

Abstract
CCR7 is a chemokine receptor expressed on the surfaces of T cells, B cells, and mature dendritic cells that controls cell migration in response to the cognate ligands CCL19 and CCL21. CCR7 is critical for the generation of an adaptive T cell response. However, the roles of CCR7 in the host defense against pulmonary infection and innate immunity are not well understood. We investigated the role of CCR7 in the host defense against acute pulmonary infection with Pseudomonas aeruginosa. We intranasally infected C57BL/6 mice with P. aeruginosa and characterized the expression of CCR7 ligands and the surface expression of CCR7 on pulmonary leukocytes. In response to infection, expression of CCL19 and expression of CCL21 were oppositely regulated, and myeloid dendritic cells upregulated CCR7 expression. We further examined the effects of CCR7 deficiency on the inflammatory response to P. aeruginosa infection. We infected Ccr7−/− and wild-type mice with P. aeruginosa and characterized the accumulation of pulmonary leukocytes, production of proinflammatory mediators, neutrophil activation, and bacterial clearance. CCR7 deficiency led to an accumulation of myeloid dendritic cells and T cells in the lung in response to infection. CCR7 deficiency resulted in higher expression of CD80 and CD86 on dendritic cells; increased production of interleukin-12/23p40 (IL-12/23p40), gamma interferon (IFN-γ), and IL-1α; increased neutrophil respiratory burst; and, ultimately, increased clearance of acute P. aeruginosa infection. In conclusion, our results suggest that CCR7 deficiency results in a heightened proinflammatory environment in response to acute pulmonary P. aeruginosa infection and contributes to more efficient clearance.
The lung samples significant volumes of air and possesses a large surface area for efficient gas exchange; however, these characteristics present significant challenges for the maintenance of a sterile environment (25). Routine exposures to airborne pathogens are typically cleared by resident components of the innate immune system (25). More severe exposures require the recruitment of phagocytic cells and the induction of adaptive immune mechanisms to prevent local and systemic colonization (4).
Pseudomonas aeruginosa is a Gram-negative bacterium and a common etiological agent in the development of nosocomial infections and chronic respiratory infections in patients with cystic fibrosis (34). Clearance of P. aeruginosa from the lung requires the efforts of diverse cells, including recruited and resident leukocytes in addition to pulmonary epithelial cells (34). Previous studies have identified a variety of cells that act directly or indirectly through the production of soluble mediators, including antimicrobial peptides and cytokines, to effectively clear pulmonary P. aeruginosa infection. Cells implicated in the pulmonary response to P. aeruginosa infection include epithelial cells (11), neutrophils (38), alveolar macrophages (14, 21), T cells (28), natural killer T (NKT) cells (28), NK cells (42), and dendritic cells (DCs) (30). Of these, DCs remain the least investigated cell type.
Leukocyte accumulation at the site of infection is a critical and highly regulated component of immune system function. Chemokines are a family of chemoattractant cytokines, which guide migration through binding of G protein-coupled receptors on the surface of leukocytes (33). Ligation of chemokine receptors leads to leukocyte activation and migration according to varying chemokine gradients. The diverse array of chemokines and chemokine receptors act to coordinate the complex cellular interactions required to respond to various pathogens (23). The chemokine receptor CCR7 orchestrates a complex series of molecular interactions, including chemotaxis and cell activation of T cells, B cells, and mature DCs, through cognate ligands CCL19 and CCL21 (9). CCL19 and CCL21 are expressed primarily in secondary lymphoid organs, but they are also expressed in other tissues, such as the gastrointestinal tract, kidney, and lung (27, 43). Previous studies demonstrated that disruption of cellular interactions coordinated through CCR7 impaired T cell function and increased susceptibility to viral infections (10, 12, 19, 29). These results were attributed to disruption of lymph node architecture and impaired T cell and DC migration to the lymph node, which prevented efficient antigen presentation to naïve T cells. Recent reports demonstrated additional functions of CCR7 signaling beyond migration, including enhanced dendritic cell function through increased phagocytosis, cytokine production, and upregulation of costimulatory molecules (24, 46).
Previous studies have examined the role of CCR7 or its ligands in pulmonary infections (15, 17, 19), but how CCR7 shapes the immune response to pathogen exposures in the lung remains unclear. In this study, we investigated whether cellular migration and activation due to CCR7 signaling were required for the host defense against acute pulmonary P. aeruginosa infection. Specifically, we investigated whether the ligands for CCR7 are modulated in response P. aeruginosa infection and how disruption of the CCR7 receptor-ligand axis transformed the inflammatory response to infection. We found that CCR7 deficiency led to T cell and DC accumulation, increased production of inflammatory cytokines, enhanced neutrophil activation, and, ultimately, more effective clearance of P. aeruginosa.
MATERIALS AND METHODS
Mice and genotyping.
Ccr7−/− mice (Ccr7tm1R for strain, C56BL/6 background) were previously generated by Förster et al. (10). Ccr7−/− mice and C57BL/6 (B6) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Ccr7−/− mice were crossed with B6 mice, and heterozygotes were bred to produce Ccr7−/− mice and wild-type (WT) littermates. Progeny from heterozygote crosses were identified using PCR (wild-type forward primer, 5′-CGT GTC CTC GCC GTT-3′; wild-type reverse primer, 5′-CCC CGG GCA ATG TCC TGA-3′; Ccr7−/− forward primer, 5′-GTC TCC GCC TCC ATG CTT CAC C-3′; Ccr7−/− reverse primer, 5′-CTC TCG TGG GATCAT TGT TTT TCT-3′). Amplification of PCR products for both genotyping reactions was performed by denaturation at 95°C for 2 min and then 35 cycles of amplification at 95°C for 45 s, 55°C for 30 s, and 72°C for 60 s, followed by extension at 72°C for 5 min. Mice were between 8 and 12 weeks of age at the time of experimental use. Mice were housed under pathogen-free conditions in accordance with institutional guidelines, and all experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Cincinnati College of Medicine.
P. aeruginosa inoculation.
Stationary-phase P. aeruginosa, strain PAO1 (16), was used in all experiments. Isolated single colonies grown on tryptic soy agar plates were inoculated in Luria broth, followed by overnight incubation of shaken cultures at 37°C. The cultures were diluted with Luria broth to an optical density (OD) at 600 nm of 1.5, and P. aeruginosa was harvested by centrifugation (6,800 × g, 3 min), followed by three 1-ml washes and an appropriate final dilution with sterile Hanks balanced salt solution (HBSS) without Ca2+ or Mg2+. Mice were anesthetized with isoflurane and infected intranasally with a 20-μl suspension.
Bacterial enumeration.
At 16 hours after infection with 1 × 107 CFU of PAO1, Ccr7−/− mice and wild-type (WT) littermate controls were euthanized with an intraperitoneal (i.p.) injection of sodium pentobarbital (Nembutal) (150 to 200 mg/kg; Henry Schein) followed by exsanguination via severing of the posterior abdominal aorta, and the left lobes of lungs were harvested and homogenized in 1 ml of phosphate-buffered saline (PBS) with a Pyrex Tenbroeck tissue grinder. Serial dilutions of lung homogenates diluted in PBS were plated onto tryptic soy agar plates and incubated overnight at 37°C, and individual colony counts in terms of log10 CFU were determined.
Quantitative RT-PCR for CCR7 ligands.
