetic pathways for amino acids, nucleic acids, and essential enzymatic cofactors, as expected from cells grown in minimal medium. Only 10 peptides did not overlap or did not have the same orientation with the annotated CDSs (see File S5).
To summarize the transcriptional activity over regions of the genome, we calculated a transcriptional activity score (TAS) value for the annotated (a) and opposite (o) strands of annotated regions. Next, we took the proteomics results as experimental confirmation of these 471 CDS annotations and used the TAS values for these regions as training sets for setting a classification threshold, t, of 0.0754 (see Materials and Methods). Using the computed classification threshold, t, we assigned each CDS to one of four classes. (i) For the green class, a ≥ t and o < t. There is consistent transcriptional activity; i.e., transcription is detected above the threshold on the annotated strand and below on the opposite strand. (ii) For the red class, a < t and o ≥ t. Transcriptional activity is inconsistent; i.e., transcription is detected above the threshold on the opposite strand and below the threshold on the annotated strand. (iii) For the yellow class, a ≥ t and o ≥ t. Transcriptional activity is ambiguous; i.e., transcription is detected above the threshold on both strands. (iv) For the gray class, a < t and o < t. Transcriptional activity is below the threshold on both strands. This color classification is used in Fig. 1, 3, and 5. A scatter plot comparing the annotated and opposite-strand TAS values for the 5,256 CDSs is shown in Fig. 2 (see also Fig. S3 in the supplemental material) and the CDSs in each category are provided in File S6. The vast majority of the CDSs with proteomics support were classified as green (454/471), very few were classified as yellow (15/471) and gray (2/471), and none were classified as red. Therefore, we are confident that this use of proteomics data is useful for establishing a threshold and assigning classifications.
FIG. 3.

Profiles displayed using Artemis showing higher expression on the antisense strand than on the sense strand. The transcriptional profile (orange and purple) is shown above the annotated genome. An orange trace indicates transcription occurring from left to right (on the positive strand), and a purple trace represents transcription occurring from right to left (on the negative strand). (A) Transcriptional activity occurring on both strands for cmaE. (B) Evidence of expression of a previously unannotated CDS. The red region represents the CDS for PSPTO_2357, and the blue region represents the EasyGene call on the opposite strand. (C) An example of read-through activity in the psyI/psyR region.
FIG. 5.

Profiles displayed using Artemis illustrating transcriptional activity in an intergenic region. The transcriptional profile (orange and purple) is shown above the annotated genome. An orange trace indicates transcription from left to right (on the positive strand), and a purple trace represents transcription from right to left (on the negative strand). (A) The region between PSPTO_0963 and PSPTO_0964 contains the newly identified ncRNA psr1. (B) The region between PSPTO_1620 and PSPTO_1621 contains the newly identified ncRNA psr2. (C) The region between PSPTO_2739 and PSPTO_2740 contains the newly identified ncRNA psr3. The promoter motif recognized by the sigma factor RpoN is depicted in cyan.
FIG. 2.

Scatter plots of the average scores for all CDSs. In order to be considered, a CDS must have at least 50 bp whose MLU value is 31 or higher. The classification threshold is shown on both the x and y axes of each plot. The x and y axes correspond to the scores of the annotated and opposite strand of each gene, respectively. The lines at x = 0.0754 and y = 0.0754 determine the boundaries between the classification regions.
Of the CDSs annotated in the DC3000 genome that were classified, 2,017 genes were not detectable or had expression below our threshold (gray). A total of 3,009 (∼57%) of the genes demonstrated expression consistent with the annotation (green). This list includes genes previously found to be expressed under iron-limited conditions, such as those involved in iron uptake, iron binding, and iron transport (17). Also, we detected expression of 223 genes that encode known or predicted virulence-related genes (18, 48), including those involved in type III secretion (T3S) and coronatine production (see Table S3 in the supplemental material). Importantly, 818 out of 1,646 CDS annotated as hypothetical proteins were expressed under iron-limited conditions, providing evidence of expression for these gene products. Product names for expressed genes previously annotated as hypothetical or conserved hypothetical have been changed in the annotation record at GenBank to protein of unknown function and conserved protein of unknown function, respectively.
Identification of antisense activity.
A total of 124 genes demonstrated ambiguous expression in that expression on both strands was above our set threshold (yellow). Surprisingly, cmaE and cmaU (PSPTO_4708 and PSPTO_4714, respectively), which encode proteins involved in the production of coronatine (70, 76), were classified in this category (Fig. 3A; see also File S6 in the supplemental material). These genes appear to be within an operon with no apparent ORF present on the opposite strand. This category also included a number of type III genes encoding components of the T3S system: hopK1 (PSPTO_0044), hopAJ1 (PSPTO_0852), and hopAK1 (PSPTO_4101), as well as the transcriptional regulator aefR (PSPTO_3549) (60, 61). The presence of several antisense transcripts was confirmed by RT-PCR from total RNA (see Fig. S4). The ambiguous transcriptional activity may indicate that these genes have an unusual mechanism of regulation. Taken as a whole, these examples support the idea that P. syringae DC3000 has several instances where overlapping (antisense) transcription occurs.
Identification of new genes.
