

Abstract
Prevotella bryantii B14 is a member of the phylum Bacteroidetes and contributes to the degradation of hemicellulose in the rumen. The genome of P. bryantii harbors four genes predicted to encode glycoside hydrolase (GH) family 3 (GH3) enzymes. To evaluate whether these genes encode enzymes with redundant biological functions, each gene was cloned and expressed in Escherichia coli. Biochemical analysis of the recombinant proteins revealed that the enzymes exhibit different substrate specificities. One gene encoded a cellodextrinase (CdxA), and three genes encoded β-xylosidase enzymes (Xyl3A, Xyl3B, and Xyl3C) with different specificities for either para-nitrophenyl (pNP)-linked substrates or substituted xylooligosaccharides. To identify the amino acid residues that contribute to catalysis and substrate specificity within this family of enzymes, the roles of conserved residues (R177, K214, H215, M251, and D286) in Xyl3B were probed by site-directed mutagenesis. Each mutation led to a severely decreased catalytic efficiency without a change in the overall structure of the mutant enzymes. Through amino acid sequence alignments, an amino acid residue (E115) that, when mutated to aspartic acid, resulted in a 14-fold decrease in the kcat/Km for pNP-β-d-xylopyranoside (pNPX) with a concurrent 1.1-fold increase in the kcat/Km for pNP-β-d-glucopyranoside (pNPG) was identified. Amino acid residue E115 may therefore contribute to the discrimination between β-xylosides and β-glucosides. Our results demonstrate that each of the four GH3 enzymes has evolved to perform a specific role in lignopolysaccharide hydrolysis and provide insight into the role of active-site residues in catalysis and substrate specificity for GH3 enzymes.
Among the bacterial genera in the bovine rumen, Prevotella species are the most numerically abundant species by both culture counts and by analysis of 16S rRNA abundances (13, 43). These organisms contribute to the breakdown of plant protein and hemicellulose within the rumen microbial ecosystem. Despite these important physiological roles, relatively little is known about the molecular mechanisms for plant polysaccharide utilization by ruminal Prevotella species. The genomes for Prevotella bryantii B14 and P. ruminicola 23 have recently been sequenced and harbor a large number of putative glycoside hydrolase (GH) genes (http://www.tigr.org/tdb/rumenomics/). Glycoside hydrolase genes are grouped into different families according to their amino acid sequences and three-dimensional folds (8). The genome for P. bryantii B14 harbors multiple gene copies for several GH families. The presence of multiple copies of genes from a single glycoside hydrolase family suggests that these genes encode proteins with either redundant or divergent biochemical functions. The genome for P. bryantii B14 contains four GH family 3 (GH3) genes, each of which was annotated as coding for an enzyme with β-glucosidase activity. Previous efforts to identify genes that are important for xylan degradation by its relative Prevotella ruminicola 23 revealed a GH family 3 protein that was annotated as a β-glucosidase enzyme but actually exhibited β-xylosidase activity (12). Thus, the goal of the current study was to express these four GH family 3 genes from P. bryantii B14 and to characterize and compare the biochemical properties of their gene products.
GH family 3 is one of the most abundant families of carbohydrate active enzymes and includes members that possess distinct enzymatic activities, including β-d-glucosidase, β-d-xylosidase, α-l-arabinofuranosidase, and N-acetyl-β-d-glucosaminidase activities (16, 24). The broad substrate specificity in this family of enzymes has implications for their respective physiological roles, and previous studies have shown that their functions are important for the assimilation of glycosides, recycling of bacterial cell wall components, and modification of free glycosides (reviewed in reference 16). GH family 3 enzymes catalyze the hydrolysis of aryl glycosides with net retention of stereochemical configuration at the anomeric carbon, and this reaction involves a pair of residues that act as a catalytic nucleophile (Asp) and a catalytic acid/base (Glu). The catalytic nucleophile in several GH family 3 β-d-glucosidases has been identified by covalent labeling followed by mass spectrometry (2, 9, 11) and X-ray crystallography (30). The catalytic acid/base that serves to protonate the leaving-group oxygen and subsequently activate an attacking water has been identified for several GH3 β-d-glucosidases (9, 35, 40). In addition to these two critical residues, GH3 enzymes possess three additional highly conserved amino acid residues (Arg, Lys, and His) that are predicted to be involved in catalysis (27, 29). The residues that contribute to substrate specificity for this family of enzymes, however, have not been thoroughly investigated.
In this study, we report the biochemical properties of four GH family 3 enzymes from P. bryantii B14 and mutational studies of one of these enzymes. Results from this study indicate that these GH family 3 genes encode enzymes with nonredundant biochemical functions and provide important insights into residues that contribute to catalysis and substrate specificity for this glycoside hydrolase family.
MATERIALS AND METHODS
Materials.
Prevotella bryantii B14 (DSM 11371) was initially isolated by Bryant et al. (6) and was obtained from our culture collection housed at the Department of Animal Sciences, University of Illinois at Urbana-Champaign. The culture was grown on agar maintenance slants and stored in liquid nitrogen vapor at −105°C (XLC1110; MVE Cryogenics). Escherichia coli JM109 and E. coli BL21-CodonPlus (DE3) RIL competent cells and PicoMaxx high-fidelity DNA polymerase were acquired from Stratagene (La Jolla, CA). The pGEM-T TA cloning vector was purchased from Promega (Madison, WI). The pET-28a and pET-15b expression vectors and the pET-46 Ek/LIC vector kit were obtained from Novagen (San Diego, CA). NdeI, DpnI, and XhoI restriction endonucleases were purchased from New England Biolabs (Ipswich, MA). The RNAprotect bacteria reagent, RNeasy minikit with on-column DNase digestion, and QIAprep Spin miniprep kit were obtained from Qiagen (Valencia, CA). Xylooligosaccharides, cellooligosaccharides, aldouronic acids, and wheat arabinoxylan (WAX) (medium viscosity, 20 centistokes) were obtained from Megazyme (Bray, Ireland). Gel filtration standards were obtained from Bio-Rad (Hercules, CA). The iScript single-stranded cDNA synthesis kit was obtained from Bio-Rad (Hercules, CA). SYBR green master mix was obtained from Applied Biosystems (Foster City, CA). All other reagents were of the highest possible purity and were purchased from Sigma-Aldrich (St. Louis, MO).
Gene cloning, expression, and purification of P. bryantii B14 GH family 3 genes.
The genome of Prevotella bryantii B14 was sequenced by the North American Consortium for Fibrolytic Rumen Bacteria in collaboration with The Institute for Genomic Research (TIGR). Autoannotation of the partially sequenced P. bryantii B14 genome identified ORF0348 (cdxA), ORF0398 (xyl3A), ORF0911 (xyl3B), and ORF0975 (xyl3C) as genes predicted to encode β-glucosidase enzymes. The gene sequence for cdxA is available in the GenBank database (accession no. AAA86753.2).
Prevotella bryantii B14 was maintained and grown in a standard anaerobically prepared medium as described previously (23). DNA was isolated from mid-log-phase cultures by using the DNeasy blood and tissue DNA purification kit. The DNA sequences corresponding to the entire open reading frames (ORFs) of cdxA, xyl3A, xyl3B, and xyl3C were amplified from P. bryantii B14 genomic DNA with the PicoMaxx high-fidelity PCR system with the primers listed in Table 1. Putative signal sequences with predicted cleavage sites were identified for each of the four genes (CdxA, MNKKIGVFALGICLSGASMA; Xyl3A, MKLFTKYAVVAILTLPSTATYSLICA; Xyl3B, MRKKILLMCASIALVSCNN; Xyl3C, MKSKQLITLFIVSTSILSLHA) by using the SignalP v3.0 online server (14). To ensure the accumulation of recombinant protein in E. coli cells to facilitate purification, the primers were designed to clone the gene beginning with the amino acid codon just downstream of the predicted protease cleavage sites.
TABLE 1.
