

Abstract
Type IV secretion (T4S) systems are involved in several secretion processes, including secretion of virulence factors, such as toxins or transforming molecules, or bacterial conjugation whereby two mating bacteria exchange genetic material. T4S systems are generally composed of 12 protein components, three of which, termed VirB4, VirB11, and VirD4, are ATPases. VirB4 is the largest protein of the T4S system, is known to play a central role, and interacts with many other T4S system proteins. In this study, we have biochemically characterized the protein TraB, a VirB4 homologue from the pKM101 conjugation T4S system. We demonstrated that TraB is a modular protein, composed of two domains, both able to bind DNA in a non-sequence-specific manner. Surprisingly, both TraB N- and C-terminal domains can bind ATP, revealing a new degenerated nucleotide-binding site in the TraB N-terminal domain. TraB purified from the membrane forms stable dimers and is unable to hydrolyze ATP while, when purified from the soluble fraction, TraB can form hexamers capable of hydrolyzing ATP. Remarkably, both the N- and C-terminal domains display ATP-hydrolyzing activity. These properties define a new class of VirB4 proteins.
The type IV secretion (T4S) systems are widely distributed among the Gram-negative and -positive bacteria. T4S systems export proteins and DNA-protein complexes across the bacterial cell envelope to other bacteria or eukaryotic cells, generally through a process requiring direct cell-to-cell contact (10, 11, 16). T4S systems have been grouped according to sequence relatedness of machine components, with systems homologous to the archetypal VirB/VirD4 T4S system of Agrobacterium tumefaciens being classified as type IVA and those related to the Dot/Icm T4S system of Legionella pneumophila being classified as type IVB (12). T4S systems fulfill a wide variety of functions, such as mediating the conjugative transfer of plasmids and other mobile DNA elements to bacterial recipient cells or delivering protein or DNA substrates to eukaryotic cells. Another kind of T4S system-related process is DNA release or uptake, whereby DNA substrates are exchanged with the extracellular milieu (8, 16).
T4S-related machineries are used by several plant and human pathogens for the purpose of delivering virulence effectors to eukaryotic cell targets. Such pathogens include extracellular organisms such as A. tumefaciens, which is the causative agent of crown gall disease in plants, Bordetella pertussis, which is the agent responsible for whooping cough in children, and Helicobacter pylori, which is responsible for gastric ulcers and stomach cancer (3, 6, 13, 34). In addition, there are intracellular bacterial pathogens using T4S systems for their virulence, such as Brucella suis, the causative agent of brucellosis, and L. pneumoniae, the causative agent of Legionnaires' disease (5, 30).
T4S systems are generally composed of 12 protein components forming a macromolecular assembly inserted into the bacterial cell envelope. These proteins are named VirB1 to VirB11 and VirD4, based on the widely used nomenclature of the model system, the A. tumefaciens VirB/D4 T4S system (16). Three putative ATPases are key components of the T4S system: VirD4, VirB11, and VirB4. VirB4 proteins are the largest and most evolutionarily conserved proteins in T4S systems (15). VirB4 proteins are suggested to be located in the inner membrane, either directly and/or indirectly through their interactions with other components of the T4S system (14, 18, 31). An important feature of VirB4 proteins is the presence of Walker A and Walker B motifs characteristic of ATPases (29). But until very recently no nucleoside triphosphatase (NTPase) activity had been demonstrated for any VirB4 homologue. However, the VirB4 homologue of plasmid R388, the protein TrwK, has now been shown to possess an ATPase activity (2). Very little structural information is available for the VirB4 subunit family. Recently, a bioinformatics model based on the structural similarities between the Agrobacterium VirB4 C terminus and TrwB (VirD4 homolog) proposed that the VirB4 C terminus forms a discrete domain that assembles as a homohexameric ring (26), much like VirB11 and VirD4 (19, 33).
Here, we report a comprehensive biochemical study of the TraB protein, the VirB4 homologue from the pKM101 conjugation machinery. Our results suggest that TraB exists under two forms: a dimeric membrane form and a primarily hexameric soluble form. Both bind DNA nonspecifically and also ATP; however, only the hexameric form hydrolyzes ATP. TraB has a clear modular structure composed of two large contiguous domains that split the protein roughly in two. While only the C-terminal domain was known to contain a nucleotide-binding site, we establish that both domains can bind and hydrolyze ATP. Also, both cooperate to bind DNA. Our study also reveals a complex interplay between the two domains. Our results provide crucial but singular insights that suggest previously unsuspected properties of a family of proteins that play critical functions in T4S systems.
MATERIALS AND METHODS
Cloning of TraB and TraB domains.
The full-length traB gene (traBFL; amino acids [aa] 1 to 866), along with the region encoding the N-terminal domain (traBNT; aa 1 to 442) and the C-terminal domain (traBCT; aa 448 to 848) were PCR-amplified from the pKM101 plasmid and cloned into the pET151/D-TOPO vector (Invitrogen) following standard TOPO cloning protocols. Consequently, all three constructs allow the expression of N-terminally His6-tagged recombinant proteins, named TraBFL, TraBNT, and TraBCT. After DNA sequencing to check that the sequences did not contain any mutations, the four plasmids were transformed by heat shock in chemically competent Escherichia coli BL21 Star (DE3) cells (Invitrogen) for large-scale production of the recombinant proteins.
The Walker A mutants were generated by PCR amplification of the above plasmids using primers carrying the appropriate mutation. To mutate the first nucleotide-binding site (NBD1) of TraB, we used NT-RA1 (5′-TTTTTCAAGCTGGATGGCGCAACACATGACTGCGCATCAGATCGG-3′) and NT-RA2 (5′-TCTGATGCGCAGTCATGTGTTGCGCCATCCAGCTTGAAAAAAGCC-3′). This mutates residue Arg53 into Ala (mutated residues are in boldface). To mutate the second nucleotide-binding site (NBD2), we used CT-KA1 (5′-GGTATGTCGGGGGAAGGTGCGACCACGCTGCTTAACTTCCTGCTGGC-3′) and CT-KA2 (5′-GAAGTTAAGCAGCGTGGTCGCACCTTCCCCCGACATACCCGTTATTAACGC-3′). This mutates residue Lys504 into Ala (mutated residues are in boldface). After PCR, amplified plasmids were subjected to DpnI digestion in order to remove the original wild-type plasmid. After purification using an Extract-II kit (Nalgene), plasmids were transformed by heat shock in chemically competent BL21 Star (DE3) cells (Invitrogen). The presence of the mutation was verified by DNA sequencing of the isolated plasmids.
Production and purification of recombinant proteins.
E. coli strain BL21 Star (DE3) (Invitrogen) containing one of the recombinant pET151 plasmids (for TraBFL, TraBNT, or TraBCT) was grown at 37°C in Terrific Broth (Merck) supplemented with 100 μg/ml ampicillin (Sigma-Aldrich) until the culture reached an A600 of 1.2. Cultures were then shifted to 16°C for 1 h before isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM, and growth was then continued for 15 h at 16°C. Cells were harvested by centrifugation, resuspended in 20 mM Tris-HCl (pH 7.5), and stored at −20°C.