At 16 hours postinfection, mice were euthanized as described above, and the right lobes of the lungs were harvested and snap frozen in liquid nitrogen. Frozen tissue was homogenized using a Tissumizer (Tekmar Co.), and total RNA was isolated with Trizol reagent (Invitrogen). DNase treatment to remove residual DNA was performed using the Turbo DNA-free kit (Ambion). RNA absorbance at 260 nm was read using a NanoDrop (Thermo Fisher), and the A260 value was multiplied by 40 μg/ml and the appropriate dilution factor to arrive at the RNA concentration. Reverse transcription of total RNA was performed using the high-capacity cDNA Archive kit (Applied Biosystems). Probes and primers for CCL19 and RPL32 were obtained from Applied Biosystems. Quantitative reverse transcription-PCR (RT-PCR) was performed using TaqMan universal PCR master mix (Applied Biosystems) for CCL19 and RPL32 on an Applied Biosystems 7300 real-time PCR system. The following primers were used for CCL21: forward primer, 5′-CCA GCC CCA GGG AAA CAA AG-3′; reverse primer, 5′-AGG CGG GCT ACT GGG CTA TCC-3′. Quantitative RT-PCR for CCL21 was performed using Power SYBR green PCR master mix (Applied Biosystems) on an Applied Biosystems 7300 real-time PCR System. Expression of mRNA was quantified by the ΔΔCT method using RPL32 as the endogenous control.
ELISA.
For quantitation of CCL19 and CCL21 proteins, mice were euthanized at 16 h postinfection and lung tissue from the right lobes was harvested as described above. Frozen tissue was homogenized using a Tissumizer (Tekmar Co.), and total protein was isolated using T-Per reagent (Pierce Biotechnology). Sandwich enzyme-linked immunosorbent assays (ELISAs) were performed using DuoSet kits (R&D Systems) specific to CCL19 and CCL21 according to the manufacturer's protocol. Absorbance was read at 450 nm with background subtraction at 590 nm on a Tecan SpectraFluor Plus. Seven-point standard curves were generated using a four-parameter logistic curve fit (SigmaPlot 10).
Interleukin-12/23p40 (IL-12/23p40) in bronchoalveolar lavage (BAL) fluid (BALF) was measured by sandwich ELISA using a DuoSet kit (R&D Systems) according to the manufacturer's protocol. P. aeruginosa-infected and uninfected CCR7-deficient mice and wild-type littermates were euthanized, and the lungs were lavaged with two 1-ml aliquots of HBSS without Ca2+ or Mg2+ (pH 7.2, 37°C; Invitrogen). Recovered BAL fluid samples were then centrifuged (400 × g, 10 min, 4°C), and the supernatant from the first lavage was aliquoted and stored at −80°C. Absorbance was read at 450 nm with background subtraction at 590 nm on a Tecan SpectraFluor Plus. Standard curves were generated using a four-parameter logistic curve fit (SigmaPlot 10).
Characterization of lung leukocyte populations by flow cytometry.
Mice were euthanized as described above, and the lungs were voided of blood by perfusion through the right ventricle with 6 ml of PBS containing 0.6 mM EDTA. The lungs were not lavaged prior to this, thereby ensuring that leukocytes in the alveolar space were also recovered in the lung digest for flow cytometric analysis. Lungs were withdrawn aseptically from the chest cavity, washed with PBS, diced into pieces with a total volume of ≤300 μl, and digested in 5 ml of RPMI 1640 with 2.05 mM l-glutamine (HyClone) containing 175 U/ml collagenase I-A, 0.2 U/ml pancreatic elastase, 35 U/ml hyaluronidase, 20 kU/ml DNase I (Sigma-Aldrich), 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin (MP Biomedicals) for 1 h at 37°C on an orbital shaker (60 rpm). The digested lungs were sheared through 19- and 21-gauge needles and filtered through 40-μm cell strainers (BD Biosciences) to obtain a single-cell suspension. Residual red blood cells (RBCs) were lysed with RBC lysis solution (Qiagen), and cells were centrifuged in 30% Percoll (Sigma-Aldrich). Cells were washed, counted with a hemacytometer, resuspended in fluorescence-activated cell sorter (FACS) buffer (2 mM EDTA, 0.5% bovine serum albumin [BSA], and 0.05% sodium azide in PBS) to a concentration of 107 cells/ml, and incubated with purified mouse Fc Block (Becton Dickinson) at 4°C for 10 min at a concentration of 5 μg/107 cells. The following antibodies (Abs) were used for cell surface staining of leukocytes: phycoerythrin (PE)-conjugated rat anti-mouse CD8a (clone 53-6.7; eBioscience), peridinin chlorophyll protein (PerCP)-Cy5.5-conjugated hamster anti-mouse CD3e (clone 145-2C11; eBioscience), fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD4 (clone GK1.5; eBioscience), allophycocyanin (APC)-conjugated rat anti-mouse CCR7 (clone 4B12; eBioscience), FITC-conjugated Armenian hamster anti-mouse CD11c (clone NA418; eBioscience), PerCP-Cy5.5-conjugated rat anti-mouse CD11b (clone M1/70; eBioscience), FITC-conjugated rat anti-mouse B220 (clone RA3-6B2; eBioscience), PE-conjugated rat anti-mouse PDCA-1 (clone eBio129c; eBioscience), allophycocyanin-conjugated Armenian hamster anti-mouse CD80 (clone: 16-10A1; eBioscience), allophycocyanin-conjugated rat anti-mouse CD86 (clone GL1; eBioscience), FITC-conjugated rat anti-mouse F4/80 (clone BM8; eBioscience), PerCP-Cy5.5-conjugated rat anti-mouse GR-1 (clone RB6-8C5; eBioscience), allophycocyanin-conjugated rat anti-mouse CD11b (clone M1/70; eBioscience), and FITC-conjugated rat anti-mouse NKp46 (clone 29A1.4; eBioscience). All staining was performed for 30 min on ice in the dark except for staining with allophycocyanin-conjugated rat anti-mouse CCR7 and the corresponding isotype control, which were stained for 30 min in the dark at 37°C. After staining, cells were washed once with 2 ml of FACS buffer and resuspended in 500 μl of FACS buffer. Additionally, the appropriate isotype control Abs were used to set photomultiplier tube voltages. Flow cytometry was performed using a BD FACSCalibur system, and the data were analyzed using BD CellQuest Pro software or FlowJo v7.2.5 (Tree Star).
The following strategies were employed to identify lung leukocyte populations from the whole-lung digest. Lymphocytes were identified by forward-scatter (FSC) and side-scatter (SSC) properties and further divided into B cells (B220+), CD4+ T cells (CD3+ CD4+), CD8 T cells (CD3+ CD8+), NKT cells (CD3+ NK1.1+), and NK cells (NKp46+ CD3−). Dendritic cells and alveolar macrophages were identified according to the strategy described by Vermaelen and Pauwels (39). Briefly, CD11chigh cells were identified and then divided into myeloid dendritic cells (mDCs) (low autofluorescence in FL2 and FSC/SSC properties) and alveolar macrophages (high autofluorescence in FL2 and FSC/SSC properties). Myeloid dendritic cells were further divided into intraepithelial (IE) mDCs and CD11bhigh dendritic cells (CD11b mDCs) on the basis of high CD11b expression (2, 7, 36). Plasmacytoid dendritic cells were identified on the basis of coexpression of B220 and PDCA-1. Neutrophils were identified as GR-1high, CD11bhigh, F4/80-negative cells.
Cytokine profiling of BALF.