We found 106 regions with expression that was inconsistent with the genome annotation (red) (see File S6 in the supplemental material). These inconsistencies may arise due to unannotated ORFs located on the opposite strand of an annotated CDS (Fig. 3B). To help identify missed coding regions (unannotated ORFs), EasyGene, version 1.2, predictions (46, 54) for DC3000 were obtained from the Center for Biological Sequence Analysis, Technical University of Denmark (http://servers.binf.ku.dk/cgi-bin/easygene/search). This tool was selected because it uses a fundamentally different technique for gene identification than was used in the original annotation. EasyGene predictions were compared with the primary genome annotation and the LIMR profiles for genes that displayed inconsistent expression. Eleven cases were identified where expression inconsistent with the original annotation corresponded to an EasyGene gene call (Table 2). Of the 11 newly predicted ORFs, two (old locus tag PSPTO_1442, now PSPTO_5635, and old locus tag PSPTO_3093, now PSPTO_5642 and PSPTO_5643) were supported by proteomics data. The sequences of the putative ORFs were searched against the nonredundant (NR) database from NCBI using BLASTP (7). Most of these predicted genes were found to encode hypothetical proteins (Table 2). One of the predicted proteins, encoded by the new ORF antisense to PSPTO_2357, contains a conserved domain present in the Fur (ferric uptake regulator) superfamily (COG0735) and is similar to PA2384 which has been recently described as regulating genes involved in iron uptake and quorum sensing of P. aeruginosa (92). This ORF has been assigned the locus tag PSPTO_5638. This CDS is located in a region that is conserved among DC3000, P. syringae pv. phaseolicola 1448A, and P. syringae pv. syringae B728a. The protein is likely misannotated in DC3000 since it has been annotated in P. syringae pv. phaseolicola 1448A (PSPPH_2116) and P. syringae pv. syringae B728a (Psyr_2141) as a FecR protein. Another predicted protein, encoded by a new ORF antisense to PSPTO_1113, contains a conserved domain present in the PagL superfamily, which are lipases required for the deacylation of the 3-O-position fatty acid (a 3-O-deacylase) and are found in a number of other Gram-negative bacteria (30). This new ORF (assigned the locus tag PSPTO_5636) is homologous to PagL (PA4661) in P. aeruginosa and proteins that encode PagL homologs annotated in the other Pseudomonas genomes, including P. syringae B728A, P. syringae 1448a, Pseudomonas putida, and Pseudomonas fluorescens (Pseudomonas Genome Database V2). This ORF was overlooked in the original DC3000 annotation, but, importantly, our results provide evidence that it and corresponding ORFs in other pseudomonads are correctly annotated. Each of the 11 ORFs having expression inconsistent with the original annotation and corresponding to an EasyGene gene call have been assigned new locus tag numbers and the supporting evidence for the change incorporated into the GenBank annotation record.
TABLE 2.
Selected CDSs having transcriptional activity inconsistent with genome annotation
| Original locus tag | Description of original annotated CDSa | Description of EasyGene callb | Easygene coordinates (nt) | Locus tag assigned to new gene |
|---|---|---|---|---|
| PSPTO_0392 | Hypothetical protein | Hypothetical protein | 432379-432798c | PSPTO_0391 |
| PSPTO_1113 | Hypothetical protein | PagL family | 1228041-1228448 | PSPTO_5636 |
| PSPTO_1442 | Hypothetical protein | Hypothetical protein | 1581112-1581555 | PSPTO_5635 |
| PSPTO_1837 | Hypothetical protein | Hypothetical protein | 2006821-2007108 | PSPTO_5637 |
| PSPTO_2357 | Hypothetical protein | Fur-like | 2611919-2612308c | PSPTO_5638 |
| PSPTO_2512 | Hypothetical protein | Hypothetical protein | 2777027-2777347 | PSPTO_5639 |
| PSPTO_2619 | Hypothetical protein | Hypothetical protein | 2910705-2911238 | PSPTO_5640 |
| PSPTO_2682 | Hypothetical protein | Hypothetical protein | 2978065-2978625 | PSPTO_5641 |
| PSPTO_3093 | Hypothetical protein | No homology | 3476622-3477131c, 3476773-3477240c | PSPTO_5642, PSPTO_5643c |
| PSPTO_4311 | Hypothetical protein | Hypothetical protein | 4862483-4862680 | PSPTO_5644 |
| PSPTO_5429 | Hypothetical protein | Hypothetical proteins | 6174792-6175730c, 6174382-6174792c | PSPTO_5645, PSPTO_5646 |
Protein descriptions obtained from P. syringae DC3000 genome annotation.
Results of BLAST analysis of predicted protein from EasyGene call.
Overlapping ORF predictions.
Transcriptional read-through.
Several genes exhibited expression that contradicted the annotated CDS but did not contain a plausible ORF antisense to the called CDS. For example, PSPTO_3836, which encodes the quorum-sensing regulator PsyR, displayed transcriptional activity on the opposite strand, contradictory to the annotated CDS (Fig. 3C). Since psyI and psyR are convergently transcribed, the antisense transcription may be due to read-through from psyI. Several other cases where overlapping transcription is found appear in regions containing convergently transcribed genes, and there is insufficient room for transcription terminators, such as the pair PSPTO_ 0907 and PSPTO_0908 (cheB-1) and the pair PSPTO_4080 and PSPTO_4081.
Other cases of transcriptional activity inconsistent with annotation occur in small, annotated CDSs in areas where there is extensive transcription on the opposite strand. We cannot make any determination as to whether these small CDSs should be removed from the annotation because they may, in fact, be expressed at higher levels under conditions other than those we tested.
Detection of predicted ncRNAs.
In addition to classifying expression of known coding regions, we observed transcriptional activity from a number of unannotated regions. These regions could represent unannotated CDSs, or small ncRNAs. Using the LIMR profile and the previously determined classification threshold, the genes for predicted ncRNAs described in the Rfam database (33) were classified. Our results showed consistent transcriptional activity under iron-limited growth conditions for 19 of the 21 predicted ncRNAs (excluding tRNAs and rRNAs) in the DC3000 genome (Table 3; see also File S7 in the supplemental material). These ncRNAs mainly corresponded to housekeeping RNAs and riboswitches or regulatory elements within 5′ untranslated regions (UTRs). Two others, prrF2 and signal recognition particle, displayed transcriptional activity which was above the threshold on both strands. The transcriptional start sites and points of termination were determined for prrF2 and the antisense transcript to confirm expression and length. The length of the prrF2 transcript determined by 5′ and 3′ RACE was consistent with the RNA-Seq profiles. For the antisense transcript, the 5′ end corresponded with the profile, and three different termination points were identified by 3′ RACE. As shown in Fig. 4, the region of overlap of prrF2 and the antisense transcript ranged from 46 to 64 bases and spanned the terminator region of prrF2 and the conserved core sequence (85). Transcription for four ncRNAs (P11, P15, P9, and ykkC-yxxD) could not be detected under these growth conditions. These ncRNAs may not be expressed under this growth condition or were not efficiently recovered during the column purification of the RNA sample due to their predicted small size.
TABLE 3.