Oligonucleotide primers used in this study
| Primer | Orientation | Sequence (5′→3′)a |
|---|---|---|
| Cloning | ||
| CdxA (ORF0348) | Forward | 5′-catatgCAGGCTCCTCAGTTACGAGC-3′ |
| Reverse | 5′-ctcgagTTATTTATTTCTTCTCAATAAGTTAAGCG-3′ | |
| SDM1 | 5′-TAGGTGATAATGCTGCTCACATGGTTGGTACAGCTAAAC-3′ | |
| SDM2 | 5′-GTTTAGCTGTACCAACCATGTGAGCAGCATTATCACCTA-3′ | |
| Xyl3A (ORF0398) | Forward | 5′-GACGACGACAAGATGCTCATCTGCGCTGCTGAAAAG-3′ |
| Reverse | 5′-GAGGAGAAGCCCGGTTAATTTAACGTATAATGTATCTG-3′ | |
| Xyl3B (ORF0911) | Forward | 5′-GCGCcatatgCAAACTATACTTATTAATCAGCAGG-3′ |
| Reverse | 5′-CGCGctcgagTCACTTGATGACTTCAG-3′ | |
| Xyl3C (ORF0975) | Forward | 5′-GCGCcatatgATGAAAAGTAAACAACTAATAAC-3′ |
| Reverse | 5′-CGCGctcgagTTATTTTAGGTAAATAATTAATTTTTTC-3′ | |
| Mutagenesisb | ||
| Xyl3B E115A | Forward | 5′-GCTCTATTCCACGAAGCAGTGCTCTCGGGTGTT-3′ |
| Reverse | 5′-AACACCCGAGAGCACTGCTTCGTGGAATAGAGC-3′ | |
| Xyl3B E115D | Forward | 5′-GCTCTATTCCACGAAGATGTGCTCTCGGGTGTTAA −3′ |
| Reverse | 5′-TTAACACCCGAGAGCACATCTTCGTGGAATAGAGC −3′ | |
| Xyl3B R177A | Forward | 5′-CGAAATCCAAGTTTCAACGCGCTCGAAGAGTCGTATGG-3′ |
| Reverse | 5′-CCATACGACTCTTCGAGCGCGTTGAAACTTGGATTTCG-3′ | |
| Xyl3B K214A | Forward | 5′-GTGTGGGGGCTTGCAGCGCGCACTATCTCGGATATG-3′ |
| Reverse | 5′-CATATCCGAGATAGTGCGCGCTGCAAGCCCCCACAC-3′ | |
| Xyl3B H215A | Forward | 5′-GGGGGCTTGCAGCAAGGCCTATCTCGGATATGGT-3′ |
| Reverse | 5′-ACCATATCCGAGATAGGCCTTGCTGCAAGCCCCC-3′ | |
| Xyl3B M251A | Forward | 5′-CTGGAAGCAAAGCGCTGGCGCCTGGTTATCACGCTG-3′ |
| Reverse | 5′-CAGCGTGATAACCAGGCGCCAGCGCTTTGCTTCCAG-3′ | |
| Xyl3B D286A | Forward | 5′-TGGTATGGTGGTTAGTGCCTATACAGCCATAGACC-3′ |
| Reverse | 5′-GGTCTATGGCTGTATAGGCACTAACCACCATACCA-3′ | |
| Q-PCRc | ||
| GyrAQ-PCR | Forward | 5′-CCCGTGTAGTAGGTGAGGTTCTTG-3′ |
| Reverse | 5′-TCCAGTCTTGGCCCATACG-3′ | |
| CdxAQ-PCR | Forward | 5′-TTAGGAATTTGTTTGTCTGGAGCAT-3′ |
| Reverse | 5′-GCTTTCTCTTCCAACGTCATAGC-3′ | |
| Xyl3AQ-PCR | Forward | 5′-GGCCTGTGCCAAGCACTTT-3′ |
| Reverse | 5′-CGCGTGGCGAGATGTTATT-3′ | |
| Xyl3BQ-PCR | Forward | 5′-CGCTTCGAGGCAAACAGAAT-3′ |
| Reverse | 5′-GGGAACGAATAGTCACCACACA-3′ | |
| Xyl3CQ-PCR | Forward | 5′-AAACTCGGTGACTTTGATTCTGATAAC-3′ |
| Reverse | 5′-TTTGGTAGGCAAGCTGTTTATGTT-3′ |
Oligonucleotide primers were synthesized by Integrated DNA Technologies (Coralville, IA). Restriction enzyme sites are in lowercase type in the primer sequence. Underlined sequences indicate engineered codon mutations.
Residues were selected for conservative mutagenesis due to their conservation as predicted by an alignment of the primary amino acid sequences for family 3 glycoside hydrolases with biochemically defined catalytic activities, and primers were designed in accordance with the QuikChange protocol by Stratagene (La Jolla, CA).
Primers for Q-PCR were designed by using the Primer Express v3.0 program from Applied Biosystems.
During the course of the current study, our laboratory made a transition from traditional restriction enzyme-based cloning methods to ligation-independent methods. Thus, the cloning procedures employed for the four genes in the current study differ. The cdxA gene was amplified by PCR with primers engineered to incorporate 5′ NdeI and 3′ XhoI restriction sites for subsequent directional cloning. The resulting amplicon was cloned into pGEM-T via TA cloning, and a second primer set (CdxA-SDM1 and CdxA-SDM2) was then employed to introduce a silent mutation into a native NdeI restriction site in the gene (A→T at nucleotide position 2256) by site-directed mutagenesis. The resulting gene was then excised from the pGEM-T vector by digestion with NdeI and XhoI and subcloned in frame with the hexahistidine (His6)-encoding sequence of a modified pET-28a expression vector by replacing the NdeI-XhoI polylinker. The pET-28a vector was modified by replacing the gene for kanamycin resistance with that for ampicillin resistance (7).
For xyl3A, the gene was amplified with primers engineered to incorporate 5′-GACGACGACAAGA and 3′-ACCGGGCTTCTCCTC extensions to facilitate subsequent annealing with expression vector pET-46b. The resulting xyl3A amplicon was then digested with the exonuclease activity of T4 DNA polymerase, annealed with a similarly digested pET-46b vector, and introduced into E. coli JM109 cells by electroporation.
For xyl3B and xyl3C, the primer sets were engineered to incorporate 5′ NdeI and 3′ XhoI restriction sites for subsequent directional cloning. The resulting amplicons were then digested with NdeI and XhoI and cloned in frame with the His6-encoding sequence of a pET-15b expression vector by replacing the NdeI-XhoI polylinker.
Thus, the expression of all four genes resulted in N-terminal polyhistidine fusion proteins. The integrity of the cloned cdxA, xyl3A, xyl3B, and xyl3C genes was confirmed by DNA sequencing (W. M. Keck Center for Comparative and Functional Genomics at the University of Illinois at Urbana-Champaign).
The resulting plasmid constructs, pET28a-cdxA, pET46b-xyl3A, pET15b-xyl3B, and pET15b-xyl3C, were introduced into E. coli BL21 CodonPlus (DE3) RIL competent cells by heat shock. The recombinant E. coli cells were then grown overnight in lysogeny broth (LB) (10 ml) (3, 4) supplemented with ampicillin (100 μg/ml) and chloramphenicol (50 μg/ml) at 37°C with vigorous aeration. After 8 h, the starter cultures were diluted into fresh LB (1 liter) supplemented with ampicillin and chloramphenicol and cultured at 37°C with aeration until the absorbance at 600 nm reached 0.3. Gene expression was then induced by the addition of isopropyl β-d-thiogalactopyranoside (IPTG) at a final concentration of 0.1 mM, and the temperature for culturing was lowered to 16°C. After 16 h, the cells were harvested by centrifugation (4,000 × g for 15 min at 4°C), resuspended in 35 ml lysis buffer (50 mM Tris-HCl, 300 mM NaCl [pH 7.5]), and ruptured by 2 passages through an EmulsiFlex C-3 cell homogenizer from Avestin (Ottawa, Canada). The cell lysates were then clarified by centrifugation at 20,000 × g for 30 min at 4°C, and the recombinant proteins were purified by using a two-step purification scheme with an AKTAxpress fast protein liquid chromatograph (FPLC; GE Healthcare) by coupling nickel affinity chromatography (5-ml HisTrap FF column; GE Healthcare) with buffer exchange (HiPrep 26/10 desalting column; GE Healthcare). The proteins were then eluted from the HisTrap column with a Tris-HCl elution buffer (50 mM Tris-HCl, 300 mM NaCl, 250 mM imidazole [pH 7.5]) and exchanged into a Tris-HCl protein storage buffer (50 mM Tris-HCl, 150 mM NaCl [pH 7.5]). Aliquots of eluted fractions were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) according to a method described previously by Laemmli (33), and protein bands were visualized by staining with Coomassie brilliant blue G-250.
The protein concentrations were quantified by absorbance spectroscopy according to methods described previously by Gill and von Hippel (21), with the following extinction coefficients: 92,710 M−1 cm−1, 130,640 M−1 cm−1, 96,615 M−1 cm−1, and 123,650 M−1 cm−1 for CdxA, Xyl3A, Xyl3B, and Xyl3C, respectively. The absorbance value for each protein solution was measured at 280 nm with a NanoDrop 1000 apparatus from Thermo Fisher Scientific Inc. (Waltham, MA).
Hydrolysis of para-nitrophenyl-linked sugars.