Mice were euthanized, and the lungs were lavaged with two 1-ml aliquots of HBSS without Ca2+ or Mg2+ (pH 7.2, 37°C; Invitrogen). Recovered bronchoalveolar lavage (BAL) fluid (BALF) samples were centrifuged (400 × g, 10 min, 4°C), and the supernatant from the first lavage was aliquoted and stored at −80°C. Cytokine (gamma interferon [IFN-γ], IL-1α, monocyte chemoattractant protein 1 [MCP-1], tumor necrosis factor alpha [TNF-α], and granulocyte-macrophage colony-stimulating factor [GM-CSF]) concentrations in the BAL fluid supernatants were determined by enzyme-linked immunosorbent assay (ELISA) using Milliplex Multiplex kits (Millipore, Billerica, MA) according to the manufacturer's protocol. In a 96-well multiscreen filter plate, 25 μl of sample in duplicate was incubated with 25 μl of antibody-coated beads overnight at 4°C on a plate shaker. The plates were then washed two times on a vacuum apparatus, and 25 μl of secondary antibody was added and incubated at room temperature for 1 h while shaking. Finally, 25 μl of streptavidin-RPE was added directly to the secondary antibody and incubated for 30 min at room temperature with shaking. Plates were then washed two more times, and 150 μl of sheath fluid was added. Plates were shaken for 5 min and then read using Luminex technology on the Bio-Plex (Bio-Rad, Hercules, CA). Concentrations were calculated from standard curves using recombinant proteins and expressed in pg/ml. The cytokine analysis was conducted by the Cytokine and Mediator Measurement Core laboratory at Cincinnati Children's Hospital Medical Center.
Flow cytometric analysis of oxidative burst.
Mice were euthanized, and the lungs were lavaged with two 1-ml aliquots of HBSS without Ca2+ or Mg2+ (pH 7.2, 37°C; Invitrogen). The BAL fluid samples recovered from the first 1-ml lavage were centrifuged (400 × g, 10 min, 4°C) to recover the supernatant. The cell pellet from the first lavage was resuspended with the fluid from the second lavage and centrifuged to pellet the cells. The cells were washed with HBSS and counted using a hemacytometer before being resuspended at 106 cells/ml in HBSS-1 μM dihydrorhodamine-123 (DHR-123). Cells were incubated at 37°C for 15 min and placed on ice. Flow cytometry analysis was performed immediately, as described above. DHR-123 is preferentially oxidized by H2O2, yielding rhodamine-123, which is detected in FL1 on a BD FACSCalibur (41). Alveolar macrophages and neutrophils were identified according to autofluorescence in FL2/FL3 (neutrophils, low autofluorescence; alveolar macrophage, high autofluorescence) and forward- and side-scatter properties. Peripheral blood was harvested by cardiac puncture using a 3-ml syringe and a 21-gauge needle. Red blood cells were lysed with RBC lysis solution (Qiagen). The cells were washed with HBSS and counted using a hemacytometer before being resuspended in HBSS-1 μM dihydrorhodmine-123 as described above. Following the 15-min incubation at 37°C, phorbol myristate acetate (PMA) was added to a final concentration of 10 μM, and cells were incubated at 37°C for an additional 10 minutes. Cells were placed on ice, and flow cytometry analysis was performed as described above.
Statistical analyses.
Data are presented as means ± standard errors of the means (SEM). Statistically significant differences between groups were identified by Student's t test, and a P value of <0.05 was considered statistically significant.
RESULTS
CCL19 and CCL21 are oppositely regulated following P. aeruginosa infection in WT and Ccr7−/− mice.
We examined the pulmonary expression and regulation of CCR7 ligands to investigate the functional significance of the CCR7 receptor-ligand axis. We identified constitutive pulmonary expression of both CCR7 ligands at the RNA and protein levels. CCL19 transcripts in the lung were increased approximately 6-fold at 16 h postinfection (Fig. 1A). CCL19 protein was increased in accordance with mRNA levels (202.5 ng/ml versus 361.4 ng/ml) (Fig. 1B). In contrast, CCL21 transcripts and protein were reduced in the lung after P. aeruginosa infection (66%; 2,356.7 ng/ml versus 1,653.4 ng/ml, respectively) (Fig. 1A and B).
FIG. 1.
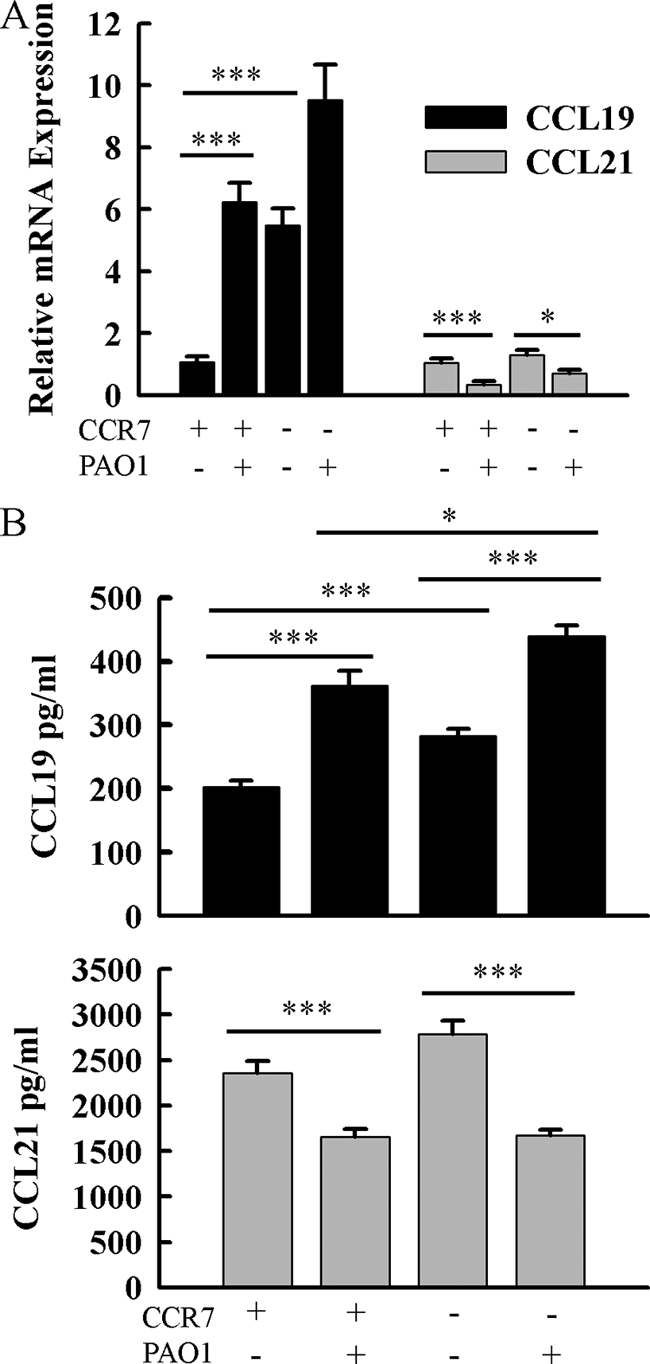
Characterization of CCL19 and CCL21 expression in the lung in response to P. aeruginosa infection and CCR7 deficiency. (A) Total RNA was isolated from lung homogenates of infected and uninfected Ccr7−/− and WT mice. CCL19 and CCL21 transcripts were assayed by quantitative RT-PCR and normalized to Rpl32. (B) CCL19 and CCL21 proteins were assayed by sandwich ELISA. Total protein was isolated from lung homogenates of infected and uninfected Ccr7−/− and WT mice. Data are presented as means ± SEM (n = 5 to 12 mice/group). *, **, and ***, P < 0.05, P < 0.01, and P < 0.001, respectively.