Selected list of small ncRNAs having consistent or ambiguous transcriptional activity
| ncRNA or riboswitcha | Ig regionb | Genomic coordinatesc |
|---|---|---|
| rsmY | PSPTO_0506/PSPTO_0507 | 555344-555465 |
| P26 | PSPTO_0618/PSPTO_0619 | 678273-678338 |
| prrF1 | PSPTO_0973/PSPTO_0974 | 1059034-1058888c |
| yybP-ykoY | PSPTO_1145/PSPTO_1146 | 1255819-1255678c |
| t44 | PSPTO_1533/PSPTO_1535 | 1693384-1693536 |
| PrrB_RsmZ | PSPTO_1566/PSPTO_1567 | 1728406-1728566 |
| Cobalamin | PSPTO_1707/PSPTO_1708 | 1878136-1878350 |
| FMN | PSPTO_1840/PSPTO_1841 | 2009078-2008912c |
| Cobalamin | PSPTO_3150/PSPTO_3151 | 3542313-3542537c |
| Cobalamin | PSPTO_3150/PSPTO_3151 | 3542594-3542808 |
| prrF2* | PSPTO_3156/PSPTO_3157 | 3549457-3549603 |
| Cobalamin | PSPTO_3255/PSPTO_3256 | 3679925-3680123 |
| SRP_bact* | PSPTO_3653/PSPTO_3654 | 4116112-4116013c |
| P16 | PSPTO_3823/PSPTO_3824 | 4332812-4333007 |
| RNaseP | PSPTO_4471/PSPTO_4418 | 4986712-4986420c |
| S15 | PSPTO_4487/PSPTO_4488 | 5055007-5054896c |
| tmRNA | PSPTO_4516/PSPTO_4517 | 5086183-5086569 |
| P24 | PSPTO_4792/PSPTO_4793 | 5433294-5433053c |
| TPP | PSPTO_4976/PSPTO_4977 | 5648734-5648630c |
| 6S | PSPTO_5226/PSPTO_5227 | 5949055-5949233 |
| P1 | PSPTO_5309/PSPTO_5310 | 6038511-6038690 |
ncRNA or riboswitch designation according to Rfam (accessed October 2007 [http://www.sanger.ac.uk/Software/Rfam/]). *, expression classified as ambiguous (yellow; see Results).
The CDSs flanking the ncRNA.
Coordinates of the predicted ncRNA in the DC3000 genome taken from Rfam. c, the ncRNA is transcribed on the negative strand of the genome.
FIG. 4.

(A) Genetic location of prrF2 (black arrow) and antisense RNA (black arrow) between PSPTO_3156 and PSPTO_3157 (gray arrows) in DC3000. Rho-independent terminators are indicated by stem-loop structures. (B) Nucleotide sequence of the region containing prrF2 and the antisense RNA. Only one strand is shown. The Rho-independent terminator is underlined and represented in italics. The validated transcriptional start sites for prrF2 and the antisense transcript are indicated by bent arrows. The 3′ end for prrF2 is represented by a straight arrow pointing downward. The 3′ ends of the antisense RNA are represented by straight arrows pointing upward. The region of maximum overlap is shown in boldface.
We also classified regions corresponding to several ncRNAs that have been predicted for P. syringae DC3000 and described in the literature but not included in the Rfam database. For example, we found transcriptional activity located between PSPTO_0343 and polI (see File S8 in the supplemental material). This region is predicted to contain an ncRNA with similarity to the Spot 42 (spf) ncRNA of Escherichia coli (32). Also, transcriptional activity between PSPTO_3698 and PSPTO_3699 was detected. This region is predicted to encode the ncRNA rsmX (43). The transcriptional start sites for these two putative ncRNAs were mapped (see Table S1), confirming the expression and strandedness of these ncRNAs.
Identification of novel RpoN-regulated ncRNAs.
Two additional regions were identified that appear to encode novel ncRNAs, based on the fact that these areas exhibited transcriptional activity but do not contain plausible ORFs. One ncRNA (which we designated psr1) is located in an intergenic region between PSPTO_0964, which encodes a putative NtrC-like sigma-54 (RpoN) dependent transcriptional regulator, and PSPTO_0963 (Fig. 5A; see also File S9 in the supplemental material). The other ncRNA (psr2) is located between PSPTO_1621 and PSPTO_1622 (Fig. 5B). These areas had been recently identified to contain a conserved RNA motif, termed gamma-150 (84). We confirmed the expression and determined the size for both ncRNAs to be ∼380 nucleotides by Northern blot analysis, which is significantly larger than the conserved motif of ∼180 nt (data not shown). Independent transcriptional start sites for the ncRNAs and downstream genes, PSPTO_0963 (pcnB) and PSPTO_1621, were also identified (see Table S1). These results are consistent with the predicted sizes obtained from the LIMR profile and demonstrate that the ncRNAs are transcribed independently of neighboring genes.
We noticed that psr1 was located upstream of pcnB. This gene has been predicted to be regulated by RpoN in P. fluorescens (39). Also, it has been reported for P. putida that an RpoN promoter element is located upstream of PP4697, which encodes the poly(A) polymerase, pcnB (19). Since the predicted RpoN binding sites are located more than 500 base pairs upstream of pcnB, we hypothesized that RpoN may instead regulate the ncRNA psr1. RpoN recognizes and binds to a −24/−12-type promoter with the following sequence: 5′-YTGGCACG-N4-TTGCW-3′, with the bold G and C positioned at −24 and −12 relative to the start of transcription (12). A conserved sequence which appears to be an RpoN binding site was identified directly upstream of psr1 and psr2 (Fig. 6A).
FIG. 6.

(A) Alignment of the upstream region of psr1, psr2, and psr3. The alignment was generated with ClustalW (72). Conserved nucleotides are marked with an asterisk. The mapped transcriptional start for psr1 and psr2 and the predicted transcriptional start for psr3 are marked as +1. The shaded gray boxes at −24 and −12 represent the predicted sigma-54 (RpoN) promoter element. (B) Genomic organization of the psr3 gene region in P. syringae DC3000, P. syringae 1448A, and P. syringae B728a. Similar colors represent conserved proteins. In P. syringae DC3000, PSPTO_2741 and PSPTO_2740 correspond to the ISPsy4 transposase. The predicted RpoN promoter element is represented by the green octagon. The magenta arrow represents the gamma-150 ncRNA motif.
A third putative ncRNA found in the DC3000 is also thought to be part of the gamma-150 family (84). This ncRNA (named psr3) is predicted to be located between PSPTO_2739 and PSPTO_2740 (an insertion [IS] element) (Fig. 5C). We did not detect transcriptional activity or an RpoN binding site near this region. However, upon closer inspection we found transcriptional activity and a putative RpoN binding site upstream of the ncRNA motif on the other side of an IS element (Fig. 5A and C). Our data suggest that the three ncRNAs, which have previously been predicted to be part of the same family and share the same motif (gamma-150), are regulated by RpoN. However, psr3, which appears to have been disrupted by an IS element, is most likely nonfunctional in P. syringae DC3000.