The enzyme-catalyzed hydrolysis of para-nitrophenyl (pNP)-linked monosaccharide substrates was assayed by using a thermostated Synergy II multimode microplate reader from BioTek Instruments Inc. (Winooski, VT). A library of pNP substrates was screened for activity, including pNP-α-l-arabinopyranoside, pNP-α-l-arabinofuranoside, pNP-β-d-fucopyranoside, pNP-α-l-fucopyranoside, pNP-α-d-galactopyranoside, pNP-β-d-galactopyranoside, pNP-α-d-glucopyranoside, pNP-β-d-glucopyranoside (pNPG), pNP-β-d-maltopyranoside, pNP-α-d-maltopyranoside, pNP-α-d-mannopyranoside, pNP-β-d-mannopyranoside, pNP-α-l-rhamnopyranoside, pNP-β-d-xylopyranoside (pNPX), and pNP-β-d-cellobioside. The substrates (1 mM) in citrate buffer (100 μl) (50 mM sodium citrate, 150 mM NaCl [pH 5.5]) were incubated at 37°C in the presence or absence of CdxA (6 nM), Xyl3A (0.5 μM), Xyl3B (9 nM), or Xyl3C (0.5 μM) for 30 min, and the level of pNP release was determined by continuously monitoring the absorbance at 400 nm. The path length correction feature of the instrument was employed to convert the absorbance values recorded to correspond to a 1-cm path length. The extinction coefficient for pNP at pH 5.5 and at a wavelength of 400 nm was measured to be 0.673 mM−1 cm−1. In preliminary studies, we determined that the enzymatic properties of Xyl3B were unaffected by the amino-terminal hexahistidine fusion peptide (data not shown). Therefore, in subsequent analyses, the hexahistidine-tagged proteins were used.
Hydrolysis of neutral and acidic oligosaccharides.
To assess the capacity of P. bryantii GH family 3 proteins to hydrolyze neutral xylo- and cellooligosaccharides, the enzymes were incubated with cellohexaose or xylohexaose, and the products were analyzed by high-performance liquid chromatography (HPLC). Initial biochemical studies with pNP-linked sugar substrates revealed that the optimal pH for the four proteins was 5.5 (see Fig. S2 in the supplemental material). Reaction mixtures (450 μl) were prepared in citrate buffer (50 mM sodium citrate, 150 mM NaCl [pH 5.5]) with xylohexaose or cellohexaose (60 μg/ml, final concentration) at 37°C, and reactions were initiated by the addition of CdxA, Xyl3A, Xyl3B, or Xyl3C (100 nM, final concentration) to the mixture. At regular time intervals (0 min, 15 min, 30 min, and 3 h), 100-μl aliquots were removed, and the reactions were terminated by the addition of 300 μl of 0.1 M NaOH to the mixture.
For analysis of aldouronic acid hydrolysis, the enzymes (0.5 μM, final concentration) were incubated with the aldouronic acid mixture (60 μg/ml, final concentration) in citrate buffer (50 mM sodium citrate, 150 mM NaCl [pH 5.5]) at 37°C. After 16 h, 100-μl aliquots were removed, and the reactions were terminated by the addition of 300 μl 0.1 M NaOH to the mixture.
The oligosaccharide compositions of the neutral and acidic oligosaccharide mixtures following enzymatic hydrolysis were then assessed by high-performance anion-exchange chromatography (HPAEC) with a pulsed amperometric detector (PAD) with a System Gold HPLC instrument from Beckman Coulter (Fullerton, CA) equipped with a CarboPac PA1 guard column (4 by 50 mm) and a CarboPac PA1 analytical column (4 by 250 mm) from Dionex Corporation (Sunnyvale, CA) and a Coulochem III electrochemical detector from ESA Biosciences (Chelmsford, MA). Monomeric xylose (X1), glucose (G1), xylooligosaccharides (X2 to X6), and cellooligosaccharides (G2 to G6) were used as standards. Oligosaccharides were resolved by using a mobile phase of 100 mM NaOH with a linear gradient ending with 125 mM sodium acetate (NaOAc) over 25 min.
Growth of P. bryantii B14 and analysis of gene expression.
P. bryantii B14 was grown anaerobically in a chemically defined medium modified from that described previously by Griswold and Mackie (23) (see Table S1 in the supplemental material) with either glucose (0.18%, wt/vol) or wheat arabinoxylan (0.15%, wt/vol) as the sole carbohydrate source. Cultures were grown in butyl rubber-stoppered serum bottles, which were modified to incorporate a sidearm tube to allow turbidity measurements at 600 nm by use of a Spectronic 20D+ spectrophotometer from Thermo Fisher Scientific Inc. (Waltham, MA). At the mid-log phase of growth (optical density at 600 nm [OD600] of 0.2), 30 ml of culture was removed and combined with 2 volumes of RNAprotect bacterial reagent, and RNA was purified by using the RNeasy kit with the optional on-column DNase treatment step. The RNA was converted to single-stranded cDNA by use of the iScript cDNA synthesis kit. Quantitative PCR (Q-PCR) experiments were performed by use of SYBR green master mix with single-stranded cDNA as the template and the primers listed for Q-PCR in Table 1. Reaction mixtures (20 μl) were prepared in 384-well PCR plates with a final primer concentration of 500 nM, and amplification was monitored by use of a LightCycler 480 real-time PCR system from Roche Applied Science (Indianapolis, IN).
To determine the amplification efficiency for each primer set, a plot of the cycle number at the crossing point versus the amount of cDNA per reaction for a set of 6 cDNA dilutions ranging from 0.05 ng to 50 ng per reaction was constructed. The slope of the line was determined and the amplification efficiency (E) was calculated with equation 1:
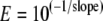 |
(1) |
The relative expression ratio (R) was then calculated for each gene by use of equation 2, where E is the amplification efficiency for each primer set, CP is the cycle number at the crossing point, and the reference gene was the DNA gyrase subunit A gene (gyrA):
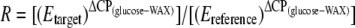 |
(2) |
Multiple-sequence alignment.
To generate multiple-sequence alignments, the indicated sequences were imported into ClustalW (http://align.genome.jp/) and aligned by using the Blosum62 matrix. The multiple-sequence alignment file was then entered into the ESPript V2.2 alignment program (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi) to apply shading and boxing schemes.
Site-directed mutagenesis.
Mutagenesis was performed by use of the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA). First, mutagenic primers were engineered with the desired mutation in the center of the primer and ∼15 bases of correct sequence on either side (Table 1). Reaction mixtures were prepared according to the manufacturer's instructions, with pET15b-xyl3B as the DNA template for the generation of the Xyl3B mutants. After cycling of the reaction mixture 18 times in a PCR thermal cycler, the mixture was digested with DpnI, and the resulting DNA was transformed into electrocompetent E. coli DH5α cells by electroporation. The mutant genes were sequenced to ensure that the appropriate mutations were introduced, while the rest of the gene sequences remained unchanged. Expression and purification of the mutant recombinant proteins were performed as described above for wild-type (WT) Xyl3B.
Protease sensitivity assays.
Xyl3B wild-type and mutant enzymes (2 mg/ml, final concentration) were incubated in Tris buffer (50 mM Tris-HCl, 150 mM NaCl, 50 mM dithiothreitol [DTT] [pH 8.0]) with various concentrations of trypsin (0 μg/ml, 3.1 μg/ml, 25 μg/ml, and 100 μg/ml) at 25°C. After 1 h, the reactions were terminated by the addition of an equal volume of 2× SDS sample buffer (4% [wt/vol] SDS, 100 mM Tris-HCl, 0.4 mg bromophenol blue/ml, 0.2 M DTT, 20% [vol/vol] glycerol) to the mixture. The samples were boiled for 5 min and resolved by SDS-PAGE (12% [wt/vol] acrylamide) (70 V for 20 min and then 200 V for an additional 40 min). The gels were soaked in fixing solution (25% isopropanol, 10% [vol/vol] acetic acid) for 15 min and then placed into Coomassie stain (10% [vol/vol] acetic acid, 0.06 mg/ml Coomassie brilliant blue G-250). After 10 h, the gels were destained for 4 h with 10% (vol/vol) acetic acid, rinsed twice with double-distilled water (ddH2O), and then photographed with a G:Box gel dock imaging system from Syngene (Frederick, MD).
Circular-dichroism spectroscopy of wild-type and mutant Xyl3B.
Circular-dichroism (CD) spectra were collected for wild-type and mutant Xyl3B in the far-UV range by utilizing a J-815 circular-dichroism spectropolarimeter from Jasco (Easton, MD). A rectangular cuvette with a total volume of 350 μl and a path length of 0.1 cm was used for each assay. The CD spectra of wild-type and mutant Xyl3B (1.25 μM, final concentration) in phosphate buffer (50 mM sodium phosphate, pH 5.5) were recorded from 190 nm to 260 nm at a scan rate of 50 nm/s with a wavelength step of 0.1 nm and with 5 accumulations.