CCR7 deficiency resulted in increased basal expression of CCL19 transcripts and protein, but CCL21 expression was not different than that in WT mice (Fig. 1A and B). Following P. aeruginosa infection of Ccr7−/− mice, CCR7 ligands were regulated similarly to those in WT mice. CCL19 transcripts were modestly increased after infection (P = 0.098) (Fig. 1A), and CCL19 protein was significantly increased (282.2 ng/ml versus 438.7 ng/ml) (Fig. 1B). Moreover, CCL21 transcripts and protein levels were also reduced in Ccr7−/− mice following P. aeruginosa infection (Fig. 1).
A greater proportion of mDCs in the lung express CCR7 after P. aeruginosa infection.
CCR7 expression is regulated in response to immune stimulation, and increased receptor expression is known to modulate cellular activation (26, 29). Therefore, we performed an extensive characterization of CCR7 expression on pulmonary leukocytes in uninfected and P. aeruginosa-infected WT mice; these data are summarized in Table 1. CCR7 expression was detectable only on T cells, B cells, and dendritic cells in both infected and uninfected mice. The proportion of myeloid dendritic cells (mDCs) expressing CCR7 increased approximately 8-fold after infection (Table 1). Although CCR7 expression on the surface of CD8+ T cells increased after infection, CCR7 expression on the surface of CD4+ T cells decreased (Table 1). However, the proportion of both T cell subsets expressing CCR7 was unchanged after infection. The majority of B cells expressed CCR7, and infection had no effect on the proportion or amount of CCR7 expressed (Table 1).
TABLE 1.
Flow cytometric analysis of CCR7 expression on the surface of pulmonary leukocytes from uninfected and P. aeruginosa-infected mice
| Cells | % CCR7+a in: |
MFI of CCR7+ subsetb in: |
||
|---|---|---|---|---|
| Uninfected mice | PAO1-infected mice | Uninfected mice | PAO1-infected mice | |
| mDCse | 3.31 ± 0.76 | 23.90 ± 4.23d | 520.08 ± 19.17 | 610.60 ± 75.80 |
| CD11b | 2.99 ± 0.85 | 31.02 ± 5.74d | 388.63 ± 64.71 | 561.62 ± 58.36 |
| IE | 3.45 ± 0.74 | 22.64 ± 4.66d | 557.78 ± 24.77 | 657.20 ± 99.32 |
| Plasmacytoid DCs | 9.38 ± 0.77 | NDc | 29.30 ± 1.33 | ND |
| CD8+ T cells | 80.37 ± 0.96 | 76.80 ± 2.71 | 109.71 ± 2.41 | 161.31 ± 14.51d |
| CD4+ T cells | 82.13 ± 0.76 | 74.08 ± 3.70 | 203.83 ± 5.73 | 185.01 ± 2.81d |
| B cells | 60.40 ± 1.42 | 67.36 ± 3.30 | 29.28 ± 0.12 | 23.75 ± 0.26 |
Values are presented as means ± SEM (n = 4 or 5 mice/group) for the percentage of each leukocyte population that expressed CCR7.
Values are presented as means ± SEM (n = 4 or 5 mice/group) for the mean fluorescence intensity of the CCR7-positive subset of each leukocyte population.
ND, not determined. The recovery of plasmacytoid dendritic cells in the lung digest after infection was not sufficient for reliable statistics.
Significantly different (P < 0.05) from value for the uninfected control group as calculated by Student's t test.
Myeloid dendritic cells are defined by high CD11c expression, low autofluorescence in FL2, and FSC/SSC properties. mDCs are divided into two populations, intraepithelial (IE) mDCs and CD11b mDCs, on the basis of high CD11b expression.
Numbers of lung leukocyte populations from uninfected and P. aeruginosa-infected Ccr7−/− mice.
We hypothesized that the lungs of Ccr7−/− mice would have altered numbers of T cells, B cells, and DCs because subsets of these populations express CCR7 and because CCL19 and CCL21 are constitutively expressed in the lung. Therefore, we performed flow cytometric analysis to identify leukocyte populations in whole-lung digests and calculated absolute numbers using the total number of lung leukocytes recovered. CD4+ and CD8+ T cells accumulated in the lungs as a result of CCR7 deficiency (Fig. 2A). However, CCR7 deficiency had no effect on plasmacytoid DC (data not shown), mDC, or B cell accumulation in uninfected mice (Fig. 2A and B).
FIG. 2.

Enumeration of pulmonary leukocyte populations. Lung cells were harvested from uninfected and infected Ccr7−/− and WT mice. Leukocytes were isolated and stained with the following antibodies: FITC-conjugated B220, PerCP-Cy5.5-conjugated CD3e, PE-conjugated CD8a, FITC-conjugated CD4, FITC-conjugated CD11c, and PerCP-Cy5.5-conjugated CD11b. The numbers of myeloid dendritic cell populations (A) and lymphocyte populations (B) isolated from lung digests were enumerated by multiplying percentages obtained by flow cytometry by absolute counts. Data are presented as means ± SEM (n = 4 to 12 mice/group). *, **, and ***, P < 0.05, P < 0.01, and P < 0.001, respectively.
Furthermore, we hypothesized that CCR7 deficiency would lead to changes in the accumulation of T cells, B cells, and dendritic cells in response to infection. The absolute number of mDCs was increased in response to P. aeruginosa infection in WT mice (Fig. 2B). CCR7 deficiency resulted in a greater increase in mDCs in response to infection (Fig. 2B). Total B cells and T cells were reduced in WT mice after infection. T cell numbers were not reduced in response to infection in Ccr7−/− mice (Fig. 2A). However, CCR7 deficiency did not result in a difference in B cells in P. aeruginosa-infected mice (Fig. 2A).
CCR7 deficiency had no effect on the accumulation of other lung leukocyte populations (i.e., NK cells, alveolar macrophages, NKT cells, and neutrophils) in uninfected or P. aeruginosa-infected mice (data not shown).
Ccr7−/− mDCs express higher levels of costimulatory molecules after P. aeruginosa infection.
A previous study concluded that CCR7 signaling increases the expression of costimulatory molecules on the surface of DCs (24). Therefore, we investigated whether CCR7 deficiency leads to impaired upregulation of costimulatory molecules on mDCs after P. aeruginosa infection. Basal expression of costimulatory molecules on mDCs was intermediate and was not affected by CCR7 deficiency (data not shown). CCR7 deficiency resulted in an increase in the mean fluorescence intensity of the costimulatory molecules CD80 and CD86 on the surface of highly CD80/CD86 positive mDCs (i.e., activated) after P. aeruginosa infection (Fig. 3A). CCR7 deficiency had no effect on the proportion of mDCs expressing high levels of costimulatory molecules in response to P. aeruginosa infection (Fig. 3C). However, CCR7 deficiency led to a greater absolute number of mDCs that express higher levels of costimulatory molecules in response to infection.
FIG. 3.