All purification steps were carried out at 4°C. TraBCT is soluble while TraBFL and TraBNT partition between soluble forms in the cytoplasm and membrane forms in the inner membrane. TraBCT and the soluble forms of TraBFL and TraBNT were purified from cytoplasmic extracts as follows. The cells were defrosted and resuspended in a buffer (3 ml per g of cell paste) containing 20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 1 mM β-mercaptoethanol (βME), and one tablet of EDTA-free protease inhibitor cocktail (Roche). After cells were broken by two passages through an EmulsiFlex-C5 homogenizer and DNA was fragmented by sonication, the lysate was clarified by centrifugation at 18,000 rpm for 45 min in a Sorvall SS-34 rotor. The clarified lysate was loaded onto a HisTrap HP (high-performance) 5-ml column (GE Healthcare) equilibrated in buffer Asol (20 mM Tris-HCl [pH 7.5], 300 mM NaCl, 1 mM βME; sol indicates buffer used to purify the soluble forms of TraB proteins) plus 4% of buffer Bsol (20 mM Tris-HCl [pH 7.5], 300 mM NaCl, 1 mM βME, 500 mM imidazole). The column was then washed with 100 ml of buffer Asol plus 8% buffer Bsol. Finally the proteins still bound to the column were eluted in a gradient from 8% to 100% of buffer Bsol in 100 ml. Eluted fractions containing the protein of interest were pooled and concentrated in less than 4 ml before being loaded onto a HiPrep 16/60 Sephacryl S-300 HR column (Amersham) equilibrated in the gel filtration (GF) buffer GFsol containing 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, and 1 mM βME or in a buffer with acetate, termed GFacetate-sol and containing 50 mM HEPES-NaOH (pH 7.0), 75 mM potassium acetate, 2 mM magnesium acetate, 10% (wt/vol) glycerol, and 0.1 mM EDTA (see also the Results and Discussion section for the naming of the gel filtration buffers). The protein of interest eluted as a single peak. Fractions under this peak were pooled.
TraBFL and TraBNT were also purified from the membranes. When proteins were purified from membrane extracts, the following protocol was applied. The cells were defrosted and resuspended in a buffer (3 ml per g of cell paste) containing 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1 mM βME, and one tablet of EDTA-free protease inhibitor cocktail (Roche). After cells were broken by two passages through an EmulsiFlex-C5 homogenizer and DNA was fragmented by sonication, unbroken cells were removed by centrifugation at 14,000 rpm for 10 min in a Sorvall SS-34 rotor. Total membranes were pelleted by ultracentrifugation (45 min at 100,000 × g at 4°C) and resuspended in buffer EB (20 mM Tris-HCl [pH 7.5], 50 mM NaCl, 1 mM βME, 1% [vol/vol] Triton X-100) supplemented with one tablet of EDTA-free protease inhibitor cocktail (Roche). Membrane-embedded proteins were extracted during 1 h at 4°C. The membrane extract was further clarified by ultracentrifugation (30 min at 100,000 × g at 4°C). Triton X-100 was used only for extraction; then it was replaced by the hydrogenated Triton X-100 [Triton X-100(H); Calbiochem] that does not absorb UV light. We further used a concentration of 0.01% Triton X-100(H) (0.16 mM) because this is below the critical micelle concentration (CMC) of the detergent (0.2 to 0.9 mM), thus avoiding the formation of detergent micelles. The cleared extract was loaded onto a HisTrap HP 5-ml column (GE Healthcare) equilibrated in buffer Amb [20 mM Tris-HCl (pH 7.5(, 300 mM NaCl, 1 mM βME, 0.01% Triton X-100(H); mb indicates the buffer used to purify the membrane forms of TraB proteins] plus 4% of buffer Bmb [20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 1 mM βME, 0.01% Triton X-100(H), 500 mM imidazole]. The column was then washed with 100 ml of buffer Amb plus 6% buffer Bmb. Finally, the proteins still bound to the column were eluted in a gradient from 6% to 100% of buffer Bmb in 100 ml. Eluted fractions containing either TraBFL or TraBNT were pooled and concentrated in less than 4 ml before being loaded onto a HiPrep 16/60 Sephacryl S-300 HR column (Amersham) equilibrated in either buffer GFmb containing 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1 mM βME, and 0.01% Triton X-100(H) or buffer GFacetate-mb containing 50 mM HEPES-NaOH (pH 7.0), 75 mM potassium acetate, 2 mM magnesium acetate, 10% (wt/vol) glycerol, 0.1 mM EDTA, and 0.01% Triton X-100(H) (see also the Results and Discussion section for the naming of the gel filtration buffers). The proteins TraBFL and TraBNT both eluted as a single peak. Fractions under this peak were pooled.
The apparent molecular mass of proteins eluted from the gel filtration column was deduced from a calibration carried out with low- and high-molecular-weight calibration kits (Amersham Biosciences). In addition to dynamic light scattering (DLS; described below), the molecular weights of the various multimeric forms were assessed using 3 to 12% or 4 to 16% blue native (BN)-PAGE (Invitrogen). Determination of protein concentration was carried out by using either the theoretical absorption coefficients (mg/ml·cm) at 280 nm as obtained with the program ProtParam at the EXPASY server (available on the World Wide Web at www.expasy.ch/tools) or a Bio-Rad protein assay reagent (Bio-Rad).
ATPase assays.
Two sets of assays were carried out to assess the ATPase activity of all purified proteins. For the first set of assays, we used the proteins in GFsol for the soluble forms or those in GFmb for the membrane forms. The ATPase assays were carried out using an Innova Bioscience kit, which required that the assay be performed in a final buffer containing 50 mM Tris-HCl (pH 7.4), 25 mM NaCl, 2.5 mM MgCl2, and 0.5 mM ATP (the enzyme concentration was 70 μM [in monomer equivalent]).