Data acquisition was coordinated by using Jasco Spectra Manager v1.54A software. Raw data files were uploaded onto the DICHROWEB online server (http://www.cryst.bbk.ac.uk/cdweb/html/home.html) and analyzed by use of the CDSSTR algorithm with reference set 4, which is optimized for the analysis of data recorded in the range of 190 nm to 240 nm (36).
Determination of steady-state kinetic values for wild-type and mutant Xyl3B.
Kinetic studies of the wild-type and mutant Xyl3B proteins were performed by using a thermostated Synergy II multimode microplate reader from BioTek Instruments Inc. (Winooski, VT). pNPX or pNPG (0 to 46.25 mM) was incubated in citrate reaction buffer (100 μl) (50 mM sodium citrate, 150 mM NaCl [pH 5.5]) at 37°C in a 96-well flat-bottom microtiter plate, and reactions were initiated by the addition of wild-type or mutant Xyl3B to the mixture. In order to obtain initial velocities of the enzyme-catalyzed reactions, the protein concentrations were adjusted such that the slopes of the resulting progress curves (change in absorbance per unit of time) were linear. The hydrolysis of pNPX was continuously monitored by recording the UV signal at 400 nm. Initial rate data were then plotted against the substrate concentration, and kinetic values were estimated by applying a nonlinear curve fit using GraphPad Prism v5.02 from GraphPad Software (San Diego, CA). The extinction coefficient for para-nitrophenol at pH 5.5 and at a wavelength of 400 nm was measured to be 0.673 mM−1 cm−1. For mutants with very low activities, the apparent kcat [kcat(apparent)] was determined at a 46.25 mM pNPX concentration and an enzyme concentration of 10 μM.
Nucleotide sequence accession numbers.
The sequences for xyl3A, xyl3B, and xyl3C have been deposited in the NCBI GenBank database under accession numbers GU564512.1, GU564513.1, and GU564514.1, respectively.
RESULTS
Identification, cloning, and expression of Prevotella bryantii B14 GH family 3 genes.
To identify putative GH family 3 genes in P. bryantii B14, the genome sequence of the bacterium was uploaded onto the Rapid Annotation Using Subsystem Technology (RAST) online server (1), and a BLASTp search of the annotated genome was performed by using the barley ExoI enzyme (28) as a query. This search revealed the presence of four putative GH3 genes (ORF0348, ORF0398, ORF0911, and ORF0975), which were each annotated as encoding β-d-glucosidase enzymes (Table 2). ORF0348 was previously cloned and expressed in E. coli cells, and its biochemical properties were characterized by Wulff-Strobel and Wilson (46). This recombinant protein was found to have cellodextrinase and cyanoglycosidase activities; thus, the protein was named CdxA. The genomic context for the four enzymes is summarized in Fig. S1 in the supplemental material. CdxA is located just downstream of a putative GH family 5 endomannanase, and ORF975 is located just upstream of a putative xylose transporter, whereas ORF398 and ORF911 do not appear to be linked to other genes with predicted roles in polysaccharide degradation.
TABLE 2.
Domain architecture and predicted activities for GH family 3 proteins from Prevotella bryantii B14
 |
Open reading frame assignments were made by TIGR following autoannotation of the draft genome assembly for Prevotella bryantii B14.
Functional domains were assigned by utilizing the Pfam online server (http://www.sanger.ac.uk/Software/Pfam/) (17). Domain hits were included if the expect value (E value) was lower than 1 × 10−8.
Open reading frame functional annotations were made by TIGR following autoannotation of the draft genome assembly for Prevotella bryantii B14. ORF0348 was cloned and biochemically characterized by Wulff-Strobel and Wilson (46).
For experimental evidence, see reference 46.
The domain architectures for the coding sequences were analyzed by use of the Pfam online server (17). These studies suggested that all four proteins possess two conserved domains, as with most GH family 3 proteins (24). However, the analysis also predicted that ORF0398 and ORF0975 encode proteins that possess a PA14 domain insertion within the C-terminal GH family 3 domains (Table 2). It is predicted that the PA14-like insertion sequence may represent a novel carbohydrate binding module (41). These bioinformatic analyses suggested that the four GH family 3 genes encode proteins with redundant biochemical functions. To test this prediction, we cloned and expressed the four GH family 3 genes as recombinant polyhistidine fusion proteins in E. coli. The gene products were purified by metal affinity chromatography, and the predicted molecular masses (ORF0348, 86 kDa; ORF0398, 99 kDa; ORF0911, 86 kDa; ORF0975, 96 kDa) were in agreement with the sizes of the purified proteins estimated by comparisons with molecular weight resolved by SDS-PAGE (Fig. 1A).
FIG. 1.

GH family 3 genes from P. bryantii B14 encode functional enzymes. (A) Purification of recombinant GH3 proteins. The eluate from nickel-chelate chromatography was analyzed by 12% SDS-PAGE, followed by Coomassie brilliant blue G-250 staining. MW, molecular weight (in thousands). (B to E) Hydrolysis of pNP-linked sugars. CdxA (B), Xyl3A (C), Xyl3B (D), and Xyl3C (E) were assessed for their capacities to hydrolyze several pNP-linked sugars by UV spectroscopy. pNPX, pNPA, pNPG, and pNPG2 represent pNP-β-d-xylopyranoside, pNP-α-l-arabinopyranoside, pNP-β-d-glucopyranoside, and pNP-β-d-cellobioside, respectively.
Prevotella bryantii B14 GH family 3 genes encode enzymes with nonredundant biochemical functions.
To determine the substrate specificities for the gene products of ORF0348, ORF0398, ORF0911, and ORF0975, the recombinant proteins were screened for activity with a library of α- and β-para-nitrophenol (pNP)-linked sugars. The putative β-d-glucosidase proteins were incubated in the presence of the derivatized sugars, and the reactions were monitored spectrophotometrically. These experiments revealed that ORF0348 (CdxA) catalyzed the release of pNP from pNP-β-d-glucopyranoside and pNP-β-d-cellobioside (Fig. 1B). When ORF0398 was incubated with the derivatized sugars, pNP was released from pNP-β-d-xylopyranoside (Fig. 1C). When ORF0911 was incubated with the derivatized sugars, pNP was released from pNP-β-d-xylopyranoside, and in addition, low hydrolytic activity was observed with pNP-α-l-arabinofuranoside (Fig. 1D). When ORF0975 was incubated with the derivatized sugars, pNP was released from pNP-β-d-xylopyranoside, pNP-β-d-glucopyranoside, and pNP-β-d-cellobioside, although this activity was much lower than the activity detected for the three other proteins (Fig. 1E). These results indicated that each of the four putative GH family 3 genes encodes a functional glycoside hydrolase enzyme. Furthermore, these data suggest that each of the four GH family 3 proteins in Prevotella bryantii B14 possesses a unique substrate specificity.
Prevotella bryantii B14 GH family 3 proteins release monosaccharides from plant cell wall polysaccharides.
Initial data suggested that the four GH3 enzymes have activity with β-1,4-linked xylosides or glucosides (Fig. 1B to E). To evaluate whether the four GH family 3 proteins may contribute to the hydrolysis of plant cell wall polysaccharides that contain glucose or xylose, each protein (0.1 μM) was incubated with cellohexaose or xylohexaose (60 μg/ml each), and the reaction products were monitored over time by HPAEC. During the course of the reaction, CdxA (ORF0348) converted cellohexaose to glucose and cellobiose (Fig. 2A). In addition, CdxA converted xylohexaose to a mixture of shorter xylooligosaccharides, although this activity was much lower than the hydrolytic activity with cellohexaose. These data are in support of previous findings that identified ORF0348 as a cellodextrinase (CdxA) (46).
FIG. 2.

Hydrolysis of cellohexaose and xylohexaose by P. bryantii B14 GH family 3 enzymes. The hydrolysis of cellohexaose and xylohexaose was assessed by incubating the enzyme with each substrate and then resolving the products by high-performance anion-exchange chromatography (HPAEC) followed by detection with a pulsed amperometric detector (PAD). The hydrolysis products were identified by comparisons of peaks with retention times of purified substrates. Abbreviations for oligosaccharides are as follows: glucose through cellohexaose, G1 to G6; xylose through xylohexaose, X1 to X6.
In contrast to the case for CdxA, the gene products of ORF0398, ORF0911, and ORF0975 did not hydrolyze cellohexaose to cellobiose and glucose, although some products, mainly G4 and G5, were detectable. These proteins were, however, capable of converting xylohexaose to a mixture of shorter xylooligosaccharides and xylose (Fig. 2C to E). These results suggested that the major enzymatic activity of each of the gene products of ORF0398, ORF0911, and ORF0975 is β-xylosidase activity, and thus, the genes were designated xyl3A, xyl3B, and xyl3C, respectively.