CD11c+ dendritic cells from Ccr7−/− mice infected with P. aeruginosa express higher levels of costimulatory molecules CD80 and CD86 than those from WT mice. (A) Lungs from Ccr7−/− and WT mice were digested at 16 h after infection with P. aeruginosa. Lung leukocytes were isolated and stained with the following Abs: FITC-conjugated CD11c, PerCP-Cy5.5-conjugated CD11b, and either allophycocyanin-conjugated CD80 or allophycocyanin-conjugated CD86. Myeloid dendritic cells (mDCs) were identified by high CD11c expression, low autofluorescence in FL2, and FSC and SSC properties. CD11b mDCs and IE mDCs were divided on the basis of high CD11b expression. The mean fluorescence intensities (MFI) of cells expressing high levels of CD80 or CD86 from each population are presented as means ± SEM (n = 5 mice/group). * and **, P < 0.05 and P < 0.01, respectively, compared to WT. (B) Representative histograms show expression profiles of costimulatory molecules gated on total mDCs. Heavy black lines represent Ccr7−/−, dotted lines represent WT, and gray shaded histograms represent pooled mDCs from infected Ccr7−/− and WT mice stained with the appropriate APC-conjugated isotype control. (C) The percentage of each population of mDCs expressing high levels of CD80 or CD86 is represented as open circles. Horizontal lines represent the mean for each group.
IL-12/23p40, IFN-γ, and IL-1α are increased in Ccr7−/− mice in response to P. aeruginosa infection.
We hypothesized that greater absolute numbers of T cells and more highly activated mDCs would lead to increased production of soluble inflammatory mediators. Therefore, we measured cytokine and chemokine levels in bronchoalveolar lavage fluid. CCR7 deficiency resulted in a 15-fold increase in IFN-γ in response to infection (Fig. 4A). IL-1α and IL-12/23p40 levels were increased approximately 2-fold due to CCR7 deficiency in response to infection (Fig. 4A). CCR7 deficiency had no effect on the increased production of TNF-α, MCP-1, or GM-CSF in response to infection (Fig. 4B). CCR7 deficiency resulted in a 2-fold increase in basal production of IL-12/23p40 and GM-CSF (Fig. 4). IFN-γ, TNF-α, and MCP-1 were undetectable in uninfected mice of either strain (data not shown). These data suggest that CCR7 deficiency leads to a heightened proinflammatory environment in response to P. aeruginosa infection.
FIG. 4.

Ccr7−/− mice have increased levels of IL-12/23p40, IFN-γ, and IL-1α in the BALF following P. aeruginosa infection compared to WT mice. Bronchoalveolar lavage was performed at 16 h after infection on Ccr7−/− mice and WT mice and on uninfected control mice. IL-12/23p40 in BALF was assayed by sandwich ELISA. IFN-γ, IL-1α, TNF-α, MCP-1, and GM-CSF were assayed by a Luminex bead-based assay. Data are presented as means ± SEM (n = 5 to 10 mice/group) *, **, and ***, P < 0.05, P < 0.01, and P < 0.001, respectively, compared to WT.
Neutrophils recovered from the BALF of Ccr7−/− mice exhibit enhanced respiratory burst activity after infection with P. aeruginosa.
We examined whether CCR7 deficiency leads to increased phagocytic cell activation in response to P. aeruginosa infection by measuring the respiratory burst activity of cells recovered from BALF. CCR7 deficiency led to an increase in neutrophil respiratory burst in response to infection (Fig. 5A). Alveolar macrophage respiratory burst was modestly increased in Ccr7−/− mice in response to infection (P = 0.063) (Fig. 5A). Nonspecific stimulation of neutrophils recovered from peripheral blood of Ccr7−/− and WT mice revealed no differences in respiratory burst, which demonstrated that CCR7 deficiency had no direct effect on the capacity of neutrophils to become activated (Fig. 5B).
FIG. 5.

Neutrophils recovered after P. aeruginosa infection from the BALF of Ccr7−/− mice have increased respiratory burst compared to those from WT mice. (A) Ccr7−/− and WT mice were lavaged at 16 h after infection with P. aeruginosa. Cells recovered from BALF were incubated with dihydrorhodamine-123, and respiratory burst was assayed by measuring conversion of dihydrorhodamine-123 to fluorescent rhodamine-123 by flow cytometry. Neutrophils and macrophages were identified according to FSC and SSC characteristics. Data presented as means ± SEM (n = 6 mice/group). **, P < 0.01 compared to WT. (B) Respiratory burst of peripheral blood neutrophils and macrophages from Ccr7−/− and WT mice in response to PMA stimulation. Data are presented as means ± SEM (n = 4 mice/group). (C) Representative histograms showing intensity of rhodamine-123 staining of cells recovered from the BALF of infected mice. Heavy black lines, Ccr7−/−; dotted lines, WT; shaded gray histograms, pooled cells recovered from BALF of infected Ccr7−/− and WT mice without dihydrorhodamine-123 staining.
CCR7-deficient mice clear pulmonary P. aeruginosa infection more effectively.
We infected Ccr7−/− and WT mice intranasally with 1 × 107 CFU of P. aeruginosa to investigate the functional consequences of CCR7 deficiency in the host response to P. aeruginosa. We examined the bacterial load in the lungs at three time points after infection: 0 h, 4 h, and 16 h. Clearance of P. aeruginosa from the lungs of Ccr7−/− mice was significantly increased compared to that of WT littermates at 16 h postinfection (Fig. 6). However, clearance of P. aeruginosa was not different at 4 h postinfection. P. aeruginosa CFU recovered from the lungs of WT and Ccr7−/− mice immediately after intranasal inoculation indicated no difference in dose. CCR7 deficiency had no effect on the relative cellularity recovered from the BALF or on the kinetics of neutrophil recruitment at 4 or 16 h postinfection (data not shown). These data suggest that CCR7 deficiency results in an enhanced host defense against P. aeruginosa infection.
FIG. 6.
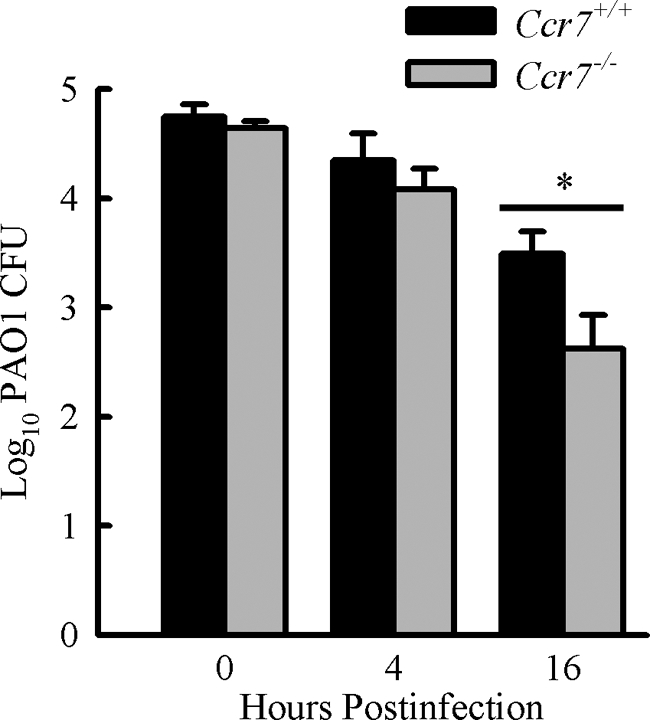
CCR7 deficiency leads to increased pulmonary clearance of acute P. aeruginosa respiratory infection. Ccr7−/− and WT mice were intranasally infected with 1 × 107 CFU of P. aeruginosa, and bacterial CFU in the lungs were assessed at 0, 4, and 16 h postinfection. Data are presented as means ± SEM (0 h, n = 4 mice/group; 4 h, n = 3 to 5 mice/group; 16 h, n = 9 to 12 mice/group). *, P < 0.05.