For the second set of ATPase assays, we used the proteins in GFacetate-sol for the soluble forms and those in GFacetate-mb for the membrane forms. ATPase activity was monitored using a coupled-enzyme assay (24). A total of 44 μl of TraBFL (3.3 μM), TraBNT (7.5 μM), and TraBCT (10.3 μM) in GFacetate-sol (for the soluble forms of the three proteins) or in GFacetate-mb (for the membrane forms of TraBFL and TraBNT) was incubated in 400 μl of ATP assay buffer, consisting of 50 mM PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)]-NaOH (pH 6.45), 75 mM potassium acetate, 5% (wt/vol) glycerol, 10 mM magnesium acetate, 1 mM potassium chloride, 1 mM dithiothreitol, 0.1 mM EDTA, 0.5 mM phosphoenolpyruvate, 0.25 mM NADH, 60 μg/ml pyruvate kinase, 60 μg/ml lactate dehydrogenase, and 0.0625 to 10 mM ATP. The reaction mixtures were preincubated at 37°C for 2 min, after which the ATPase assay was started by the addition of various concentrations of ATP. Activity was measured by the decrease in NADH absorbance at 340 nm for 15 min at 37°C in a UV-visible spectrophotometer (CARY 3; Varian) and the slopes (absorbance/min) were calculated by the program Kinetics-CARY. All the data sets were fitted to a Michaelis-Menten equation:
 |
(1) |
where Vi is the initial velocity or specific ATPase activity (mol of ATP hydrolyzed per min per mol of enzyme [monomer equivalent]), Vmax is the maximum velocity of the enzyme at substrate saturation, S represents the concentration of ATP (mM), and Km is the substrate concentration at which the initial velocity reaches half of its maximum value (Vmax/2) and represents the Michaelis constant. To better evaluate the enzymatic parameters of the various TraB domains, we used a Lineweaver-Burk plot and fitted the data sets to a linear regression:
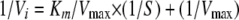 |
(2) |
Note that adding 0.01% Triton to the soluble form of TraBFL did not affect its ATPase activity; thus, 0.01% Triton does not affect ATPase activity of TraB proteins.
DLS.
Dynamic light-scattering experiments were performed with a DynaPro-801 (Protein Solutions) at room temperature. All samples were filtered prior to the measurements (Millex syringe filters; 0.22-μm pore size; Millipore Corp.). The hydrodynamic radius was deduced from translational diffusion coefficients using the Stokes-Einstein equation. Diffusion coefficients were inferred from the analysis of the decay of the scattered intensity autocorrelation function. All calculations were performed using the software provided by the manufacturer (Dynamics, version 5.25.44).
DNA mobility assay.
Proteins (in GFsol or GFmb) at various concentrations and DNA (pKM101 plasmid or PCR product) were mixed and incubated for 10 min at room temperature. A small volume of loading buffer (0.005% bromophenol blue-20% [wt/vol] sucrose) was added to the sample before it was loaded on a native agarose gel prepared using 0.3% or 0.4% agarose dissolved in SYBR Safe DNA gel stain (Invitrogen) in 0.5× TBE (Tris-Borate-EDTA) buffer to visualize the free DNA and the protein-DNA complex bands. Gels were electrophoresed at 100 V for 90 min to 2 h at room temperature in 0.5× TBE buffer. Free DNA and DNA-protein complex bands were visualized under a UV lamp.
Limited trypsin proteolysis.
Protein and DNA (pKM101 plasmid) were preincubated for 10 min on ice in buffer GFmb or GFsol prior to the addition of trypsin (0.3 μg/ml). The reactions were allowed to proceed on ice for 60 min. At various time points (0, 15, 30, 45, and 60 min), an aliquot was removed from the reaction mixture, and NuPAGE lithium dodecyl sulfate (LDS; 4×) sample buffer (Invitrogen) was added. Samples were boiled for 10 min and then loaded on a Nu-PAGE (4 to 12%) gel (Invitrogen) and electrophoresed for 35 min at 200 V in NuPAGE morpholinepropanesulfonic acid (MOPS)-SDS running buffer (Invitrogen). After electrophoresis the gel was stained with SimplyBlue SafeStain (Invitrogen).
Fluorescence measurements.
The fluorescent ATP nucleotide analogue 3′(2′)-O-(2,4,6-trinitrophenyl)-ATP (TNP-ATP) was purchased from Molecular Probes. Fluorescence measurements were performed on a Hitachi F2500 Fluorescence Spectrophotometer, and all the data were processed using the software FL Solutions F-2500. The excitation wavelength was set at 410 nm, and the emission wavelength was scanned in the 470- and 650-nm range. TNP-ATP binding was calculated from the fluorescence maxima determined graphically. The temperature of the sample was maintained at 20°C by circulating thermostatically controlled water through the cuvette holder. For determination of the dissociation constant of TNP-ATP (KdTNP-ATP), 1 μM (monomer concentration) protein solutions (in GFsol or GFmb) were titrated with TNP-ATP. The Kd values for MgATP (KdATP) were determined by displacement of protein-bound TNP-ATP. Protein solutions (1 μM) were incubated for 20 s with either 30 μM (for TraBFL) or 15 μM (for TraBNT and TraBCT) TNP-ATP. MgATP aliquots were then added from 0.1 M stock solution incrementally, and fluorescence was measured after incubation for 20 s. All spectra were corrected for buffer fluorescence and for dilution (never exceeding 5% of the original volume). Titration curve fitting was accomplished using ProFit for Mac OS X, version 6.1.4 (Quantum Soft) with the following quadratic equation in the case of increasing TNP-ATP fluorescence (21):
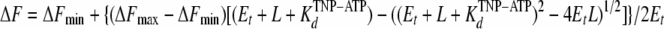 |
(3) |
where ΔF represents the relative fluorescence intensity, ΔFmin is the relative fluorescence intensity at the start of the titration, ΔFmax is the relative fluorescence intensity at a saturating concentration of TNP-ATP (L), Et is the total concentration of protein (monomer equivalent), and KdTNP-ATP is the apparent dissociation constant of the protein-TNP-ATP complex.
In the case of displacement of bound TNP-ATP by ATP, the following quadratic equation was used (21):
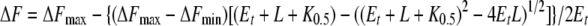 |
(4) |
where ΔFmax is the relative fluorescence intensity at the start of the titration, and ΔFmin is the relative fluorescence intensity at saturating concentration of MgATP, K0.5 represents here the amount of MgATP necessary to displace half the amount of bound TNP-ATP, and L represents the MgATP concentration. We then used the K0.5 value obtained from the displacement experiments and the following equation to calculate the apparent dissociation constant of the protein-MgATP complex (KdATP):
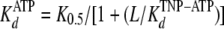 |
(5) |
where L represents the TNP-ATP concentration at the start of the titration.
RESULTS AND DISCUSSION
Identification and production of TraB domains.
TraB, like other VirB4 proteins, contains a nucleoside triphosphate (NTP)-binding site encompassing the Walker A and Walker B motifs located in the second half of the protein sequence (Fig. 1A, NBD). This C-terminal region (residues 448 to 848) is also the most conserved among VirB4 proteins and, as already suggested by Middleton et al. (26) might be similar in structure to the soluble domain of TrwB, a VirD4 homolog (19; see also Fig. S1 in the supplemental material). Therefore, we decided to clone and express individually the N- and C-terminal domains of TraB, together with the full-length protein (Fig. 1 and Materials and Methods). The three proteins were expressed in E. coli as N-terminally His6-tagged recombinant proteins. The C-terminal domain of TraB (TraBCT) was purified from the soluble fraction (see Materials and Methods). However, because a distinct single transmembrane domain (TM) in the N-terminal region of TraB is predicted (http://www.sbc.su.se/∼miklos/DAS/), the full-length TraB (TraBFL) and the N-terminal domain of TraB (TraBNT) were initially purified from the membrane fraction. However, we noticed that both TraBFL and TraBNT partitioned equally in the soluble and membrane fractions, and thus soluble forms of both TraBFL and TraBNT were also purified (see Materials and Methods). TraBCT was never found in the membrane fraction. All proteins were purified to homogeneity using the same two-step purification strategy (Fig. 1B and Materials and Methods).