Since psr3 appears to be disrupted by an IS element in DC3000, the genomic region containing this ncRNA was checked for disruption by an IS element in other sequenced P. syringae strains. It appears that DC3000 is the only strain in which psr3 has been disrupted (Fig. 6B). Also, predicted RpoN binding sites are found upstream of the psr1, psr2, and psr3 genes in P. syringae DC3000, 1448A, and B728a, suggesting for these pseudomonads that the ncRNAs are regulated by RpoN (data not shown).
DISCUSSION
This paper presents a genome-scale transcriptomics survey of P. syringae DC3000. Our results show that the combination of strand-specific RNA-Seq, proteomics, and computational methods yields biologically meaningful results from a single sample, and many of these results were independently validated using 5′ RACE, Rfam data, or Northern blot analysis. It has been shown that by analyzing a single RNA sample, RNA-Seq can be extremely informative (50, 77). The incorporation of additional biological data (proteomics) enabled identification of regions that displayed unusual transcriptional activity, such as those areas with antisense activity or transcriptional activity that contradicted genome annotation, in an efficient and effective manner.
To summarize our method, we used an enriched RNA sample from P. syringae DC3000 to characterize the transcriptome using the Illumina Genome Analyzer sequencing technology. Computational analyses were developed to generate profiles from the sequence data. We used proteomics data to help identify transcriptionally active regions and used these to compute an optimal threshold for detection. Genes were classified based upon whether their transcriptional activity was consistent or inconsistent with current annotation, or if their activity was ambiguous or not detectable. Although our study incorporated proteomics data for setting the classification threshold, other methods exist for establishing a threshold. For example, the threshold can be set manually, as in reference 53. Alternatively, sets of genes with known transcriptional activity under the chosen conditions can be used to construct training sets for setting the classification threshold.
For a variety of bacteria, including, E. coli, Bacillus subtilis, Corynebacterium glutamicum, P. fluorescens, and P. aeruginosa, the presence of cis-encoded ncRNAs expressed within or antisense to protein coding sequences has been reported (24-26, 40, 41, 47, 64, 66, 67, 80, 81, 90, 91). Strand-specific RNA-Seq is proving to be a powerful approach for identification of these regions in bacteria (49). We identified 124 cases in which both the annotated sense and the antisense demonstrated transcriptional activity. This is the first report of overlapping sense/antisense transcription for DC3000. A surprising finding was the discovery of overlapping antisense transcripts for genes regulated by the HrpL regulon (hopAJ1) and genes that encode proteins involved in the biosynthesis of coronatine (cmaE and cmaU). At this time we do not know if these antisense transcripts silence gene expression, lead to alternative processing of the mRNA, or target the sense transcript for selective degradation. However, they may have a significant role because of the importance of T3S and coronatine production to virulence in P. syringae. Further experiments to determine the transcriptional control of the antisense RNAs and the environmental/growth conditions under which they are expressed will be needed to understand the biological significance.
Many reported cases of antisense transcription are thought to represent 3′ read-through (13, 22, 34) from convergently transcribed genes, and we detected this pattern as well. For example, the quorum-sensing regulator psyR (PSPTO_3863) appears to have read-through from psyI (PSPTO_3864). These genes are arranged convergently and overlap by 69 bp. This arrangement has been reported for only one other pseudomonad, P. syringae pv. maculicola (27) but is also found in other phytopathogenic and plant-associated bacteria, where it has been hypothesized to play a role in the regulation of the expression of these two genes (27, 63). Our data raise the possibility that transcription of psyI may impact expression of psyR under specific conditions such as those described here. Thus, our results confirm and extend observations of antisense transcripts described by others although the significance of this phenomenon remains to be determined.
Our results demonstrate that directional RNA-Seq is an effective approach for confirming the expression of ncRNAs. Computational methods are often used to predict these transcripts, but RNA-Seq provides direct evidence of strand-specific transcriptional activity in intergenic regions. The DC3000 genome shows transcriptional activity in a number of intergenic regions. Several of these regions represent previously predicted ncRNAs (Table 3) (32, 33, 43). We should note that the RNA isolation procedure used here does not efficiently purify small RNA molecules (or is not optimized for the isolation of small RNA molecules); therefore, it is likely that we did not detect a complete representation of ncRNAs expressed under the conditions evaluated here. Interestingly, the region containing one ncRNA, prrF2, shows significant transcriptional activity on both strands, suggesting the presence of an antisense transcript. prrF2 belongs to a family of Fur-regulated ncRNAs that have been described in P. aeruginosa (85). The two prrF ncRNAs are almost identical and, in contrast to those of P. aeruginosa, are located in separate areas of the genome in DC3000. We have previously shown that prrF2 demonstrates iron-dependent expression (17), as has been reported for P. aeruginosa. The mRNA targets of prrF2 in DC3000 have not yet been determined, so the impact of antisense activity on iron homeostasis or any other biological process is unclear. Perhaps the role of the antisense ncRNA is to specifically regulate the transcriptional levels of prrF2. Overall, our identification of an antisense transcript of an ncRNA reveals additional complexity with respect to gene regulation and iron homeostasis in DC3000.
We also discovered three previously unannotated ncRNA genes (psr1, psr2, and psr3). These areas had been recently identified to contain a conserved RNA motif (84). We confirmed their expression, size, and existence under iron-limited conditions and demonstrated that the transcripts were distinct from the flanking genes. In particular, independent start sites were mapped for pcnB and psr1, clarifying the organization of this region proposed by Jones et al. (39). In addition, the observation of a putative RpoN promoter region upstream of these ncRNAs suggests that RpoN may regulate these ncRNAs.
RpoN, also known as sigma 54 or sigma N, is an alternative sigma factor that regulates transcription of genes encoding proteins involved in diverse functions, including the utilization of alternative carbon and nitrogen sources, nitrogen fixation, and the expression of virulence determinants (59, 87). In the pseudomonads, RpoN regulates expression of genes involved in alginate, flagellin, and pilin synthesis (14, 37, 74). RpoN is a key regulator of biocontrol activity in P. fluorescens (58), and for phytopathogenic bacteria such as P. syringae, RpoN is an important virulence regulator and is required for the elicitation of the hypersensitive response (35) and modulating pathogenicity-related genes, such as production of coronatine (1). We are continuing studies to elucidate the mechanism of regulation of these putative RpoN-regulated ncRNAs in P. syringae and determine their precise role in the cellular functions and/or pathogenesis of this bacterium. Since several of the ncRNAs are conserved among the pseudomonads, an analysis of their regulation and role in pathogenesis can provide information about global gene regulation in other pseudomonads.