DISCUSSION
CCR7 is a critical facilitator of the interactions between DCs and T cells, which are necessary for the rapid induction of an effective T cell-mediated adaptive response. CCR7 expression guides cells toward increasing concentrations of CCL19 and CCL21, which are expressed predominantly in secondary lymphoid organs. CCL19 and CCL21 expression has been identified in peripheral organs, such as the lung (27, 43), but their role in host defense is not fully understood. Previous studies employing models of pulmonary infection have shown that CCR7 deficiency leads to impaired host defense (1, 6, 15, 18, 19, 22, 31). However, we demonstrated that CCR7 deficiency results in the accumulation of T cells and activated mDCs in lungs in response to P. aeruginosa infection, which alters the inflammatory environment causing increased production of IL-1α, IL-12/23p40, and IFN-γ, resulting in an enhanced neutrophil antimicrobial capacity and more efficient clearance.
Our results demonstrate that CCL19 expression and CCL21 expression are differently regulated, leading to an increase in CCL19 and a decrease in CCL21 in the lung after P. aeruginosa infection. Earlier studies examining the effects of CCL19 and CCL21 binding to CCR7 identified similarities in affinity, Ca2+ flux, and chemotaxis (32, 35). However, subsequent studies demonstrated that binding of CCL19 leads to receptor desensitization, greater ERK1/2 recruitment, and increased mitogen-activated protein kinase (MAPK) signaling (20, 45). How these differences in signaling translate into functional differences between ligation of CCR7 by CCL19 or CCL21 is not fully understood. Our data suggest that CCR7 deficiency leads to T cell accumulation in the lung and that T cell egress in response to P. aeruginosa infection is CCR7 mediated. These results are in congruence with previous studies showing that memory T cells migrate to the periphery, where they require CCR7 expression to continue to lymph nodes before reentering circulation (3, 5). Our results suggest that P. aeruginosa infection promotes T cell migration out of the lung, and we speculate this is accomplished in part through shifting the balance of CCR7 ligand expression toward CCL19 and away from CCL21. CCL21 expression in the lung has been identified in perivascular lymphatic vessels and peribronchial regions (17). Although CCL19 expression has been identified in the lung, the localization of CCL19 expression has not been determined. However, limiting the expression of CCL19 in the lung to the lymphatic vessels would provide a mechanism by which increasing CCL19 expression relative to CCL21 expression would promote CCR7-mediated migration out of the lung. It is interesting to note that CCL19 expression in the lung is substantially higher in Ccr7−/− mice, which may suggest a homeostatic, albeit futile, response to alleviate the accumulation of T cells in Ccr7−/− mice. It remains to be fully examined whether biasing expression toward CCL19 or CCL21 is a means to dynamically modulate leukocyte migration out of the lung.
A substantial proportion of mDCs expressed CCR7 only after P. aeruginosa infection in our model, and CCR7 deficiency resulted in an accumulation of mDCs only in response to P. aeruginosa infection. This is in agreement with previous reports that conclude that CCR7 and its ligands are required for the migration of activated dendritic cells from the periphery to the lymph node (10, 17, 29). However, other studies have investigated additional roles for CCR7 in dendritic cell function other than migration and concluded that CCR7 signaling is critical for full maturation of licensed dendritic cells (1, 24). These two studies used plt mice, which are a naturally occurring mutant strain that lack CCL19 and expresses CCL21 only at reduced levels in some peripheral organs. Our results clearly indicate that CCR7 signaling is not required for upregulation of costimulatory molecules in response to infection and, furthermore, that mDCs from Ccr7−/− mice actually express higher levels of costimulatory molecules than those from WT mice. This discrepancy is presumably due to the use of plt mice as opposed to Ccr7−/− mice. It is reasonable to suggest that basal signaling of CCR7 on licensed mDCs suppresses full maturation of mDCs and that binding of CCL19 and CCL21 overcomes this suppression. This would provide an explanation for why mDCs expressing CCR7 in a largely ligand-free environment exhibit impaired maturation but maturation of CCR7-deficient mDCs is promoted.
Our data suggest that CCR7 deficiency leads to enhanced clearance of pulmonary P. aeruginosa infection. CCR7 deficiency did not result in increased recruitment of neutrophils in response to P. aeruginosa infection; however, neutrophils recovered from the BALF of Ccr7−/− mice possess greater antimicrobial capacity in response to infection. We were unable to identify expression of CCR7 on the surface of neutrophils from either uninfected or P. aeruginosa-infected mice, and ex vivo stimulation demonstrated that neutrophils from Ccr7−/− and WT mice do not differ in their capacity to become activated. These results suggest that increased neutrophil activation in Ccr7−/− mice is not a cell autonomous effect. We measured levels of regulatory cytokines that may affect neutrophil activation to address this issue and observed increased expression of IL-1α, IFN-γ, and IL-12/23p40 in Ccr7−/− mice upon P. aeruginosa infection. The stimulatory effects of IFN-γ on neutrophil activity are well documented (reviewed in references 8 and 44), and the proinflammatory effects of IL-12, which are primarily due to the stimulation of IFN-γ production, have been examined in detail (reviewed in reference 37). Attributing the increased production of IL-12 and IFN-γ in infected Ccr7−/− mice to specific cell types remains unresolved. However, it is plausible to suggest that mDCs, which are more numerous and more activated in Ccr7−/− mice after infection, are responsible for the increased production of IL-12. Dendritic cells are known to produce large amounts of IL-12 early in the course of infection, which stimulates other cells, mainly T cells, NKT cells, and NK cells, to produce IFN-γ (37). Our previous studies identified NK cells as the predominate source of IFN-γ following acute pulmonary P. aeruginosa infection (42). It would reasonable to expect that this would be the case for Ccr7−/− mice, although the accumulation of T cells in Ccr7−/− mice presents the possibility that activation of this population could be responsible in part for the increased production of IFN-γ.
Our results contrast with those of previous studies that have examined the role of CCR7 in infection. Many studies employed viral pathogens (6, 15, 18, 19, 31), while other studies investigated infections by Listeria monocytogenes (22) or Leishmania donovani (1). Each of these studies indicated that clearance of infection is delayed or impaired in plt or Ccr7−/− mice. The effective clearance of infection in the previous studies requires the generation of a pathogen-specific, adaptive T cell response. P. aeruginosa infection differs in that immunocompetent mice clear infection with impunity within 48 to 72 h, before a specific adaptive T cell response can be generated. Our results suggest that CCR7 deficiency is protective for host defense to infection when the initiation of an adaptive T cell response to a novel pathogen is dispensable, presenting the possibility of therapeutic benefit in delaying the expression of CCR7 on activated DCs in certain clinical situations. A recent study described the ability of 9-cis-retinoid and fenretinide to inhibit upregulation of CCR7 on DCs and described their potential uses as an immunosuppressant (40). Conversely, our results suggest that in certain contexts, blocking CCR7 expression may result in immunostimulation. It would be of interest to explore whether pharmacological intervention to temporarily delay CCR7 expression would enhance the host response to infection without significantly impairing the initiation of an adaptive T cell response.