For several of the reactions, monosaccharides did not accumulate despite the evident depolymerization of the xylohexaose or cellohexaose substrate (CdxA, xylohexaose; Xyl3A, cellohexaose; Xyl3B, cellohexaose; Xyl3C, cellohexaose). Several GH family 3 enzymes were previously reported to exhibit transglycosylase activities (10, 32, 45), and it is possible that the lack of monosaccharide accumulation in the above-mentioned reactions is a result of transglycosylation side reactions.
Xyl3B and Xyl3C possess unique substrate specificities.
Natural xylan polymers may have a variety of different substituents on the xylose backbone chain, including arabinofuranosyl, acetyl, and 4-O-methyl glucuronyl groups. To test whether substituents on the xylan chain may affect the β-xylosidase activity of Xyl3B and Xyl3C, the proteins were incubated with aldouronic acids in the presence or absence of a GH family 67 α-glucuronidase enzyme, which was cloned from Prevotella bryantii B14 (Agu67A), and the hydrolytic products were analyzed by HPAEC. The aldouronic acid mixture was obtained from Megazyme and, according to the vendor, was constructed by controlled acid hydrolysis of 4-O-methyl glucuronoxylan. The cloning, expression, purification, and biochemical characterization of Agu67A will be published elsewhere.
When the aldouronic acid mixture was incubated with Agu67A alone, 4-O-methyl glucuronic acid (MGA) was released (peak at 16.08 min), and xylooligosaccharides (X2, 8.79 min; X3, 9.62 min; X4, 10.62 min; X5, 11.56 min), xylose (5.67 min), and two additional peaks (15.31 min and 15.69 min) were detected (Agu67A) (Fig. 3). In the presence of Xyl3B alone, the level of xylose in the hydrolysate was increased compared to that of the control. In the presence of Xyl3C alone, the level of xylose was also increased compared to that of the control. In addition, the shoulders of the two peaks centered at 15.31 min and 15.69 min disappeared, resulting in symmetrical peaks being observed at these positions (compare peaks from Xyl3C hydrolysates to peaks from control and also Xyl3B hydrolysates). When Agu67A and Xyl3B were incubated together with the aldouronic acid mixture, a large amount of xylose was released, and MGA was detected; however, the two peaks at 15.31 min and 15.69 min remained. When Agu67A and Xyl3C were incubated together with the aldouronic acid mixture, a large amount of xylose was released, 4-O-methyl glucuronic acid was detected, and the two peaks at 15.31 min and 15.69 min disappeared (Fig. 3B). In the Agu67A/Xyl3C mixture, small peaks that corresponded to xylobiose and xylotriose were detected, indicating that the cleavage of these two xylooligosaccharides by Xyl3C was incomplete. These results show that although Xyl3B and Xyl3C both possess β-xylosidase activity, they exhibit different specificities for components of the aldouronic acid mixture.
FIG. 3.

Xyl3B and Xyl3C exhibit differences in specificity for 4-O-methyl glucuronic acid-substituted xylooligosaccharides. Shown are data for the hydrolysis of aldouronic acids. P. bryantii B14 Xyl3B (A) or Xyl3C (B) was incubated with aldouronic acids in the presence or absence of a GH family 67 α-glucuronidase enzyme, which was cloned from Prevotella bryantii B14 (Agu67A), and the hydrolytic products were analyzed by HPAEC-PAD. The hydrolysis products were identified by comparisons of peaks with retention times of purified substrates.
Expression of xyl3A is induced during growth of P. bryantii B14 on xylan compared to glucose.
Three of the GH family 3 genes from P. bryantii B14 encode enzymes with β-xylosidase activity. To evaluate whether the expression of these genes is influenced by the carbohydrate growth source, P. bryantii B14 was cultured with glucose or soluble wheat arabinoxylan (WAX), and the relative levels of gene expression for each of the GH3 genes were compared after the expression values were normalized to values for a housekeeping gene (gyrA). P. bryantii B14 grew rapidly in the presence of either glucose or wheat arabinoxylan as the sole carbohydrate source (Fig. 4A), with specific growth rates (μ) of 0.59 h−1 and 0.62 h−1, respectively. The relative expression ratios for the four genes during the growth of P. bryantii B14 on WAX compared to the growth on glucose were as follows: cdxA, 0.56 ± 0.04; xyl3A, 45 ± 11; xyl3B, 1.4 ± 0.3; xyl3C, 0.96 ± 0.3 (Fig. 4B). Of the four genes, the expression of xyl3A was induced during growth on WAX compared to glucose, whereas the expression levels for xyl3B and xyl3C were constitutive. Despite the fact that three of the P. bryantii B14 GH3 genes encode β-xylosidase enzymes, these results suggest that there are differences in the transcriptional regulation of these genes.
FIG. 4.

Expression of GH3 genes during growth of P. bryantii B14 with wheat arabinoxylan and glucose. (A) P. bryantii B14 was cultured in a chemically defined growth medium with either wheat arabinoxylan (0.15%, wt/vol) or glucose (0.18%, wt/vol) as the sole carbohydrate source, and the OD600 values over time were monitored. (B) At an optical density of 0.2 (mid-log phase of growth), cells were harvested, RNA was extracted, and quantitative reverse transcription-PCR experiments were performed as described in Materials and Methods. Four technical replicates of the Q-PCR were performed for each of three independent biological replicates, and data are reported as means ± standard errors from the mean.
Mutational analysis of Xyl3B reveals residues that contribute to catalysis and substrate specificity.
To identify amino acid residues that contribute to the catalysis and substrate specificity of GH3 enzymes, Xyl3B was chosen for mutational analysis due to its less complex domain architecture (Table 2). Amino acid sequence alignments were constructed with biochemically characterized GH3 enzymes (see Fig. S3 in the supplemental material), and these alignments revealed the presence of five distinct motifs that contain conserved amino acid residues. Within these motifs, nine amino acid residues (Gly119, Arg171, Arg177, Glu180, Asp185, Lys214, His215, Met251, and Asp286) were conserved throughout all the sequences that were aligned. To assess whether these conserved amino acid residues map to the active site of Xyl3B, a three-dimensional homology model based upon the available crystal structure for the barley β-glucan exohydrolase (ExoI) was constructed by use of the ModWeb online server for protein structure modeling (15). The homology model for Xyl3B exhibited a Cα root mean square deviation (RMSD) value of 17 Å compared to the barley crystal structure. However, of the nine conserved amino acids identified in the sequence alignment, five residues mapped to within 5 Å of the transition state analog bound in the active site of the model. Furthermore, the side-chain RMSD values for these residues were appreciably lower than the overall Cα RMSD for the homology model (Arg177, 1.3 Å; Lys214, 0.51 Å; His215, 0.55 Å; Met251, 0.64 Å; Asp286, 0.31 Å), indicating that the spatial conservation of these residues is much higher than that of other residues in the protein. In the homology model, an additional amino acid residue (Glu115) that aligns with Asp120 from the barley enzyme was identified (see Fig. S3 in the supplemental material). In ExoI, Asp120 makes short hydrogen bond contacts with the 4′ and 6′ hydroxyl groups of glucose (Fig. 5A); thus, we hypothesized that the longer glutamic acid residue in Xyl3B could be important for conferring xylosidase activity to Xyl3B because xylose lacks the 6′ hydroxyl group found in glucose (Fig. 5B).
FIG. 5.

GH family 3 active-site residues. (A) Active-site residues for the β-glucan exohydrolase (ExoI) from barley (Hordeum vulgare) bound to glucose are shown (PDB accession no. 1EX1). The residues in parentheses are the corresponding residues in Xyl3B that align with the barley ExoI residues. (B) Predicted model for discrimination between xylosides and glucosides by GH3 enzymes. Asp120 from barley ExoI forms hydrogen bond contacts with the 4′OH and 6′OH groups of glucose. Glu115 from P. bryantii B14 Xyl3B is predicted to form hydrogen bond contacts with the 4′OH of xylose and may discriminate between glucose and xylose on the basis of steric interactions.
For each of the amino acid positions indicated above, the residue was mutated to alanine to investigate whether these residues were important for catalysis. In addition, a Glu115→Asp mutation was generated to test the predicted role of this residue in discriminating between xylosides and glucosides by Xyl3B.
To ensure that the amino acid substitutions had not resulted in gross structural changes, the protease sensitivity patterns of wild-type and mutant forms of Xyl3B were compared. These experiments revealed that the Xyl3B E115A, E115D, R177A, K214A, H215A, M251A, and D286A mutants all exhibited sensitivity to tryptic proteolysis (see Fig. S4 in the supplemental material) similar to that of wild-type Xyl3B. To further evaluate whether mutations in Xyl3B had any influence on the protein structure, the proportions of secondary structural elements between the mutant and wild-type proteins were compared by circular-dichroism spectroscopy. These experiments revealed that the overall secondary structures for the mutant and wild-type Xyl3B proteins were similar (see Table S2 in the supplemental material).