Comparison of the genomic region containing psr3 in the other pseudomonads revealed that an IS element inserted into this ncRNA, separating the predicted RpoN promoter region from the structural portion of psr3. Unfortunately, since we do not yet have any functional information concerning these ncRNAs, it is difficult to speculate on the physiological consequences of the disruption. Since these ncRNAs belong to the same family, we hypothesize that psr3 is no longer required for this plant pathogen or that psr3 is associated with pathogenesis or host-specific factors. Alternatively, the ncRNAs may be functionally redundant, and therefore inactivation of one may not have any biological significance. Possibly, the functions of the ncRNAs may be additive to give more precise control of targets, as has been reported for the ncRNAs qrr1-5 in Vibrio harveyi (75). Nonetheless, it will be important to determine if the ncRNAs are expressed at different degrees or under differing environmental conditions to understand their role in gene regulation.
In summary, we describe the development of a new, rapid technique, based upon mRNA sequencing, for characterizing bacterial transcriptomes. The strand specificity, coupled with our computational methods, gives a global qualitative analysis that enables large-scale validation of gene expression, identification of candidate transcriptional start sites, confirmation of expression of genes encoding hypothetical proteins, and discovery of nonannotated genes and ncRNAs. Newly identified genes and ncRNAs as well as expression data for previously identified hypothetical genes have been incorporated in the DC3000 annotation record at GenBank and are now available for informed transitive annotation of similar genes in other genome sequences. Finally, strand-specific sequencing in combination with a unique classification scheme of transcriptional activity effectively revealed unusual and interesting cases of gene expression, generating intriguing hypotheses regarding gene expression for future investigations.
Supplementary Material
Acknowledgments
We thank Nola Pellegrini for mapping transcriptional start sites. We also thank Barbara Hover and Jie Zhao at the DNA Microarray Core Facility at Cornell University for analysis of RNA samples.
Footnotes
Published ahead of print on 26 February 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Alarcon-Chaidez, F. J., L. Keith, Y. Zhao, and C. L. Bender. 2003. RpoN (σ54) is required for plasmid-encoded coronatine biosynthesis in Pseudomonas syringae. Plasmid 49:106-117. [DOI] [PubMed] [Google Scholar]
- 2.Albrecht, M., C. M. Sharma, R. Reinhardt, J. Vogel, and T. Rudel. 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res. 38:868-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alter, O., P. O. Brown, and D. Botstein. 2003. Generalized singular value decomposition for comparative analysis of genome-scale expression data sets of two different organisms. Proc. Natl. Acad. Sci. U. S. A. 100:3351-3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alter, O., P. O. Brown, and D. Botstein. 2000. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. U. S. A. 97:10101-10106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alter, O., and G. H. Golub. 2004. Integrative analysis of genome-scale data by using pseudoinverse projection predicts novel correlation between DNA replication and RNA transcription. Proc. Natl. Acad. Sci. U. S. A. 101:16577-16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alter, O., and G. H. Golub. 2005. Reconstructing the pathways of a cellular system from genome-scale signals by using matrix and tensor computations. Proc. Natl. Acad. Sci. U. S. A. 102:17559-17564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 8.Applied Biosystems. 2007. iTRAQ reagents chemistry reference guide, PN 4351918. Applied Biosystems, Foster City, CA. http://docs.appliedbiosystems.com/search.taf.
- 9.Applied Biosystems. 2004. iTRAQ reagents, PN 4350831. Applied Biosystems, Foster City, CA. http://docs.appliedbiosystems.com/search.taf.
- 10.Argaman, L., R. Hershberg, J. Vogel, G. Bejerano, E. G. Wagner, H. Margalit, and S. Altuvia. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 11:941-950. [DOI] [PubMed] [Google Scholar]
- 11.Barnes, A., P. Nurse, and R. S. Fraser. 1979. Analysis of the significance of a periodic, cell size-controlled doubling in rates of macromolecular synthesis for the control of balanced exponential growth of fission yeast cells. J. Cell Sci. 35:41-51. [DOI] [PubMed] [Google Scholar]
- 12.Barrios, H., B. Valderrama, and E. Morett. 1999. Compilation and analysis of sigma(54)-dependent promoter sequences. Nucleic Acids Res. 27:4305-4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berka, R. M., J. Hahn, M. Albano, I. Draskovic, M. Persuh, X. Cui, A. Sloma, W. Widner, and D. Dubnau. 2002. Microarray analysis of the Bacillus subtilis K-state: genome-wide expression changes dependent on ComK. Mol. Microbiol. 43:1331-1345. [DOI] [PubMed] [Google Scholar]
- 14.Boucher, J. C., M. J. Schurr, and V. Deretic. 2000. Dual regulation of mucoidy in Pseudomonas aeruginosa and sigma factor antagonism. Mol. Microbiol. 36:341-351. [DOI] [PubMed] [Google Scholar]
- 15.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 16.Breitling, R., P. Armengaud, and A. Amtmann. 2005. Vector analysis as a fast and easy method to compare gene expression responses between different experimental backgrounds. BMC Bioinformatics 6:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bronstein, P. A., M. J. Filiatrault, C. R. Myers, M. Rutzke, D. J. Schneider, and S. W. Cartinhour. 2008. Global transcriptional responses of Pseudomonas syringae DC3000 to changes in iron bioavailability in vitro. BMC Microbiol. 8:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buell, C. R., V. Joardar, M. Lindeberg, J. Selengut, I. T. Paulsen, M. L. Gwinn, R. J. Dodson, R. T. Deboy, A. S. Durkin, J. F. Kolonay, R. Madupu, S. Daugherty, L. Brinkac, M. J. Beanan, D. H. Haft, W. C. Nelson, T. Davidsen, N. Zafar, L. W. Zhou, J. Liu, Q. P. Yuan, H. Khouri, N. Fedorova, B. Tran, D. Russell, K. Berry, T. Utterback, S. E. Van Aken, T. V. Feldblyum, M. D'Ascenzo, W. L. Deng, A. R. Ramos, J. R. Alfano, S. Cartinhour, A. K. Chatterjee, T. P. Delaney, S. G. Lazarowitz, G. B. Martin, D. J. Schneider, X. Y. Tang, C. L. Bender, O. White, C. M. Fraser, and A. Collmer. 2003. The complete genome sequence of the Arabidopsis and tomato pathogen Pseudomonas syringae pv. tomato DC3000. Proc. Natl. Acad. Sci. U. S. A. 100:10181-10186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cases, I., D. W. Ussery, and V. de Lorenzo. 2003. The σ54 regulon (sigmulon) of Pseudomonas putida. Environ. Microbiol. 5:1281-1293. [DOI] [PubMed] [Google Scholar]