Another factor that may contribute to the discrepancy between our results and studies that demonstrated an impaired or delayed clearance of infection due to CCR7 deficiency is the nature of P. aeruginosa and how it interacts with the host. Unlike the pathogens employed in the studies mentioned above (1, 6, 15, 18, 19, 22, 31), P. aeruginosa is an extracellular pathogen. Recently, a study by Hartigan et al. examined the role of CCR7 in host defense against Aspergillus fumigatus (13), which is also an extracellular pathogen. Employing a murine model of invasive aspergillosis, the authors concluded that CCR7 deficiency results in increased survival and clearance of infection. In agreement with our results, the authors concluded that the enhanced host defense associated with CCR7 deficiency is primarily the result of more beneficial regulation of the inflammatory response by dendritic cells (13). More research is necessary to examine how broadly applicable our results are to other infections caused by extracellular pathogens. However, our results suggest that the host defense to extracellular pathogens is better served by dendritic cells that remain in situ to help shape the response than by dendritic cells migrating to the lymph node. It is interesting to speculate why migration to the lymph node is the default response of dendritic cells to infection when clearly there are some occasions when it is beneficial for dendritic cells to remain at the site of infection. The results from our model of acute P. aeruginosa infection identify a potential target for therapeutic intervention, with the goal being the retention of dendritic cells at the site of infection.
Our results underscore the dual role of dendritic cells in innate and adaptive immunity and present a framework for how CCR7 biases dendritic cell function toward the promotion of one or the other. It remains to be ascertained whether these results are more broadly applicable to other infections and whether therapy based on delaying CCR7 expression can by employed without severely undermining the adaptive response. However, our results elucidate a previously unappreciated aspect of CCR7 function in host defense and identify a new potential target for therapeutic intervention.
Acknowledgments
This work was supported by University of Cincinnati Center for Environmental Genetics grant P30-ES06096-02 (to M.T.B.), NIH grant R01 ES015036 (to M.T.B.), and NIH grant T32 ES016646 (to G.T.M.).
We thank Alvaro Puga for graciously providing access to the BD FACSCalibur flow cytometer. We thank Erica Eppert and Katherine Rooney for critical reading of the manuscript. We are grateful for thoughtful discussion from Lisa R. Young.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 22 February 2010.
REFERENCES
- 1.Ato, M., A. Maroof, S. Zubairi, H. Nakano, T. Kakiuchi, and P. M. Kaye. 2006. Loss of dendritic cell migration and impaired resistance to Leishmania donovani infection in mice deficient in CCL19 and CCL21. J. Immunol. 176:5486-5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beaty, S. R., C. E. Rose, Jr., and S.-S. J. Sung. 2007. Diverse and potent chemokine production by lung CD11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J. Immunol. 178:1882-1895. [DOI] [PubMed] [Google Scholar]
- 3.Bromley, S. K., S. Y. Thomas, and A. D. Luster. 2005. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol. 6:895-901. [DOI] [PubMed] [Google Scholar]
- 4.Curtis, J. L. 2005. Cell-mediated adaptive immune defense of the lungs. Proc. Am. Thorac. Soc. 2:412-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Debes, G. F., C. N. Arnold, A. J. Young, S. Krautwald, M. Lipp, J. B. Hay, and E. C. Butcher. 2005. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol. 6:889-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Debes, G. F., K. Bonhagen, T. Wolff, U. Kretschmer, S. Krautwald, T. Kamradt, and A. Hamann. 2004. CC Chemokine receptor 7 expression by effector/memory CD4+ T cells depends on antigen specificity and tissue localization during influenza A virus infection. J. Virol. 78:7528-7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.del Rio, M.-L., J.-I. Rodriguez-Barbosa, J. Bolter, M. Ballmaier, O. Dittrich-Breiholz, M. Kracht, S. Jung, and R. Förster. 2008. CX3CR1+c-kit+ bone marrow cells give rise to CD103+ and CD103− dendritic cells with distinct functional properties. J. Immunol. 181:6178-6188. [DOI] [PubMed] [Google Scholar]
- 8.Ellis, T. N., and B. L. Beaman. 2004. Interferon-γ activation of polymorphonuclear neutrophil function. Immunology 112:2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Förster, R., A. C. Davalos-Misslitz, and A. Rot. 2008. CCR7 and its ligands: balancing immunity and tolerance. Nat. Rev. Immunol. 8:362-371. [DOI] [PubMed] [Google Scholar]
- 10.Förster, R., A. Schubel, D. Breitfeld, E. Kremmer, I. Renner-Müller, E. Wolf, and M. Lipp. 1999. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99:23-33. [DOI] [PubMed] [Google Scholar]
- 11.Giannoni, E., T. Sawa, L. Allen, J. Wiener-Kronish, and S. Hawgood. 2006. Surfactant proteins A and D enhance pulmonary clearance of Pseudomonas aeruginosa. Am. J. Respir. Cell Mol. Biol. 34:704-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gunn, M. D., S. Kyuwa, C. Tam, T. Kakiuchi, A. Matsuzawa, L. T. Williams, and H. Nakano. 1999. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 189:451-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartigan, A. J., J. Westwick, G. Jarai, and C. M. Hogaboam. 2009. CCR7 deficiency on dendritic cells enhances fungal clearance in a murine model of pulmonary invasive aspergillosis. J. Immunol. 183:5171-5179. [DOI] [PubMed] [Google Scholar]
- 14.Hashimoto, S., J. F. Pittet, K. Hong, H. Folkesson, G. Bagby, L. Kobzik, C. Frevert, K. Watanabe, S. Tsurufuji, and J. Wiener-Kronish. 1996. Depletion of alveolar macrophages decreases neutrophil chemotaxis to Pseudomonas airspace infections. Am. J. Physiol. Lung Cell. Mol. Physiol. 270:L819-L828. [DOI] [PubMed] [Google Scholar]
- 15.Heer, A. K., N. L. Harris, M. Kopf, and B. J. Marsland. 2008. CD4+ and CD8+ T cells exhibit differential requirements for CCR7-mediated antigen transport during influenza infection. J. Immunol. 181:6984-6994. [DOI] [PubMed] [Google Scholar]
- 16.Holloway, B. W., V. Krishnapillai, and A. F. Morgan. 1979. Chromosomal genetics of Pseudomonas. Microbiol. Mol. Biol. Rev. 43:73-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itakura, M., A. Tokuda, H. Kimura, S. Nagai, H. Yoneyama, N. Onai, S. Ishikawa, T. Kuriyama, and K. Matsushima. 2001. Blockade of secondary lymphoid tissue chemokine exacerbates Propionibacterium acnes-induced acute lung inflammation. J. Immunol. 166:2071-2079. [DOI] [PubMed] [Google Scholar]
- 18.Junt, T., E. Scandella, R. Förster, P. Krebs, S. Krautwald, M. Lipp, H. Hengartner, and B. Ludewig. 2004. Impact of CCR7 on priming and distribution of antiviral effector and memory CTL. J. Immunol. 173:6684-6693. [DOI] [PubMed] [Google Scholar]