The catalytic properties of wild-type Xyl3B and the seven mutant proteins were compared through determination of their steady-state kinetic properties in the presence of pNP-β-d-xylopyranoside (pNPX) and pNP-β-d-glucopyranoside (pNPG). The results revealed that each of the six residues, when mutated to alanine, resulted in mutant enzymes that exhibited large losses in catalytic efficiency (kcat/Km) relative to the wild-type enzyme. These losses were manifested in large decreases in the magnitudes of the turnover numbers (kcat) for the E115A, R177A, K214A, H215A, and D286A mutants and modest changes in the Km values for the enzymes, except for a large increase in the Km for the M251A mutation (Table 3).
TABLE 3.
Steady-state kinetic parameters for Xyl3B wild-type and mutant enzymes
| Enzyme | Mean value ± SEMa |
|||||
|---|---|---|---|---|---|---|
|
pNP-β-d-Xylopyranoside |
pNP-β-d-Glucopyranoside |
|||||
| kcat (s−1) | Km (mM) | kcat/Km (s−1 mM−1) | kcat (s−1) | Km (mM) | kcat/Km (s−1 mM−1) | |
| Xyl3B WT | 250 ± 10 | 8.4 ± 1 | 30 ± 4 | 19 ± 0.6 | 22 ± 1 | 0.86 ± 0.05 |
| Xyl3B E115A | 0.35 ± 0.02 | 28 ± 3 | (1.3 ± 0.2) × 10−2 | (8.7 ± 0.5) × 10−2 | 39 ± 4 | (2.2 ± 0.3) × 10−3 |
| Xyl3B E115D | 87 ± 5 | 39 ± 4 | 2.2 ± 0.2 | 16 ± 0.4 | 17 ± 1 | 0.94 ± 0.06 |
| Xyl3B R177Ab | 0.17 ± 0.01 | 8.8 ± 1 | (1.9 ± 0.2) × 10−2 | ND | ND | ND |
| Xyl3B K214Ab | 0.10 ± 0.01 | 5.7 ± 0.9 | (1.8 ± 0.3) × 10−2 | ND | ND | ND |
| Xyl3B H215A | 4.0 ± 0.2 | 19 ± 1 | 0.21 ± 0.02 | (3.8 ± 0.4) × 10−2 | 23 ± 4 | (1.7 ± 0.3) × 10−3 |
| Xyl3B M251Ab | 75 ± 6 | 100 ± 10 | 0.75 ± 0.1 | ND | ND | ND |
| Xyl3B D286Ab,c | (1.20 ± 0.3) × 10−2 | ND | ND | ND | ND | ND |
The data are reported as means ± standard errors from the mean for three independent experiments.
Kinetic parameters for pNPG were not determined because the activity was below the limit of sensitivity for the assay.
The reported kcat value is kcat(apparent), as Km values were not determined; rates for this mutant were determined at a 46.25 mM pNPX concentration and thus represent a lower boundary on kcat.
DISCUSSION
Prevotella bryantii B14 harbors four GH3 genes, each of which is predicted to encode a β-glucosidase enzyme. The presence of multiple GH3 genes in P. bryantii B14 raises the important question of whether these genes encode enzymes with redundant activities or whether they have each evolved to perform unique functions within the cell. This gene family is one of the largest glycoside hydrolase families identified, and many organisms that metabolize oligo- or polysaccharides possess multiple GH3 genes. As an example, an analysis of the 18 available genomes for Bacteroides spp. (which are the closest relatives of Prevotella spp.) revealed an average of 12 GH3 genes per genome, with the Bacteroides uniformis genome harboring 23 putative GH3 genes. Autoannotation of GH3 genes almost universally assigns the function of β-glucosidase activity to the corresponding gene products. The consequence of this is that many genes within this family are incorrectly annotated, and this limits the ability to assess the role of these genes within the context of the metabolic repertoire of the organisms in which these genes reside. This is particularly evident within the human gut microbiome, where a recent survey identified GH3 as the most abundant GH family present in this microbial ecosystem (22). Despite the high prevalence of this gene family within the human gut microbial community, the role of these genes in this gut consortium is currently unknown. The GH3 family also abounds in the rumen microbial ecosystem (5), and as demonstrated by the present study, it is highly represented in ruminal Prevotella spp. In both the human and rumen ecosystems, these genes are involved in energy capture from host-derived or nondigestible dietary components. An understanding of the functional diversity of GH family 3 gene products in a Prevotella sp. may lead to inferences that are also applicable to organisms within the human gut ecosystem.
Thus, the goal of the current study was 2-fold: (i) to characterize the substrate specificities of GH3 enzymes from P. bryantii B14 and (ii) to identify amino acid residues that contribute to catalysis and substrate specificity for a representative member, Xyl3B. Xyl3B was chosen due to its less complex domain architecture (Table 2), rendering it more amenable to homology modeling based on known crystal structures for members of this family.
Results from the current study clearly indicate that these four genes encode functional glycoside hydrolases and, furthermore, that each of these enzymes possesses unique substrate specificities. CdxA was previously found to have cellodextrinase activity (46), and the results from the current study support this conclusion. P. bryantii B14 is not cellulolytic; however, this bacterium can rapidly ferment cellooligosaccharides obtained via cross-feeding with cellulolytic rumen bacteria (42). During growth on cellodextrins, longer-chain oligosaccharides are hydrolyzed extracellularly to cellotriose or cellobiose prior to transport (42). Thus, CdxA may function to depolymerize cellodextrins, the products of which can subsequently be metabolized by P. bryantii B14.
β-Xylosidases release xylose units from the nonreducing ends of xylooligosaccharides produced by the action of endoxylanase enzymes. In nature, these xylooligosaccharides are commonly substituted with acetyl, arabinofuranosyl, or 4-O-methyl glucuronyl groups, and the presence of these groups likely influences the capacity of β-xylosidases to release xylose from these substrates. The specificity of GH family 3 β-xylosidases for substituted xylooligosaccharides has not been comprehensively studied, although previous studies reported that a GH3 β-xylosidase (Bxl1) from Trichoderma reesei could release xylose from aldouronic acids in which the penultimate xylose of the reducing end was substituted with 4-O-methyl glucuronic acid (25, 37). We hypothesize that similar to T. reesei Bxl1, Xyl3C may cleave xylose from the nonreducing end of aldouronic acids substituted with 4-O-methyl glucuronic acid at the penultimate xylose residue; however, Xyl3B lacks this capacity. A confirmation of this hypothesis will require the testing of enzymes on purified substrates in the future. One striking difference between Xyl3C and Xyl3B is their domain organizations, with Xyl3C harboring a putative PA14 insertion sequence within the C-terminal (α/β)6 sandwich domain (Table 2). It is predicted that PA14 domains may serve a carbohydrate binding function (41, 47), and it is possible that this domain could be responsible for the differences in specificity observed between Xyl3C and Xyl3B. Xyl3A also has a PA14 domain; however, this enzyme exhibits a cleavage pattern similar to that of Xyl3B (data not shown), which suggests that factors other than the presence of a PA14 domain contribute to the unique activity of Xyl3C. This conclusion may be supported by the fact that Bxl1 from T. reesei does not possess a PA14 domain.
Xylanase activity in P. bryantii B14 is inducible during growth on xylans (18-20, 38, 39), and the current study shows that xyl3A is induced during growth on wheat arabinoxylan compared to glucose, whereas cdxA, xyl3B, and xyl3C are not. Although xyl3A, xyl3B, and xyl3C each encode an enzyme with β-xylosidase activity, the differential expression of these genes further supports the biochemical data, which indicate that each gene has unique functional roles within the cell.
A gene cluster with a GH43 β-xylosidase and a GH10 endoxylanase was previously identified for P. bryantii B14 (19), and these enzymes were shown to function synergistically to produce xylose from xylan substrates. Although the GH43 β-xylosidase was shown to convert xylopentaose to xylose (20), the activity on substituted xylans was not evaluated. Our study has identified three additional enzymes with β-xylosidase activity from P. bryantii B14. The presence of four enzymes (one GH43 and three GH3 enzymes) that exhibit β-xylosidase activity in this bacterium is somewhat unexpected. However, it is possible that each of these enzymes has evolved to hydrolyze xylosidic linkages adjacent to substitutions, and thus, these enzymes may function together mutualistically to break down heterogeneous xylan substrates present in ingested forage. Another possibility is that yet-to-be-identified xylose-containing oligosaccharide substrates are encountered within the rumen and that these enzymes have evolved to release fermentable sugars from these substrates.