FIG. 1.

Isolation of TraB domains. (A) Schematic representation of the domain structure of TraB. N, N terminus; C, C terminus; TM, putative transmembrane domain; NBD, NTP binding domain; TraBFL, residues 1 to 866; TraBNT, residues 1 to 442; TraBCT, residues 448 to 848. (B) SDS-NuPAGE 4 to 12% showing the purified proteins after gel filtration. Lane 1, TraBFL (102 kDa); lane 2, TraBNT (55 kDa); and lane 3, TraBCT (48.8 kDa). Molecular mass markers are indicated on the left side of the gel (kDa).
Oligomerization state of TraBFL, TraBNT, and TraBCT.
In SDS-PAGE the proteins migrate at their expected molecular masses: 102 kDa for TraBFL, 55 kDa for TraBNT, and 48.8 kDa for TraBCT (Fig. 1B). However, gel filtration analysis indicated that the proteins are oligomeric. Oligomerization was tested under two buffer conditions established at the final gel filtration stage of the purification. These conditions together with the cell compartment from which the proteins originate (cytosol or membrane) are referred to in Materials and Methods and thereafter as GFsol, GFacetate-sol, GFmb, and GFacetate-mb, where sol and mb refer to the soluble cytosolic and membrane forms of the proteins, respectively, and acetate indicates that the buffer contains acetate. The membrane form of TraBFL in GFmb is primarily dimeric, as assessed by BN-PAGE and gel filtration (see Fig. S2A in the supplemental material) although higher-order oligomers (tetramers and hexamers) are also observed using BN-PAGE. The fractions containing the dimers of membrane-extracted TraBFL were selected. These dimers are stable over time and were used for subsequent studies of the membrane form of TraBFL. TraBNT purified from the membrane fraction is overwhelmingly dimeric, in GFmb, although large aggregates eluting in the void volume are observed, indicating a tendency of the protein to aggregate (see Fig. S2B). The oligomerization state of TraBFL or TraBNT in GFacetate-mb was not investigated.
TraBCT and the soluble forms of TraBFL and TraBNT were all dimeric in GFsol (see results for TraBCT in Fig. S2C in the supplemental material). All three were, however, hexameric in GFacetate-sol, as assessed using gel filtration (data not shown) and DLS (Table 1). Thus, we conclude that the soluble forms of TraBCT, TraBFL, and TraBNT transition between at least two forms, dimeric and hexameric, which are in a dynamic equilibrium where one form is favored over the other depending on solution conditions, notably the presence or absence of acetate ions. Whether the membrane forms of TraBFL and TraBNT also transition between two oligomeric states remains to be investigated. It is, however, interesting that these proteins do not display ATPase activities even in the presence of acetate ions (see “ATP hydrolysis of TraB proteins” below), suggesting that dimeric TraBFL and TraBNT might not be able to transition to higher oligomeric forms. This could be caused by the presence of detergent in the GFacetate-mb buffer, but it was shown that detergent does not disrupt ATPase activities of the soluble TraB protein forms (see “ATP hydrolysis of TraB proteins” below).
TABLE 1.
DLS data of the soluble TraB-derived proteins in GFacetate
| Protein (NBD1 and/or NBD2)a | MMCalc (kDa)b | Hexamer MMCalc (kDa)c | MMDLS (kDa)d | Radius (nm)e | Polydispersity (nm [%]) |
|---|---|---|---|---|---|
| TraBFL (WT/WT) | 102.6 | 615.6 | 593.0 | 9.1 | 3.5 (38.1) |
| TraBFL (WT/KA) | 102.6 | 615.6 | 603.4 | 9.2 | 3.1 (33.6) |
| TraBFL (RA/WT) | 102.6 | 615.6 | 666.9 | 9.6 | 3.8 (39.6) |
| TraBFL (RA/KA) | 102.6 | 615.6 | |||
| TraBNT (WT) | 54.6 | 327.6 | 365.6 | 7.4 | 2.9 (39.0) |
| TraBNT (RA) | 54.6 | 327.6 | |||
| TraBCT (WT) | 48.8 | 292.8 | 348.5 | 7.3 | 2.3 (32.4) |
| TraBCT (KA) | 48.8 | 292.8 | 353.3 | 6.9 | 2.3 (24.2) |
The presence or absence of nucleotide-binding domain mutation(s) is shown in parentheses for the full-length TraB and fragments. WT, wild type; RA, R53A mutation in NBD1; KA, K504A mutation NBD2.
MMCalc, theoretical molecular mass calculated from amino acid sequence.
The hexamer molecular mass was calculated as MMCalc × 6.
Molecular mass experimentally determined by DLS.
Estimation of the hydrodynamic radius of the particle.
These results contrast with those obtained on the full-length TrwK protein, the VirB4 homologue in the R388 plasmid system, for which no membrane form has been identified. TrwK was found to be soluble and primarily monomeric although a minor hexameric form accounting for 5% of the total species was ascribed as the ATP-hydrolyzing form (2). Also, no TrwK fragment has been studied.
Because TrwK has already been the subject of extensive studies, we decided to focus subsequent work on TraB and TraB fragments primarily (but not exclusively; see ATPase assay results below), specifically, on the dimeric, membrane forms of TraBFL and TraBNT (purified in GFmb) and on the dimeric soluble form of TraBCT (purified in GFsol).
Interaction of TraB-derived constructs with DNA.
Because TraB is a component of a T4S system transporting DNA, we investigated whether TraB could bind DNA. First, we showed that TraBFL (membrane form in GFmb) is protected from proteolysis degradation after being incubated in the presence of the pKM101 DNA (Fig. 2A). This result suggested that TraB was able to interact with the pKM101 DNA. In a second approach, we demonstrated that TraBFL could shift the migration of the pKM101 DNA on a native agarose gel in a concentration-dependent manner (results not shown). However, this interaction is not DNA sequence specific since TraBFL could also shift the migration of a random PCR product (Fig. 2B). We conclude that TraBFL can bind DNA nonspecifically. Then, we asked which of the two TraB domains is responsible for DNA binding. Interestingly, both the N-terminal (TraBNT; membrane form in GFmb) and C-terminal (TraBCT; dimeric soluble form in GFsol) domains of TraB were able to bind either the pKM101 DNA (results not shown) or a random PCR product (Fig. 2B). In this assay, an unrelated protein (bovine serum albumin [BSA]) was unable to bind the PCR product (Fig. 2B). In conclusion, we demonstrated that TraB can bind DNA in a non-sequence-specific manner and that both the N-terminal and C-terminal domains are involved in binding. Interestingly, although the end shift positions are similar for all three proteins, saturation occurs more rapidly for TraBCT than for either TraBFL or TraBNT. This indicates an influence of TraBNT on DNA binding of TraBCT.
FIG. 2.