- 19.Kocks, J. R., H. Adler, H. Danzer, K. Hoffmann, D. Jonigk, U. Lehmann, and R. Förster. 2009. Chemokine receptor CCR7 contributes to a rapid and efficient clearance of lytic murine {gamma}-herpes virus 68 from the lung, whereas bronchus-associated lymphoid tissue harbors virus during latency. J. Immunol. 182:6861-6869. [DOI] [PubMed] [Google Scholar]
- 20.Kohout, T. A., S. L. Nicholas, S. J. Perry, G. Reinhart, S. Junger, and R. S. Struthers. 2004. Differential desensitization, receptor phosphorylation, {beta}-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J. Biol. Chem. 279:23214-23222. [DOI] [PubMed] [Google Scholar]
- 21.Kooguchi, K., S. Hashimoto, A. Kobayashi, Y. Kitamura, I. Kudoh, J. Wiener-Kronish, and T. Sawa. 1998. Role of alveolar macrophages in initiation and regulation of inflammation in Pseudomonas aeruginosa pneumonia. Infect. Immun. 66:3164-3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kursar, M., U. E. Höpken, M. Koch, A. Köhler, M. Lipp, S. H. E. Kaufmann, and H.-W. Mittrücker. 2005. Differential requirements for the chemokine receptor CCR7 in T cell activation during Listeria monocytogenes infection. J. Exp. Med. 201:1447-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackay, C. R. 2001. Chemokines: immunology's high impact factors. Nat. Immunol. 2:95-101. [DOI] [PubMed] [Google Scholar]
- 24.Marsland, B. J., P. Bättig, M. Bauer, C. Ruedl, U. Lässing, R. R. Beerli, K. Dietmeier, L. Ivanova, T. Pfister, L. Vogt, H. Nakano, C. Nembrini, P. Saudan, M. Kopf, and M. F. Bachmann. 2005. CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity 22:493-505. [DOI] [PubMed] [Google Scholar]
- 25.Martin, T. R., and C. W. Frevert. 2005. Innate immunity in the lungs. Proc. Am. Thorac. Soc. 2:403-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martín-Fontecha, A., S. Sebastiani, U. E. Höpken, M. Uguccioni, M. Lipp, A. Lanzavecchia, and F. Sallusto. 2003. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J. Exp. Med. 198:615-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagira, M., T. Imai, K. Hieshima, J. Kusuda, M. Ridanpaa, S. Takagi, M. Nishimura, M. Kakizaki, H. Nomiyama, and O. Yoshie. 1997. Molecular cloning of a novel human CC chemokine secondary lymphoid-tissue chemokine that is a potent chemoattractant for lymphocytes and mapped to chromosome 9p13. J. Biol. Chem. 272:19518-19524. [DOI] [PubMed] [Google Scholar]
- 28.Nieuwenhuis, E. E. S., T. Matsumoto, M. Exley, R. A. Schleipman, J. Glickman, D. T. Bailey, N. Corazza, S. P. Colgan, A. B. Onderdonk, and R. S. Blumberg. 2002. CD1d-dependent macrophage-mediated clearance of Pseudomonas aeruginosa from lung. Nat. Med. 8:588-593. [DOI] [PubMed] [Google Scholar]
- 29.Ohl, L., M. Mohaupt, N. Czeloth, G. Hintzen, Z. Kiafard, J. Zwirner, T. Blankenstein, G. Henning, and R. Förster. 2004. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity 21:279-288. [DOI] [PubMed] [Google Scholar]
- 30.Pene, F., B. Zuber, E. Courtine, C. Rousseau, F. Ouaaz, J. Toubiana, A. Tazi, J.-P. Mira, and J.-D. Chiche. 2008. Dendritic cells modulate lung response to Pseudomonas aeruginosa in a murine model of sepsis-induced immune dysfunction. J. Immunol. 181:8513-8520. [DOI] [PubMed] [Google Scholar]
- 31.Rangel-Moreno, J., J. E. Moyron-Quiroz, L. Hartson, K. Kusser, and T. D. Randall. 2007. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc. Natl. Acad. Sci. U. S. A. 104:10577-10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riol-Blanco, L., N. Sanchez-Sanchez, A. Torres, A. Tejedor, S. Narumiya, A. L. Corbi, P. Sanchez-Mateos, and J. L. Rodriguez-Fernandez. 2005. The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. J. Immunol. 174:4070-4080. [DOI] [PubMed] [Google Scholar]
- 33.Rot, A., and U. H. von Andrian. 2004. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 22:891-928. [DOI] [PubMed] [Google Scholar]
- 34.Sadikot, R. T., T. S. Blackwell, J. W. Christman, and A. S. Prince. 2005. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am. J. Respir. Crit. Care Med. 171:1209-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sullivan, S. K., D. A. McGrath, D. Grigoriadis, and K. B. Bacon. 1999. Pharmacological and signaling analysis of human chemokine receptor CCR-7 stably expressed in HEK-293 cells: high-affinity binding of recombinant ligands MIP-3beta and SLC stimulates multiple signaling cascades. Biochem. Biophys. Res. Commun. 263:685-690. [DOI] [PubMed] [Google Scholar]
- 36.Sung, S.-S. J., S. M. Fu, C. E. Rose, Jr., F. Gaskin, S.-T. Ju, and S. R. Beaty. 2006. A major lung CD103 ({alpha}E)-beta7 integrin-positive epithelial dendritic cell population expressing langerin and tight junction proteins. J. Immunol. 176:2161-2172. [DOI] [PubMed] [Google Scholar]
- 37.Trinchieri, G. 2003. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3:133-146. [DOI] [PubMed] [Google Scholar]
- 38.Tsai, W. C., R. M. Strieter, B. Mehrad, M. W. Newstead, X. Zeng, and T. J. Standiford. 2000. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect. Immun. 68:4289-4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vermaelen, K., and R. Pauwels. 2004. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A 61:170-177. [DOI] [PubMed] [Google Scholar]
- 40.Villablanca, E. J., D. Zhou, B. Valentinis, A. Negro, L. Raccosta, L. Mauri, A. Prinetti, S. Sonnino, C. Bordignon, C. Traversari, and V. Russo. 2008. Selected natural and synthetic retinoids impair CCR7- and CXCR4-dependent cell migration in vitro and in vivo. J. Leukoc. Biol. 84:871-879. [DOI] [PubMed] [Google Scholar]
- 41.Walrand, S., S. Valeix, C. Rodriguez, R. Ligot, J. Chassagne, and M.-P. Vasson. 2003. Flow cytometry strudy of polymorphonuclear neutrophil oxidative burst: a comparison of three fluorescent probes. Clin. Chim. Acta 331:103-110. [DOI] [PubMed] [Google Scholar]
- 42.Wesselkamper, S. C., B. L. Eppert, G. T. Motz, G. W. Lau, D. J. Hassett, and M. T. Borchers. 2008. NKG2D is critical for NK cell activation in host defense against Pseudomonas aeruginosa respiratory infection. J. Immunol. 181:5481-5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshida, R., T. Imai, K. Hieshima, J. Kusuda, M. Baba, M. Kitaura, M. Nishimura, M. Kakizaki, H. Nomiyama, and O. Yoshie. 1997. Molecular cloning of a novel human CC chemokine EBI1-ligand chemokine that is a specific functional ligand for EBI1, CCR7. J. Biol. Chem. 272:13803-13809. [DOI] [PubMed] [Google Scholar]
- 44.Young, H. A., and K. J. Hardy. 1995. Role of interferon-gamma in immune cell regulation. J. Leukoc. Biol. 58:373-381. [PubMed] [Google Scholar]
- 45.Zidar, D. A., J. D. Violin, E. J. Whalen, and R. J. Lefkowitz. 2009. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. U. S. A. 106:9649-9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ziegler, E., M. Oberbarnscheidt, S. Bulfone-Paus, R. Förster, U. Kunzendorf, and S. Krautwald. 2007. CCR7 signaling inhibits T cell proliferation. J. Immunol. 179:6485-6493. [DOI] [PubMed] [Google Scholar]