Despite the diversity in function and considerable variation in amino acid sequences among members of this family of enzymes (24), there is a core set of conserved amino acid residues (see Fig. S3 in the supplemental material) that make hydrogen bond contacts to the substrate in the −1 subsite (44) (Fig. 5A). Mutation of the five highly conserved residues Arg177, Lys214, His215, Met251, and Asp286 to alanine resulted in large decreases in activity for Xyl3B (Table 3), although the structure of the mutant enzymes remained relatively unperturbed (Fig. S4 and Table S2). These results support the role of Arg177, Lys214, and His215 in hydrogen bonding to the hydroxyl groups of the xylose moiety bound in the active site. The activity of the Asp286→Ala mutation was too low for the Km to be accurately determined; thus, the apparent kcat was estimated. The decrease in the catalytic activity of this mutant of approximately 4 to 5 orders of magnitude (Table 3) coupled with the observation that this residue aligns with previously identified catalytic nucleophilic residues in GH3 enzymes (2, 9, 11, 31, 34, 35, 40) provides support for the assignment of Asp286 as the catalytic nucleophile for Xyl3B.
The crystal structure of the GH3 β-glucan exohydrolase (ExoI) from barley revealed that Asp120 forms a hydrogen bond to the 4′OH and 6′OH groups of the glucose substrate bound at the −1 subsite (26). The pentose sugar β-d-xylopyranose does not possess the additional CH2OH substituent from C5 that is present in the hexose sugar β-d-glucopyranose; thus, it is possible that amino acid residues positioned at this site could function to discriminate between the hexose and the pentose on the basis of steric interactions (12). Amino acid sequence alignments of Xyl3B with other biochemically characterized GH3 enzymes revealed the presence of a glutamic acid residue (Glu115) that aligned with Asp120 from the barley enzyme (see Fig. S3 in the supplemental material). The mutation of this glutamic acid residue to alanine resulted in a large decrease in the kcat and a relatively small increase in the Km with both pNPX and pNPG as substrates (Table 3), which suggests that Glu115 is localized in the active site of Xyl3B and plays a role in catalysis. The mutation of Glu115 to aspartic acid resulted in a decrease in the catalytic efficiency (kcat/Km) of Xyl3B with pNPX as a substrate but a slight increase in the catalytic efficiency with pNPG as a substrate. This 15-fold change in substrate selectivity [(kcat/Km)pNPX/(kcat/Km)pNPG = 35 for WT Xyl3B; (kcat/Km)pNPX/(kcat/Km)pNPG = 2.3 for E115D mutant Xyl3B] for pNPG compared to that for pNPX suggests that this residue contributes to determining the substrate specificity for Xyl3B (Fig. 5B). The fact that the E115D mutant remains slightly more selective for pNPX than for pNPG indicates that additional residues within the protein likely contribute to substrate discrimination.
All the substrates that GH3 enzymes hydrolyze possess either a β-d-xylose, α-l-arabinose, β-d-glucosamine, or β-d-glucose residue at the −1 subsite (16). Among these sugars, the stereochemical configurations at the C1, C2, and C3 positions are absolutely conserved. The four residues that are highly conserved in GH family 3 enzymes (Arg-Lys-His-Asp) that participate directly in catalysis (Asp) or in hydrogen-bonding interactions (Arg-Lys-His) are located in the (α/β)8 barrel domain. The results reported in the current study support the hypothesis that Arg177, Lys214, and His215 form a highly conserved, core set of charged amino acid residues in GH3 enzymes and that they make hydrogen bond contacts with the sugar residue at the −1 subsite. Furthermore, our data suggest that differences in specificity at the −1 subsite for members of this enzyme family likely arise as a result of changes in amino acid residues at positions that surround the C4, C5, and C6 positions of these sugars.
GH family 3 is one of the largest GH families, with 3,384 curated sequences currently in the Pfam database (http://pfam.sanger.ac.uk/). Of the bacterial genes identified in a recent survey of the human distal gut microbiome, GH family 3 was the most highly represented GH family (22). Therefore, the importance of this gene family in the metabolic repertoire of gut bacteria cannot be overemphasized. The results from the current study indicate that many of these genes are likely to encode enzymes with unique, nonredundant biochemical properties and that care should be taken when attempting to assign physiological functions solely from primary amino acid sequences. Our studies have provided insight into the roles of specific amino acids in catalysis and substrate selectivity; however, future studies of the role of additional amino acids within these proteins will prove invaluable for furthering our understanding of the metabolic versatility of members of this large gene family. Furthermore, structural studies of representative members in complex with substrates will help to unravel the molecular mechanisms underlying substrate discrimination in this family of enzymes.
Supplementary Material
Acknowledgments
This research was supported by the Energy Biosciences Institute. The research of D.D. was partially supported by a James R. Beck fellowship in microbiology at the University of Illinois and by an NIH NRSA fellowship (fellowship no. 1F30DK084726).
We thank Charles M. Schroeder, M. Ashley Spies, Shosuke Yoshida, Yejun Han, Michael Iakiviak, Young-Hwan Moon, and Xiaoyun Su of the Energy Biosciences Institute for valuable scientific discussions and Farhan Quader and Amara Hussain for technical assistance. We also thank members of the North American Consortium for Genomics of Fibrolytic Ruminal Bacteria for the partial genome sequence of P. bryantii B14.
Footnotes
Published ahead of print on 26 February 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Aziz, R. K., D. Bartels, A. A. Best, M. DeJongh, T. Disz, R. A. Edwards, K. Formsma, S. Gerdes, E. M. Glass, M. Kubal, F. Meyer, G. J. Olsen, R. Olson, A. L. Osterman, R. A. Overbeek, L. K. McNeil, D. Paarmann, T. Paczian, B. Parrello, G. D. Pusch, C. Reich, R. Stevens, O. Vassieva, V. Vonstein, A. Wilke, and O. Zagnitko. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bause, E., and G. Legler. 1974. Isolation and amino acid sequence of a hexadecapeptide from the active site of beta-glucosidase A3 from Aspergillus wentii. Hoppe-Seylers Z. Physiol. Chem. 355:438-442. [DOI] [PubMed] [Google Scholar]
- 3.Bertani, G. 2004. Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186:595-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertani, G. 1951. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J. Bacteriol. 62:293-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brulc, J. M., D. A. Antonopoulos, M. E. Miller, M. K. Wilson, A. C. Yannarell, E. A. Dinsdale, R. E. Edwards, E. D. Frank, J. B. Emerson, P. Wacklin, P. M. Coutinho, B. Henrissat, K. E. Nelson, and B. A. White. 2009. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. U. S. A. 106:1948-1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryant, M. P., N. Small, C. Bouma, and H. Chu. 1958. Bacteroides ruminicola n. sp. and Succinimonas amylolytica; the new genus and species; species of succinic acid-producing anaerobic bacteria of the bovine rumen. J. Bacteriol. 76:15-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cann, I. K., S. Ishino, M. Yuasa, H. Daiyasu, H. Toh, and Y. Ishino. 2001. Biochemical analysis of replication factor C from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 183:2614-2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantarel, B. L., P. M. Coutinho, C. Rancurel, T. Bernard, V. Lombard, and B. Henrissat. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233-D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chir, J., S. Withers, C. F. Wan, and Y. K. Li. 2002. Identification of the two essential groups in the family 3 beta-glucosidase from Flavobacterium meningosepticum by labelling and tandem mass spectrometric analysis. Biochem. J. 365:857-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crombie, H. J., S. Chengappa, A. Hellyer, and J. S. Reid. 1998. A xyloglucan oligosaccharide-active, transglycosylating beta-D-glucosidase from the cotyledons of nasturtium (Tropaeolum majus L) seedlings—purification, properties and characterization of a cDNA clone. Plant J. 15:27-38. [DOI] [PubMed] [Google Scholar]
- 11.Dan, S., I. Marton, M. Dekel, B. A. Bravdo, S. He, S. G. Withers, and O. Shoseyov. 2000. Cloning, expression, characterization, and nucleophile identification of family 3, Aspergillus niger beta-glucosidase. J. Biol. Chem. 275:4973-4980. [DOI] [PubMed] [Google Scholar]