Interaction between TraB and DNA. (A) Limited proteolysis of TraBFL in the absence or presence of DNA. TraBFL purified in GFmb (23.1 μg) and the pKM101 plasmid (1.94 μg) were preincubated together in buffer GFmb (see Materials and Methods) for 10 min on ice prior to the addition of trypsin (0.3 μg/ml). The reactions were allowed to proceed on ice for the indicated periods of time. The reactions were carried out in the presence (+) or absence (−) of the pKM101 plasmid. The arrow indicates TraBFL. Molecular mass markers are indicated on the left side of the gel. (B) Interaction of TraB and TraB fragments with DNA. Native 0.3% agarose gel shift electrophoresis was used to analyze the association of the different TraB domains with a random PCR product DNA: TraBFL purified in GFmb (a), TraBNT purified in GFmb (b), TraBCT purified in GFsol (c), and BSA (d). The amounts of protein (nmol) are indicated above each gel, and the amount of DNA was 350 ng in each reaction mixture. The arrows on each gel indicate the unshifted position of the PCR product DNA.
TraB-derived proteins can bind ATP.
Until now, only one conserved nucleotide-binding site has been identified in TraB, and the site is located in its C-terminal domain (Fig. 1). This prompted us to investigate whether TraBCT could bind ATP. We used fluorescence spectroscopy to monitor the interaction of the fluorescent ATP analogue TNP-ATP with the protein. Upon protein binding, the fluorescence emission intensity of TNP-ATP increases considerably, with the absolute magnitude dependent on the specific protein environment; thus, it has been widely used to characterize ATP binding by a number of proteins (9, 25). As expected, we showed that TraBCT (soluble dimeric form in GFsol) was able to bind TNP-ATP, as demonstrated by the remarkable enhancement of the emission intensity of TNP-ATP in the presence of TraBCT (Fig. 3A) together with a blue shift (from 552 to 546 nm) of the wavelength of maximal emission. Moreover, bound TNP-ATP could be dislodged from its binding site, at least partially, by excess ATP (Fig. 3B), indicating that the reported TNP-ATP-enhanced emission intensity is caused by binding to a bona fide ATP-binding site (21, 32).
FIG. 3.

ATP-binding to TraB and TraB fragments. (A) TraB and TraB fragments bind TNP-ATP. The fluorescent ATP analogue TNP-ATP displayed enhanced fluorescence upon binding by TraBFL purified in GFmb (a), TraBNT purified in GFmb (b), and TraBCT purified in GFsol (c). Fluorescence spectra 1 to 4 were taken from the following samples: spectrum 1, protein (5 μM); spectrum 2, TNP-ATP (23 μM); spectrum 3, protein (5 μM) plus TNP-ATP (23 μM); spectrum 4, protein (5 μM) plus TNP-ATP (23 μM) in the presence of ATP (0.5 mM). Fluorescence intensities are expressed in arbitrary units. These curves are the averages of three independent experiments. (d) All spectra 3 were subtracted from spectrum 2 to obtain the fluorescence contribution of the TNP-ATP bound to the protein. FL, TraBFL; NT, TraBNT; CT, TraBCT; NT+CT, fluorescence expected from the sum of TraBNT and TraBCT. The arrow shows the decrease in fluorescence intensity between FL and NT+CT. (B) Measurement of binding equilibrium parameters. (a) Fluorescence-monitored titration of TNP-ATP binding to TraB proteins. Successive aliquots of TNP-ATP stock solutions were added to protein samples (1 μM) of TraBFL (○), TraBNT (*), or TraBCT (▪) in buffer GFmb for TraBFL and TraBNT and in buffer GFsol for TraBCT, and the fluorescence intensity (excitation, 410 nm; emission, 545 nm) was recorded after each addition. Each plotted value represents the difference in fluorescence intensity between the TraB protein titration and the blank titration. The lines represent the best fit to the data generated using equation 3 in Materials and Methods: solid line, TraBFL; dashed line, TraBNT; dotted line, TraBCT. See Materials and Methods for further details. (b) Displacement of bound TNP-ATP by ATP in TraB proteins. Successive aliquots of MgATP stock solutions were added to a solution containing TraB proteins (1 μM) TraBFL (○), TraBNT (*), or TraBCT (▪) and TNP-ATP (15 μM for TraBNT and TraBCT; 30 μM for TraBFL) in buffer GFmb for TraBFL and TraBNT and in buffer GFsol for TraBCT. The fluorescence intensity (excitation, 410 nm; emission, 545 nm) was recorded after each addition. Each plotted value represents the difference in fluorescence intensity between the TraB protein titration and the blank titration. The lines represent the best fit to the data generated using equation 4 in Materials and Methods and are as identified above. See Materials and Methods for further details.
TraBCT represents just one half of the full-length TraB (TraBFL). So, we wanted to check if the rest of the protein could influence the ATP-binding property of the C-terminal domain of TraB. We used fluorescence spectroscopy to monitor the interaction of TNP-ATP with TraBFL and TraBNT, both purified from the membrane fraction (i.e., in GFmb). Similarly to TraBCT, TraBFL was able to bind TNP-ATP (Fig. 3A), albeit with a slightly different absolute magnitude that could indicate a different protein environment around the bound TNP-ATP. Furthermore, TNP-ATP binding was efficiently competed by adding an excess of ATP (Fig. 3A). Surprisingly, TraBNT also binds TNP-ATP, a binding reaction that was also competed by an excess of ATP (Fig. 3A). The latter result was unexpected, given that the only reported nucleotide-binding site in VirB4 proteins is located in the C-terminal half (Fig. 1).
The three TraB proteins (the soluble dimeric TraBCT in GFsol and the membrane TraBFL and TraBNT in GFmb) were next titrated with increasing amounts of TNP-ATP, and the results were fitted to equation 3 of Materials and Methods (Fig. 3B), from which apparent dissociation constants for TNP-ATP (KdTNP-ATP) could be derived. Equation 3 is derived from a single-site binding model, and the protein concentration is in monomer equivalent. The values for KdTNP-ATP were 6.91 ± 0.79 μM for TraBFL, 1.22 ± 0.30 μM for TraBNT, and 0.51 ± 0.08 μM for TraBCT (Table 2). Thus, as observed for DNA binding, we observe a functional interaction between the two domains which, when they come together in the full-length protein, results in enhanced binding affinity for TNP-ATP.
TABLE 2.