- 20.Cheung, F., B. J. Haas, S. M. Goldberg, G. D. May, Y. Xiao, and C. D. Town. 2006. Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics 7:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cloonan, N., A. R. Forrest, G. Kolle, B. B. Gardiner, G. J. Faulkner, M. K. Brown, D. F. Taylor, A. L. Steptoe, S. Wani, G. Bethel, A. J. Robertson, A. C. Perkins, S. J. Bruce, C. C. Lee, S. S. Ranade, H. E. Peckham, J. M. Manning, K. J. McKernan, and S. M. Grimmond. 2008. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 5:613-619. [DOI] [PubMed] [Google Scholar]
- 22.Courcelle, J., A. Khodursky, B. Peter, P. O. Brown, and P. C. Hanawalt. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Croucher, N. J., M. C. Fookes, T. T. Perkins, D. J. Turner, S. B. Marguerat, T. Keane, M. A. Quail, M. He, S. Assefa, J. Bahler, R. A. Kingsley, J. Parkhill, S. D. Bentley, G. Dougan, and N. R. Thomson. 2009. A simple method for directional transcriptome sequencing using Illumina technology. Nucleic Acids Res. 37:e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duhring, U., I. M. Axmann, W. R. Hess, and A. Wilde. 2006. An internal antisense RNA regulates expression of the photosynthesis gene isiA. Proc. Natl. Acad. Sci. U. S. A. 103:7054-7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eiamphungporn, W., and J. D. Helmann. 2009. Extracytoplasmic function sigma factors regulate expression of the Bacillus subtilis yabE gene via a cis-acting antisense RNA. J. Bacteriol. 191:1101-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eiamphungporn, W., and J. D. Helmann. 2008. The Bacillus subtilis σM regulon and its contribution to cell envelope stress responses. Mol. Microbiol. 67:830-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elasri, M., S. Delorme, P. Lemanceau, G. Stewart, B. Laue, E. Glickmann, P. M. Oger, and Y. Dessaux. 2001. Acyl-homoserine lactone production is more common among plant-associated Pseudomonas spp. than among soilborne Pseudomonas spp. Appl. Environ. Microbiol. 67:1198-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira, A. O., C. R. Myers, J. S. Gordon, G. B. Martin, M. Vencato, A. Collmer, M. D. Wehling, J. R. Alfano, G. Moreno-Hagelsieb, W. F. Lamboy, G. DeClerck, D. J. Schneider, and S. W. Cartinhour. 2006. Whole-genome expression profiling defines the HrpL regulon of Pseudomonas syringae pv. tomato DC3000, allows de novo reconstruction of the Hrp cis clement, and identifies novel coregulated genes. Mol. Plant Microbe Interact. 19:1167-1179. [DOI] [PubMed] [Google Scholar]
- 29.Fouts, D. E., R. B. Abramovitch, J. R. Alfano, A. M. Baldo, C. R. Buell, S. Cartinhour, A. K. Chatterjee, M. D'Ascenzo, M. L. Gwinn, S. G. Lazarowitz, N. C. Lin, G. B. Martin, A. H. Rehm, D. J. Schneider, K. van Dijk, X. Tang, and A. Collmer. 2002. Genomewide identification of Pseudomonas syringae pv. tomato DC3000 promoters controlled by the HrpL alternative sigma factor. Proc. Natl. Acad. Sci. U. S. A. 99:2275-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geurtsen, J., L. Steeghs, J. T. Hove, P. van der Ley, and J. Tommassen. 2005. Dissemination of lipid A deacylases (pagL) among gram-negative bacteria: identification of active-site histidine and serine residues. J. Biol. Chem. 280:8248-8259. [DOI] [PubMed] [Google Scholar]
- 31.Golub, G. H., and C. F. Van Loan. 1996. Matrix computations, 3rd ed. John Hopkins University Press, Baltimore, MD.
- 32.Gottesman, S., C. A. McCullen, M. Guillier, C. K. Vanderpool, N. Majdalani, J. Benhammou, K. M. Thompson, P. C. FitzGerald, N. A. Sowa, and D. J. FitzGerald. 2006. Small RNA regulators and the bacterial response to stress. Cold Spring Harb. Symp. Quant. Biol. 71:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griffiths-Jones, S., S. Moxon, M. Marshall, A. Khanna, S. R. Eddy, and A. Bateman. 2005. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33:D121-D124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helmann, J. D., M. F. Wu, P. A. Kobel, F. J. Gamo, M. Wilson, M. M. Morshedi, M. Navre, and C. Paddon. 2001. Global transcriptional response of Bacillus subtilis to heat shock. J. Bacteriol. 183:7318-7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hendrickson, E. L., P. Guevera, and F. M. Ausubel. 2000. The alternative sigma factor RpoN is required for hrp activity in Pseudomonas syringae pv. maculicola and acts at the level of hrpL transcription. J. Bacteriol. 182:3508-3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu, J., F. A. Wright, and F. Zou. 2006. Estimation of expression indexes for oligonucleotide arrays using the singular value decomposition. J. Am. Stat. Assoc. 101:41-50. [Google Scholar]
- 37.Ishimoto, K. S., and S. Lory. 1989. Formation of pilin in Pseudomonas aeruginosa requires the alternative sigma factor (RpoN) of RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 86:1954-1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jager, D., C. M. Sharma, J. Thomsen, C. Ehlers, J. Vogel, and R. A. Schmitz. 2009. Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc. Natl. Acad. Sci. U. S. A. 106:21878-21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones, J., D. J. Studholme, C. G. Knight, and G. M. Preston. 2007. Integrated bioinformatic and phenotypic analysis of RpoN-dependent traits in the plant growth-promoting bacterium Pseudomonas fluorescens SBW25. Environ. Microbiol. 9:3046-3064. [DOI] [PubMed] [Google Scholar]