- 12.Dodd, D., S. A. Kocherginskaya, M. A. Spies, K. E. Beery, C. A. Abbas, R. I. Mackie, and I. K. Cann. 2009. Biochemical analysis of a beta-D-xylosidase and a bifunctional xylanase-ferulic acid esterase from a xylanolytic gene cluster in Prevotella ruminicola 23. J. Bacteriol. 191:3328-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards, J. E., N. R. McEwan, A. J. Travis, and R. J. Wallace. 2004. 16S rDNA library-based analysis of ruminal bacterial diversity. Antonie Van Leeuwenhoek 86:263-281. [DOI] [PubMed] [Google Scholar]
- 14.Emanuelsson, O., S. Brunak, G. von Heijne, and H. Nielsen. 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2:953-971. [DOI] [PubMed] [Google Scholar]
- 15.Eswar, N., B. John, N. Mirkovic, A. Fiser, V. A. Ilyin, U. Pieper, A. C. Stuart, M. A. Marti-Renom, M. S. Madhusudhan, B. Yerkovich, and A. Sali. 2003. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 31:3375-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faure, D. 2002. The family-3 glycoside hydrolases: from housekeeping functions to host-microbe interactions. Appl. Environ. Microbiol. 68:1485-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finn, R. D., J. Mistry, B. Schuster-Bockler, S. Griffiths-Jones, V. Hollich, T. Lassmann, S. Moxon, M. Marshall, A. Khanna, R. Durbin, S. R. Eddy, E. L. Sonnhammer, and A. Bateman. 2006. Pfam: clans, Web tools and services. Nucleic Acids Res. 34:D247-D251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardner, R. G., J. E. Wells, J. B. Russell, and D. B. Wilson. 1995. The effect of carbohydrates on the expression of the Prevotella ruminicola 1,4-beta-D-endoglucanase. FEMS Microbiol. Lett. 125:305-310. [DOI] [PubMed] [Google Scholar]
- 19.Gasparic, A., R. Marinsek-Logar, J. Martin, R. J. Wallace, F. V. Nekrep, and H. J. Flint. 1995. Isolation of genes encoding beta-D-xylanase, beta-D-xylosidase and alpha-L-arabinofuranosidase activities from the rumen bacterium Prevotella ruminicola B(1)4. FEMS Microbiol. Lett. 125:135-141. [DOI] [PubMed] [Google Scholar]
- 20.Gasparic, A., J. Martin, A. S. Daniel, and H. J. Flint. 1995. A xylan hydrolase gene cluster in Prevotella ruminicola B(1)4: sequence relationships, synergistic interactions, and oxygen sensitivity of a novel enzyme with exoxylanase and beta-(1,4)-xylosidase activities. Appl. Environ. Microbiol. 61:2958-2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gill, S. C., and P. H. von Hippel. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182:319-326. [DOI] [PubMed] [Google Scholar]
- 22.Gill, S. R., M. Pop, R. T. Deboy, P. B. Eckburg, P. J. Turnbaugh, B. S. Samuel, J. I. Gordon, D. A. Relman, C. M. Fraser-Liggett, and K. E. Nelson. 2006. Metagenomic analysis of the human distal gut microbiome. Science 312:1355-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griswold, K. E., and R. I. Mackie. 1997. Degradation of protein and utilization of the hydrolytic products by a predominant ruminal bacterium, Prevotella ruminicola B(1)4. J. Dairy Sci. 80:167-175. [DOI] [PubMed] [Google Scholar]
- 24.Harvey, A. J., M. Hrmova, R. De Gori, J. N. Varghese, and G. B. Fincher. 2000. Comparative modeling of the three-dimensional structures of family 3 glycoside hydrolases. Proteins 41:257-269. [DOI] [PubMed] [Google Scholar]
- 25.Herrmann, M. C., M. Vrsanska, M. Jurickova, J. Hirsch, P. Biely, and C. P. Kubicek. 1997. The beta-D-xylosidase of Trichoderma reesei is a multifunctional beta-D-xylan xylohydrolase. Biochem. J. 321(Pt. 2):375-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hrmova, M., R. De Gori, B. J. Smith, J. K. Fairweather, H. Driguez, J. N. Varghese, and G. B. Fincher. 2002. Structural basis for broad substrate specificity in higher plant beta-D-glucan glucohydrolases. Plant Cell 14:1033-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hrmova, M., R. De Gori, B. J. Smith, A. Vasella, J. N. Varghese, and G. B. Fincher. 2004. Three-dimensional structure of the barley beta-D-glucan glucohydrolase in complex with a transition state mimic. J. Biol. Chem. 279:4970-4980. [DOI] [PubMed] [Google Scholar]
- 28.Hrmova, M., A. J. Harvey, J. Wang, N. J. Shirley, G. P. Jones, B. A. Stone, P. B. Hoj, and G. B. Fincher. 1996. Barley beta-D-glucan exohydrolases with beta-D-glucosidase activity. Purification, characterization, and determination of primary structure from a cDNA clone. J. Biol. Chem. 271:5277-5286. [DOI] [PubMed] [Google Scholar]
- 29.Hrmova, M., V. A. Streltsov, B. J. Smith, A. Vasella, J. N. Varghese, and G. B. Fincher. 2005. Structural rationale for low-nanomolar binding of transition state mimics to a family GH3 beta-D-glucan glucohydrolase from barley. Biochemistry 44:16529-16539. [DOI] [PubMed] [Google Scholar]
- 30.Hrmova, M., J. N. Varghese, R. De Gori, B. J. Smith, H. Driguez, and G. B. Fincher. 2001. Catalytic mechanisms and reaction intermediates along the hydrolytic pathway of a plant beta-D-glucan glucohydrolase. Structure 9:1005-1016. [DOI] [PubMed] [Google Scholar]
- 31.Jager, S., and L. Kiss. 2005. Investigation of the active site of the extracellular β-D-glucosidase from Aspergillus carbonarius. World J. Microbiol. Biotechnol. 21:337-343. [Google Scholar]
- 32.Kawai, R., K. Igarashi, M. Kitaoka, T. Ishii, and M. Samejima. 2004. Kinetics of substrate transglycosylation by glycoside hydrolase family 3 glucan (1→3)-beta-glucosidase from the white-rot fungus Phanerochaete chrysosporium. Carbohydr. Res. 339:2851-2857. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 34.Li, Y. K., J. Chir, and F. Y. Chen. 2001. Catalytic mechanism of a family 3 beta-glucosidase and mutagenesis study on residue Asp-247. Biochem. J. 355:835-840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li, Y. K., J. Chir, S. Tanaka, and F. Y. Chen. 2002. Identification of the general acid/base catalyst of a family 3 beta-glucosidase from Flavobacterium meningosepticum. Biochemistry 41:2751-2759. [DOI] [PubMed] [Google Scholar]
- 36.Lobley, A., L. Whitmore, and B. A. Wallace. 2002. DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics 18:211-212. [DOI] [PubMed] [Google Scholar]
- 37.Margolles-Clark, E., M. Tenkanen, T. Nakari-Setala, and M. Penttila. 1996. Cloning of genes encoding alpha-L-arabinofuranosidase and beta-xylosidase from Trichoderma reesei by expression in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 62:3840-3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyazaki, K., J. C. Martin, R. Marinsek-Logar, and H. J. Flint. 1997. Degradation and utilization of xylans by the rumen anaerobe Prevotella bryantii (formerly P. ruminicola subsp. brevis) B(1)4. Anaerobe 3:373-381. [DOI] [PubMed] [Google Scholar]
- 39.Miyazaki, K., H. Miyamoto, D. K. Mercer, T. Hirase, J. C. Martin, Y. Kojima, and H. J. Flint. 2003. Involvement of the multidomain regulatory protein XynR in positive control of xylanase gene expression in the ruminal anaerobe Prevotella bryantii B(1)4. J. Bacteriol. 185:2219-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paal, K., M. Ito, and S. G. Withers. 2004. Paenibacillus sp. TS12 glucosylceramidase: kinetic studies of a novel sub-family of family 3 glycosidases and identification of the catalytic residues. Biochem. J. 378:141-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rigden, D. J., L. V. Mello, and M. Y. Galperin. 2004. The PA14 domain, a conserved all-beta domain in bacterial toxins, enzymes, adhesins and signaling molecules. Trends Biochem. Sci. 29:335-339. [DOI] [PubMed] [Google Scholar]
- 42.Russell, J. B. 1985. Fermentation of cellodextrins by cellulolytic and noncellulolytic rumen bacteria. Appl. Environ. Microbiol. 49:572-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stevenson, D. M., and P. J. Weimer. 2007. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biotechnol. 75:165-174. [DOI] [PubMed] [Google Scholar]
- 44.Varghese, J. N., M. Hrmova, and G. B. Fincher. 1999. Three-dimensional structure of a barley beta-D-glucan exohydrolase, a family 3 glycosyl hydrolase. Structure 7:179-190. [DOI] [PubMed] [Google Scholar]
- 45.Watt, D. K., H. Ono, and K. Hayashi. 1998. Agrobacterium tumefaciens beta-glucosidase is also an effective beta-xylosidase, and has a high transglycosylation activity in the presence of alcohols. Biochim. Biophys. Acta 1385:78-88. [DOI] [PubMed] [Google Scholar]
- 46.Wulff-Strobel, C. R., and D. B. Wilson. 1995. Cloning, sequencing, and characterization of a membrane-associated Prevotella ruminicola B(1)4 beta-glucosidase with cellodextrinase and cyanoglycosidase activities. J. Bacteriol. 177:5884-5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zupancic, M. L., M. Frieman, D. Smith, R. A. Alvarez, R. D. Cummings, and B. P. Cormack. 2008. Glycan microarray analysis of Candida glabrata adhesin ligand specificity. Mol. Microbiol. 68:547-559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.