Summary of kinetic parameters of TraB, TraB fragments, and TraB mutants and comparison with TrwKa
| Protein (NBD1 and/or NBD2)b | Kinetics with Michaelis-Menten equation |
Kinetics with Lineweaver-Burk plotc |
|||
|---|---|---|---|---|---|
| Vmax (nmol min−1 mg−1) | Vmax (mol min−1 mol−1) | Km (mM) | Vmax (nmol min−1 mg−1) | Km (mM) | |
| TraBFL (WT/WT) | 126.2 ± 4.6 | 12.9 ± 0.4 | 0.59 ± 0.07 | 120.5 | 0.52 |
| TraBFL (WT/KA) | 25.0 ± 0.8 | 2.6 ± 0.1 | 0.60 ± 0.09 | 23.9 | 0.54 |
| TraBFL (RA/WT) | 30.1 ± 0.5 | 3.1 ± 0.1 | 0.31 ± 0.02 | 30.03 | 0.30 |
| TraBNT (WT) | 121.8 ± 4.3 | 6.8 ± 0.2 | 0.48 ± 0.07 | 130 | 0.49 |
| TraBCT (WT) | 45.1 ± 1.5 | 2.2 ± 0.1 | 0.62 ± 0.07 | 42.2 | 0.52 |
| TrwK | 48.4 | 0.7 | NA | NA | |
Values were determined at 37°C.
See Table 1, footnote a, for an explanation of the protein designations.
NA, not available.
Bioinformatics analysis of TraBNT and identification of its ATP-binding site.
In order to explain the ATP-binding property of TraBNT, we analyzed the protein sequence of TraB. Using BLAST or ScanProsite (http://www.expasy.ch/tools/blast/; http://www.expasy.ch/tools/scanprosite/), we could not detect a second binding site in the N-terminal domain of TraB. Thus, we sought to compare TraB with known ATP-binding proteins with two nucleotide-binding sites. ATP-binding cassette (ABC) transporters have two nucleotide-binding sites (28), but both of them are highly conserved. Conversely, the translocon protein SecA has two nucleotide-binding sites (see Fig. S3A in the supplemental material), a high-affinity site in the N-terminal domain (NBD1) and a low-affinity site in the C-terminal domain (NBD2) (27); the low-affinity site is far less conserved in its amino acids sequence than the high-affinity site. We thus performed a sequence alignment between the N-terminal domain of TraB and the C-terminal domain of SecA (see Fig. S3B). Surprisingly, the two domains exhibit a high degree of sequence identity (28%) (see Fig. S3B). Moreover, two regions in the TraB N-terminal domain align with the sequences known to form the Walker A and B motifs in the C-terminal domain of SecA (see Fig. S3B). Notably, key residues (GRXTXD) in the Walker A motif of SecA are conserved in TraBNT (20). We concluded that, like the SecA C-terminal domain, the N-terminal domain of TraB also contains a poorly conserved nucleotide-binding site. We then asked if this new feature in the N-terminal domain of TraB was conserved among the VirB4 protein family. As shown in Fig. 4, the nucleotide-binding site in the N-terminal domain of TraB seems to be poorly conserved among VirB4 proteins. Indeed, just 3 out of the 38 VirB4 homologues aligned, including TraB, present a motif similar to the SecA low-affinity NTP-binding site. Thus, we have identified a new ATP-binding site in the N-terminal domain of TraB that defines a new class of VirB4 proteins (Fig. 4; see also and Fig. S3C and S4 in the supplemental material).
FIG. 4.

Sequence alignment (Clustal W2) around the TraB NBD1 Walker A motif of VirB4 homologs. The shaded area shows the only three VirB4 homologs that contain the NBD2 SecA-like motif.
ATP hydrolysis of TraB proteins.
Recently, the first demonstration of an ATPase activity for a VirB4 homolog was described for the protein TrwK from the plasmid R388 T4S system conjugation system (2). ATPase assays of TraB and TraB fragments were carried out under two sets of conditions (see Materials and Methods), i.e., in a buffer similar to GFsol/GFmb and in a buffer similar to GFacetate-sol/GFacetate-mb.
In GFacetate-sol, TraBCT was found to hydrolyze ATP. Under such conditions, it is also hexameric (see above). TraBCT kinetic parameters for ATP hydrolysis were determined by analyzing the effect of ATP concentration on ATPase activity rates (Fig. 5). The data were fitted to the Michaelis-Menten equation 1, from which we calculated the kinetic parameters. TraBCT shows a Vmax of 2.20 ± 0.07 mol of ATP hydrolyzed per min per mol of enzyme and a Km of 0.62 ± 0.07 mM (Table 2) (note that in all calculations, the monomer-equivalent concentration was used). Subsequently, in order to assess the relevance of this ATPase activity, we constructed a Walker A derivative of TraBCT carrying a point mutation K503A (see Material and Methods), that has been shown to be of crucial importance for ATPase activity in many proteins (22, 23). The resulting mutant protein, TraBCT-KA, was purified in a similar way as the wild-type protein. DLS measurements indicated that this derivative forms hexamers of 353 kDa in GFacetate-sol (Table 1) and is devoid of ATPase activity when tested in GFacetate-sol.
FIG. 5.

Kinetic analysis of TraB and TraB domain ATPase activity. ATP hydrolysis activity was monitored by a coupled-enzyme assay (see Materials and Methods). (A) ATP hydrolysis rates are represented as a function of ATP concentration (x axis) and fitted using the Michaelis-Menten equation (represented by lines). The different data sets represent TraBFL (○ and —), TraBNT (* and - -), TraBCT (▪ and • • •), TraBFLRAWT (▾ and • - - •), and TraBFLWTKA (⋄ and - • • • -). All proteins were in GFacetate. The initial velocity rate (y axis) is expressed as mol of ATP hydrolyzed per min per mol of enzyme. (B) Lineweaver-Burk plot of the data presented in panel A. All the data sets have been fitted by linear regression.
TraBFL purified from the soluble fraction in GFacetate-sol (where it is hexameric [see above]) was also able to hydrolyze ATP. However, the kinetic parameters were significantly different from those obtained for TraBCT (Fig. 5 and Table 2). TraBFL shows a much higher Vmax than TraBCT, with a value of 12.90 ± 0.47 mol of ATP hydrolyzed per min per mol of enzyme for TraBFL, but a similar Km of 0.59 ± 0.07 mM (Table 2).
Surprisingly, soluble hexameric TraBNT in GFacetate-sol was also able to hydrolyze ATP (Fig. 5). TraBNT shows a Vmax of 6.80 ± 0.24 mol of ATP hydrolyzed per min per mol of enzyme, which is higher than that of TraBCT but lower than that of TraBFL, and a similar Km of 0.48 ± 0.07 mM compared to the other two proteins (Table 2). As mentioned previously, we identified a poorly conserved ATP-binding site in the N-terminal domain of TraB (NBD1). In order to check the specific involvement of this NBD1, we designed a point mutation replacing Arg53 by Ala (R53A) in TraBFL and in TraBNT (see Materials and Methods). The wild-type protein with the R53A mutation, TraBFLRAWT, shows a marked decrease in its ATPase activity (3.10 ± 0.24 mol of ATP hydrolyzed per min per mol of enzyme) compared to TraBFL (Table 2) while TraBNTRA no longer displays any measurable ATPase activity. These experiments confirm the specific involvement of the TraB NBD1 in the ATPase activity.