- 40.Kawano, M., L. Aravind, and G. Storz. 2007. An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol. 64:738-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawano, M., A. A. Reynolds, J. Miranda-Rios, and G. Storz. 2005. Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res. 33:1040-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.King, E. O., M. K. Ward, and D. E. Raney. 1954. Two simple media for the demonstration of pyocyanin and fluorescin. J. Lab. Clin. Med. 44:301-307. [PubMed] [Google Scholar]
- 43.Kulkarni, P. R., X. Cui, J. W. Williams, A. M. Stevens, and R. V. Kulkarni. 2006. Prediction of CsrA-regulating small RNAs in bacteria and their experimental verification in Vibrio fischeri. Nucleic Acids Res. 34:3361-3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuruvilla, F. G., P. J. Park, and S. L. Schreiber. 2002. Vector algebra in the analysis of genome-wide expression data. Genome Biol. 3:RESEARCH0011. http://genomebiology.com/2002/3/3/RESEARCH/0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lan, L., X. Deng, J. Zhou, and X. Tang. 2006. Genome-wide gene expression analysis of Pseudomonas syringae pv. tomato DC3000 reveals overlapping and distinct pathways regulated by hrpL and hrpRS. Mol. Plant Microbe Interact. 19:976-987. [DOI] [PubMed] [Google Scholar]
- 46.Larsen, T. S., and A. Krogh. 2003. EasyGene-a prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinformatics 4:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee, J. M., S. Zhang, S. Saha, S. Santa Anna, C. Jiang, and J. Perkins. 2001. RNA expression analysis using an antisense Bacillus subtilis genome array. J. Bacteriol. 183:7371-7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindeberg, M., C. R. Myers, A. Collmer, and D. J. Schneider. 2008. Roadmap to new virulence determinants in Pseudomonas syringae: insights from comparative genomics and genome organization. Mol. Plant Microbe Interact. 21:685-700. [DOI] [PubMed] [Google Scholar]
- 49.Liu, J. M., J. Livny, M. S. Lawrence, M. D. Kimball, M. K. Waldor, and A. Camilli. 2009. Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res. 37:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meyer, E., G. V. Aglyamova, S. Wang, J. Buchanan-Carter, D. Abrego, J. K. Colbourne, B. L. Willis, and M. V. Matz. 2009. Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics 10:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morin, R., M. Bainbridge, A. Fejes, M. Hirst, M. Krzywinski, T. Pugh, H. McDonald, R. Varhol, S. Jones, and M. Marra. 2008. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques 45:81-94. [DOI] [PubMed] [Google Scholar]
- 52.Mortazavi, A., B. A. Williams, K. McCue, L. Schaeffer, and B. Wold. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:621-628. [DOI] [PubMed] [Google Scholar]
- 53.Nagalakshmi, U., Z. Wang, K. Waern, C. Shou, D. Raha, M. Gerstein, and M. Snyder. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320:1344-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nielsen, P., and A. Krogh. 2005. Large-scale prokaryotic gene prediction and comparison to genome annotation. Bioinformatics 21:4322-4329. [DOI] [PubMed] [Google Scholar]
- 55.Oliver, H. F., R. H. Orsi, L. Ponnala, U. Keich, W. Wang, Q. Sun, S. W. Cartinhour, M. J. Filiatrault, M. Wiedmann, and K. J. Boor. 2009. Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs. BMC Genomics 10:641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parkhomchuk, D., T. Borodina, V. Amstislavskiy, M. Banaru, L. Hallen, S. Krobitsch, H. Lehrach, and A. Soldatov. 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37:e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Passalacqua, K. D., A. Varadarajan, B. D. Ondov, D. T. Okou, M. E. Zwick, and N. H. Bergman. 2009. Structure and complexity of a bacterial transcriptome. J. Bacteriol. 191:3203-3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pechy-Tarr, M., M. Bottiglieri, S. Mathys, K. B. Lejbolle, U. Schnider-Keel, M. Maurhofer, and C. Keel. 2005. RpoN (sigma54) controls production of antifungal compounds and biocontrol activity in Pseudomonas fluorescens CHA0. Mol. Plant Microbe Interact. 18:260-272. [DOI] [PubMed] [Google Scholar]
- 59.Potvin, E., F. Sanschagrin, and R. C. Levesque. 2008. Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 32:38-55. [DOI] [PubMed] [Google Scholar]
- 60.Quiniones, B., G. Dulla, and S. E. Lindow. 2005. Quorum sensing regulates exopolysaccharide production, motility, and virulence in Pseudomonas syringae. Mol. Plant Microbe Interact. 18:682-693. [DOI] [PubMed] [Google Scholar]
- 61.Quinones, B., C. J. Pujol, and S. E. Lindow. 2004. Regulation of AHL production and its contribution to epiphytic fitness in Pseudomonas syringae. Mol. Plant Microbe Interact. 17:521-531. [DOI] [PubMed] [Google Scholar]
- 62.Ross, P. L., Y. N. Huang, J. N. Marchese, B. Williamson, K. Parker, S. Hattan, N. Khainovski, S. Pillai, S. Dey, S. Daniels, S. Purkayastha, P. Juhasz, S. Martin, M. Bartlet-Jones, F. He, A. Jacobson, and D. J. Pappin. 2004. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell Proteomics 3:1154-1169. [DOI] [PubMed] [Google Scholar]
- 63.Salmond, G. P., B. W. Bycroft, G. S. Stewart, and P. Williams. 1995. The bacterial “enigma”: cracking the code of cell-cell communication. Mol. Microbiol. 16:615-624. [DOI] [PubMed] [Google Scholar]
- 64.Selinger, D. W., K. J. Cheung, R. Mei, E. M. Johansson, C. S. Richmond, F. R. Blattner, D. J. Lockhart, and G. M. Church. 2000. RNA expression analysis using a 30 base pair resolution Escherichia coli genome array. Nat. Biotechnol. 18:1262-1268. [DOI] [PubMed] [Google Scholar]
- 65.Shevchenko, A., M. Wilm, O. Vorm, and M. Mann. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850-858. [DOI] [PubMed] [Google Scholar]
- 66.Silby, M. W., and S. B. Levy. 2008. Overlapping protein-encoding genes in Pseudomonas fluorescens Pfo-1. PLoS Genet. 4:e1000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silby, M. W., P. B. Rainey, and S. B. Levy. 2004. IVET experiments in Pseudomonas fluorescens reveal cryptic promoters at loci associated with recognizable overlapping genes. Microbiology 150:518-520. [DOI] [PubMed] [Google Scholar]