Interestingly, when assayed in GFsol, wild-type TraBCT, which is dimeric under such conditions (see above), is unable to hydrolyze ATP. This would indicate that only hexameric forms of TraB proteins exhibit ATPase activities. Consistent with this observation, the membrane forms of TraBFL and TraBNT are unable to hydrolyze ATP in GFmb where the proteins are dimeric. The oligomeric state of these two proteins in GFacetate-mb was not investigated. However, neither TraBFL or TraBNT extracted from the membrane exhibits ATPase activity in GFacetate-mb: given that detergent [0.01% Triton X-100(H)] does not affect the ATPase activity of hexameric TraBCT (the kinetic parameters are the same when TraBCT is tested for ATP hydrolysis in the presence or absence of the detergent [data not shown]), lack of ATPase activity in membrane-extracted TraBFL or TraBNT is not caused by detergent but, instead, may reflect the fact that these proteins remain dimeric even in the presence of acetate ions. This would suggest that TraB in the membrane might be constitutively dimeric.
The effect of DNA binding on ATPase activity of TraB proteins was tested, and no difference in ATPase kinetic parameters was observed. Thus, DNA binding does not affect ATPase activity.
Conclusions.
In the study presented here, we have unraveled a number of features of VirB4 proteins that were not apparent in TrwK, the other VirB4 protein for which extensive biochemical characterization has been carried out (2). TraB partitions between the membrane and the cytoplasm, apparently adopting two distinct oligomeric states, each of which is characterized by a different ATP-hydrolyzing property. The form extracted from the membrane is dimeric and unable to hydrolyze ATP although it is able to bind DNA and nucleotide. Interestingly, in a buffer with NaCl and no acetate ions, the soluble form of TraB is also dimeric and also unable to hydrolyze ATP while in the absence of NaCl but in acetate, it is hexameric and able to hydrolyze ATP. Thus, we conclude that TraB can hydrolyze ATP only when in a hexameric state. TrwK differs from TraB in being exclusively cytoplasmic and primarily monomeric in low-salt buffer conditions. Higher oligomeric forms of TrwK were observed in acetate buffer, but only up to 5% of protein was assessed to be hexameric, a form that was hypothesized to be the ATP-hydrolyzing form. The results with TraB are therefore less ambiguous in ascribing the ATP-hydrolyzing form of the protein to the hexameric form. Interestingly, TraB is able to transition from the soluble dimeric inactive form to a soluble active hexameric form, demonstrating that the protein is highly dynamic.
VirB4 proteins are likely to be located at the base of the core machinery, embedded or associated with the inner membrane (16, 17). It is intriguing that TraB purifies as an inactive dimer when extracted from the membrane. It could be that in the physiological conditions of the cell at the cytoplasmic face of the inner membrane (characterized by a low NaCl concentration), the protein transitions to a hexamer even in the context of the membrane. However, two other mechanisms could lead to an active TraB in the membrane: (i) a change in oligomerization from dimer to hexamer caused by the association with other T4S system components; (ii) the binding of an ATPase-activating protein, leaving TraB dimeric but inserting in trans activating residues missing at the ATP-binding interface in the dimer. Regarding the former, it is possible that the association of VirB4 with the core complex might lead to a change in the oligomerization state. Possibly a VirB4-VirB11 interaction also could affect the oligomerization state of VirB4 as VirB11 is constitutively hexameric. Regarding the latter, there are examples of proteins that contain motifs known to be able to complete in trans ATPase active sites; one such motif is the well-characterized “Arg finger” of GAP proteins (1). VirB3 is known to associate with VirB4 as both are sometimes found in tandem in the same protein (4, 10). VirB3 has been proposed to locate VirB4 to the membrane for the VirB4 variants that are not endowed with transmembrane segments. However, VirB3 could also influence VirB4 oligomerization and activity in such a way that, when embedded in the entire machinery, a dimeric VirB4 could be rendered active.
Fragments of TraB recapitulate the behavior of the full-length protein in terms of oligomerization and ATP-hydrolyzing activity. Indeed, the soluble form of the C-terminal domain and both the soluble and membrane-bound forms of the N-terminal domain of TraB are dimeric and unable to hydrolyze ATP under NaCl-containing (GFsol or GFmb) buffer conditions. Moreover, the soluble forms of these domains are able to transition from dimer to hexamer depending on the salt versus acetate conditions. However, remarkably—and demonstrating a property unique to TraB among VirB4 proteins—both domains are able to bind and hydrolyze ATP. This led us to identify a novel cryptic but functional ATP-binding site in the N-terminal domain of the protein.
Another intriguing result is the ability of TraB and both of its domains to bind DNA. A previous study of DNA-binding by VirB4-like proteins reported that these proteins were unable to bind DNA (29). Until now, only the coupling protein VirD4 has been shown to bind DNA (32). The functional significance of DNA binding by TraB is difficult to square with the reported result that VirB4 does not contact DNA directly during substrate transfer by the A. tumefaciens VirB/D4 T4S system (7). This could indicate that there are some differences between T4S systems and that, in the case of conjugation by the pKM101-encoded T4S system, DNA makes contact with TraB, perhaps relaying substrate transfer from the VirD4-homolog in this system. However, in the absence of a mutation in TraB that abrogates DNA binding, it would be difficult to speculate further as to whether DNA binding by this protein plays a fundamental role in conjugation.
Supplementary Material
Acknowledgments
This work was funded by Welcome Trust grant 082227 to G.W.
Footnotes
Published ahead of print on 19 February 2010.
Supplemental material for this article may be found at http://jb.asm.org/.
The authors have paid a fee to allow immediate free access to this article.
REFERENCES
- 1.Ahmadian, M. R., P. Stege, K. Scheffzek, and A. Wittinghofer. 1997. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat. Struct. Biol. 4:686-689. [DOI] [PubMed] [Google Scholar]
- 2.Arechaga, I., A. Pena, S. Zunzunegui, M. Del Carmen Fernandez-Alonso, G. Rivas, and F. de la Cruz. 2008. ATPase activity and oligomeric state of TrwK, the VirB4 homologue of plasmid R388 type IV secretion system. J. Bacteriol. 190:5472-5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baron, C., D. O'Callaghan, and E. Lanka. 2002. Bacterial secrets of secretion: EuroConference on the biology of type IV secretion processes. Mol. Microbiol. 43:1359-1365. [DOI] [PubMed] [Google Scholar]
- 4.Batchelor, R. A., B. M. Pearson, L. M. Friis, P. Guerry, and J. M. Wells. 2004. Nucleotide sequences and comparison of two large conjugative plasmids from different Campylobacter species. Microbiology 150:3507-3517. [DOI] [PubMed] [Google Scholar]
- 5.Boschiroli, M. L., S. Ouahrani-Bettache, V. Foulongne, S. Michaux-Charachon, G. Bourg, A. Allardet-Servent, C. Cazevieille, J. P. Lavigne, J. P. Liautard, M. Ramuz, and D. O'Callaghan. 2002. Type IV secretion and Brucella virulence. Vet. Microbiol. 90:341-348. [DOI] [PubMed] [Google Scholar]
- 6.Burns, D. L. 2003. Type IV transporters of pathogenic bacteria. Curr. Opin. Microbiol. 6:29-34. [DOI] [PubMed] [Google Scholar]
- 7.Cascales, E., and P. J. Christie. 2004. Definition of a bacterial type IV secretion pathway for a DNA substrate. Science 304:1170-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cascales, E., and P. J. Christie. 2003. The versatile bacterial type IV secretion systems. Nat. Rev. Microbiol. 1:137-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho, Y. K., S. E. Rios, J. J. Kim, and H. M. Miziorko. 2001. Investigation of invariant serine/threonine residues in mevalonate kinase. Tests of the functional significance of a proposed substrate binding motif and a site implicated in human inherited disease. J. Biol. Chem. 276:12573-12578. [DOI] [PubMed] [Google Scholar]
- 10.Christie, P. J., K. Atmakuri, V. Krishnamoorthy, S. Jakubowski, and E. Cascales. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59:451-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christie, P. J., and E. Cascales. 2005. Structural and dynamic properties of bacterial type IV secretion systems (review). Mol. Membr. Biol. 22:51-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christie, P. J., and J. P. Vogel. 2000. Bacterial type IV secretion: conjugation systems adapted to deliver effector molecules to host cells. Trends Microbiol. 8:354-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Covacci, A., J. L. Telford, G. Del Giudice, J. Parsonnet, and R. Rappuoli. 1999. Helicobacter pylori virulence and genetic geography. Science 284:1328-1333. [DOI] [PubMed] [Google Scholar]
- 14.Dang, T. A., and P. J. Christie. 1997. The VirB4 ATPase of Agrobacterium tumefaciens is a cytoplasmic membrane protein exposed at the periplasmic surface. J. Bacteriol. 179:453-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernandez-Lopez, R., M. P. Garcillan-Barcia, C. Revilla, M. Lazaro, L. Vielva, and F. de la Cruz. 2006. Dynamics of the IncW genetic backbone imply general trends in conjugative plasmid evolution. FEMS Microbiol. Rev. 30:942-966. [DOI] [PubMed] [Google Scholar]
- 16.Fronzes, R., P. J. Christie, and G. Waksman. 2009. The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7:703-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fronzes, R., E. Schafer, L. Wang, H. R. Saibil, E. V. Orlova, and G. Waksman. 2009. Structure of a type IV secretion system core complex. Science 323:266-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilmour, M. W., T. D. Lawley, M. M. Rooker, P. J. Newnham, and D. E. Taylor. 2001. Cellular location and temperature-dependent assembly of IncHI1 plasmid R27-encoded TrhC-associated conjugative transfer protein complexes. Mol. Microbiol. 42:705-715. [DOI] [PubMed] [Google Scholar]
- 19.Gomis-Ruth, F. X., G. Moncalian, R. Perez-Luque, A. Gonzalez, E. Cabezon, F. de la Cruz, and M. Coll. 2001. The bacterial conjugation protein TrwB resembles ring helicases and F1-ATPase. Nature 409:637-641. [DOI] [PubMed] [Google Scholar]
- 20.Guina, T., D. Helfet-Hilliker, V. Ramamurthy, and D. Oliver. 1998. Sequence and phylogenetic analysis of the Borrelia burgdorferi secA gene. Biochim. Biophys. Acta 1371:24-30. [DOI] [PubMed] [Google Scholar]
- 21.Hormaeche, I., I. Alkorta, F. Moro, J. M. Valpuesta, F. M. Goni, and F. De La Cruz. 2002. Purification and properties of TrwB, a hexameric, ATP-binding integral membrane protein essential for R388 plasmid conjugation. J. Biol. Chem. 277:46456-46462. [DOI] [PubMed] [Google Scholar]
- 22.Hwang, Y., and M. Feiss. 1996. Mutations affecting the high affinity ATPase center of gpA, the large subunit of bacteriophage lambda terminase, inactivate the endonuclease activity of terminase. J. Mol. Biol. 261:524-535. [DOI] [PubMed] [Google Scholar]
- 23.Karst, J. C., A. E. Foucher, T. L. Campbell, A. M. Di Guilmi, D. Stroebel, C. S. Mangat, E. D. Brown, and J. M. Jault. 2009. The ATPase activity of an “essential” Bacillus subtilis enzyme, YdiB, is required for its cellular function and is modulated by oligomerization. Microbiology 155:944-956. [DOI] [PubMed] [Google Scholar]
- 24.Kreuzer, K. N., and C. V. Jongeneel. 1983. Escherichia coli phage T4 topoisomerase. Methods Enzymol. 100:144-160. [DOI] [PubMed] [Google Scholar]
- 25.Lu, N. T., and P. L. Pedersen. 2000. Cystic fibrosis transmembrane conductance regulator: the purified NBF1+R protein interacts with the purified NBF2 domain to form a stable NBF1+R/NBF2 complex while inducing a conformational change transmitted to the C-terminal region. Arch. Biochem. Biophys. 375:7-20. [DOI] [PubMed] [Google Scholar]
- 26.Middleton, R., K. Sjolander, N. Krishnamurthy, J. Foley, and P. Zambryski. 2005. Predicted hexameric structure of the Agrobacterium VirB4 C terminus suggests VirB4 acts as a docking site during type IV secretion. Proc. Natl. Acad. Sci. U. S. A. 102:1685-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell, C., and D. Oliver. 1993. Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol. Microbiol. 10:483-497. [DOI] [PubMed] [Google Scholar]
- 28.Moody, J. E., and P. J. Thomas. 2005. Nucleotide binding domain interactions during the mechanochemical reaction cycle of ATP-binding cassette transporters. J. Bioenerg. Biomembr. 37:475-479. [DOI] [PubMed] [Google Scholar]
- 29.Rabel, C., A. M. Grahn, R. Lurz, and E. Lanka. 2003. The VirB4 family of proposed traffic nucleoside triphosphatases: common motifs in plasmid RP4 TrbE are essential for conjugation and phage adsorption. J. Bacteriol. 185:1045-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy, C. R., and L. G. Tilney. 2002. The road less traveled: transport of Legionella to the endoplasmic reticulum. J. Cell Biol. 158:415-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schandel, K. A., M. M. Muller, and R. E. Webster. 1992. Localization of TraC, a protein involved in assembly of the F conjugative pilus. J. Bacteriol. 174:3800-3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schroder, G., and E. Lanka. 2003. TraG-like proteins of type IV secretion systems: functional dissection of the multiple activities of TraG (RP4) and TrwB (R388). J. Bacteriol. 185:4371-4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeo, H. J., S. N. Savvides, A. B. Herr, E. Lanka, and G. Waksman. 2000. Crystal structure of the hexameric traffic ATPase of the Helicobacter pylori type IV secretion system. Mol. Cell 6:1461-1472. [DOI] [PubMed] [Google Scholar]
- 34.Zupan, J., T. R. Muth, O. Draper, and P. Zambryski. 2000. The transfer of DNA from Agrobacterium tumefaciens into plants: a feast of fundamental insights. Plant J. 23:11-28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.