- 68.Smith, D. J., E. T. Maggio, and G. L. Kenyon. 1975. Simple alkanethiol groups for temporary blocking of sulfhydryl groups of enzymes. Biochemistry 14:766-771. [DOI] [PubMed] [Google Scholar]
- 69.Sorek, R., and P. Cossart. 2010. Prokaryotic transcriptomics: a new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 11:9-16. [DOI] [PubMed] [Google Scholar]
- 70.Strieter, E. R., F. H. Vaillancourt, and C. T. Walsh. 2007. CmaE: a transferase shuttling aminoacyl groups between carrier protein domains in the coronamic acid biosynthetic pathway. Biochemistry 46:7549-7557. [DOI] [PubMed] [Google Scholar]
- 71.Swingle, B., D. Thete, M. Moll, C. R. Myers, D. J. Schneider, and S. Cartinhour. 2008. Characterization of the PvdS-regulated promoter motif in Pseudomonas syringae pv. tomato DC3000 reveals regulon members and insights regarding PvdS function in other pseudomonads. Mol. Microbiol. 68:871-889. [DOI] [PubMed] [Google Scholar]
- 72.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tobes, R., and E. Pareja. 2005. Repetitive extragenic palindromic sequences in the Pseudomonas syringae pv. tomato DC3000 genome: extragenic signals for genome reannotation. Res. Microbiol. 156:424-433. [DOI] [PubMed] [Google Scholar]
- 74.Totten, P. A., J. C. Lara, and S. Lory. 1990. The rpoN gene product of Pseudomonas aeruginosa is required for expression of diverse genes, including the flagellin gene. J. Bacteriol. 172:389-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tu, K. C., and B. L. Bassler. 2007. Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev. 21:221-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ullrich, M., and C. L. Bender. 1994. The biosynthetic gene cluster for coronamic acid, an ethylcyclopropyl amino acid, contains genes homologous to amino acid-activating enzymes and thioesterases. J. Bacteriol. 176:7574-7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vega-Arreguin, J. C., E. Ibarra-Laclette, B. Jimenez-Moraila, O. Martinez, J. P. Vielle-Calzada, L. Herrera-Estrella, and A. Herrera-Estrella. 2009. Deep sampling of the Palomero maize transcriptome by a high throughput strategy of pyrosequencing. BMC Genomics 10:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vencato, M., F. Tian, J. R. Alfano, C. R. Buell, S. Cartinhour, G. A. DeClerck, D. S. Guttman, J. Stavrinides, V. Joardar, M. Lindeberg, P. A. Bronstein, J. W. Mansfield, C. R. Myers, A. Collmer, and D. J. Schneider. 2006. Bioinformatics-enabled identification of the HrpL regulon and type III secretion system effector proteins of Pseudomonas syringae pv. phaseolicola 1448A. Mol. Plant Microbe Interact. 19:1193-1206. [DOI] [PubMed] [Google Scholar]
- 79.Vera, J. C., C. W. Wheat, H. W. Fescemyer, M. J. Frilander, D. L. Crawford, I. Hanski, and J. H. Marden. 2008. Rapid transcriptome characterization for a nonmodel organism using 454 pyrosequencing. Mol. Ecol. 17:1636-1647. [DOI] [PubMed] [Google Scholar]
- 80.Vogel, J., V. Bartels, T. H. Tang, G. Churakov, J. G. Slagter-Jager, A. Huttenhofer, and E. G. Wagner. 2003. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res. 31:6435-6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wagner, E. G., and R. W. Simons. 1994. Antisense RNA control in bacteria, phages, and plasmids. Annu. Rev. Microbiol. 48:713-742. [DOI] [PubMed] [Google Scholar]
- 82.Wang, Z., M. Gerstein, and M. Snyder. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10:57-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weber, A. P., K. L. Weber, K. Carr, C. Wilkerson, and J. B. Ohlrogge. 2007. Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol. 144:32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weinberg, Z., J. E. Barrick, Z. Yao, A. Roth, J. N. Kim, J. Gore, J. X. Wang, E. R. Lee, K. F. Block, N. Sudarsan, S. Neph, M. Tompa, W. L. Ruzzo, and R. R. Breaker. 2007. Identification of 22 candidate structured RNAs in bacteria using the CMfinder comparative genomics pipeline. Nucleic Acids Res. 35:4809-4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wilderman, P. J., N. A. Sowa, D. J. FitzGerald, P. C. FitzGerald, S. Gottesman, U. A. Ochsner, and M. L. Vasil. 2004. Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. Proc. Natl. Acad. Sci. U. S. A. 101:9792-9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilhelm, B. T., S. Marguerat, S. Watt, F. Schubert, V. Wood, I. Goodhead, C. J. Penkett, J. Rogers, and J. Bahler. 2008. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453:1239-1243. [DOI] [PubMed] [Google Scholar]
- 87.Wosten, M. M. 1998. Eubacterial sigma-factors. FEMS Microbiol. Rev. 22:127-150. [DOI] [PubMed] [Google Scholar]
- 88.Wurtzel, O., R. Sapra, F. Chen, Y. Zhu, B. A. Simmons, and R. Sorek. 2010. A single-base resolution map of an archaeal transcriptome. Genome Res. 20:133-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang, Y., S. Zhang, K. Howe, D. B. Wilson, F. Moser, D. Irwin, and T. W. Thannhauser. 2007. A comparison of nLC-ESI-MS/MS and nLC-MALDI-MS/MS for GeLC-based protein identification and iTRAQ-based shotgun quantitative proteomics. J. Biomol. Technol. 18:226-237. [PMC free article] [PubMed] [Google Scholar]
- 90.Zemanova, M., P. Kaderabkova, M. Patek, M. Knoppova, R. Silar, and J. Nesvera. 2008. Chromosomally encoded small antisense RNA in Corynebacterium glutamicum. FEMS Microbiol Lett. 279:195-201. [DOI] [PubMed] [Google Scholar]
- 91.Zhaohui, Y., J. Xiaolin, R. Xiancai, C. Xiaoxing, and H. Fuquan. 2007. A novel strategy for systematic identification of natural antisense transcripts of Pseudomonas aeruginosa based on RNase I protection assay. Mol. Biol. (Mosk.) 41:640-646. (In Russian.) [PubMed] [Google Scholar]
- 92.Zheng, P., J. Sun, R. Geffers, and A. P. Zeng. 2007. Functional characterization of the gene PA2384 in large-scale gene regulation in response to iron starvation in Pseudomonas aeruginosa. J. Biotechnol. 132:342-352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.