

Abstract
Escherichia coli strain Nissle 1917, which has been widely used as a probiotic for the treatment of inflammatory bowel disorders, expresses a K5 capsule, the expression of which is often associated with extraintestinal and urinary tract isolates of E. coli. Previously, it had been shown that the expression of a K5 capsule by Nissle 1917 was important in mediating interactions with epithelial cells and the extent of chemokine expression. In this paper, we show that infection with Nissle 1917 induces expression of Toll-like receptor 4 (TLR4) and TLR5 in Caco-2 cells and that maximal induction of TLR5 required the K5 capsule. In addition, purified K5 polysaccharide was capable of inducing expression of TLR5 and mCD14 and potentiated the activity of both TLR4 and TLR5 agonists to increase the proinflammatory response. Infection with Nissle 1917 also increased the expression of the adaptor molecules MyD88 and TRIF, which was K5 capsule dependent. By Western blot analysis, it was possible to show that induction of interleukin-8 by Nissle 1917 was predominantly through the mitogen-activated protein (MAP) kinase pathway and that expression of the K5 capsule was important for activation of the MAP kinase pathway. This paper provides new information on the function of the K5 capsule in mediating interactions between Nissle 1917 and epithelial cells and the mechanisms that underlie the probiotic properties of Nissle 1917.
Escherichia coli strain Nissle 1917 (serotype O6:K5:H1) is apparently nonpathogenic (17, 57) and has been widely used in preventing infectious diarrheal diseases (6, 11, 20, 34), treating inflammatory bowel diseases (IBDs) such as ulcerative colitis and Crohn's disease (6, 14, 27, 28), and preventing colonization of the digestive tract of neonates by pathogens (30). There has been a growing interest in investigating the immunomodulatory effects of Nissle 1917 and the role of individual microbial components in these processes. It is known that colonization by Nissle 1917 may lead to an alteration of the hosts' cytokine repertoire, with increased levels of inteleukin-10 (IL-10), IL-12, monocyte chemoattractant protein 1 (MCP-1), MIP2α, and MIP2β (10, 52), together with increased immunoglobulin A secretion (11), lymphocyte or macrophage activation (10), modulation of CD4+ clonal expansion (46), and stimulation of antimicrobial peptide production by intestinal epithelial cells and tight junction formation (39, 62). In addition, Nissle 1917 activates γδT cells, stimulating CXCL8 and IL-6 release but inhibiting tumor necrosis factor alpha (TNF-α) secretion (18). Following activation, Nissle 1917 induced apoptosis in activated γδT cells, suggesting that Nissle 1917 is able to interact with the subset of T cells that operate at the interface between the adaptive and innate immune responses (18).
The role of individual components of Nissle 1917 in mediating the immunomodulatory responses is less clearly understood. Previously, flagellin of Nissle 1917 had been shown to induce human β-defensin expression (44), and recently, we have shown that the K5 capsule of Nissle 1917 mediates the interaction between Nissle 1917 and epithelial cells and the extent of chemokine induction (19). Recognition of microbial factors by the host will involve Toll-like receptors (TLRs) that act as signaling receptors of the innate immune system, recognizing a wide variety of molecular patterns typical for microorganisms and being able to initiate anti-infective inflammatory responses (1, 16, 31, 37, 48, 53). TLRs are selectively activated by different microbial ligands, including lipopolysaccharide (LPS), flagellin, peptidoglycans, and oligonucleotides with CpG sequences (48). In the case of intestinal epithelial cells, it was previously reported that LPS and flagellin play roles in induction of the proinflammatory response by both pathogenic and commensal bacteria via interaction of TLR4 and TLR5, respectively (2, 13, 25). Although, TLRs differ from one another by their ligand specificities, determined by the extracellular portion of the receptor, in the cytoplasm there is a common Toll-interleukin-1-related (TIR) domain (1, 48). There are two TLR signaling pathways following ligand ligation with the cell surface TLR. There is a MyD88-dependent pathway that is common to all TLRs and a MyD88-independent pathway involving TRIF that is peculiar to the TLR3 and TLR4 signaling pathways (1, 3, 48). Signal transduction results in downstream activation of transcription factors, like NF-κB and AP-1, which leads to an upregulation of proinflammatory cytokines and chemokines, such as TNF-α and IL-8 (48). In the case of the AP-1-mediated pathway, activation of AP-1 is preceded by the mitogen-activated protein kinase (MAPK) activation pathway (29, 58).
Recently, we showed that the K5 capsule plays a key part in mediating the interaction between Nissle 1917 and epithelial cells and the extent of the chemokine response (19). However, the mechanisms by which the K5 capsule elicited this response were unknown. In this paper, we show that Nissle 1917 induces TLR2, -4, and -5 expression, with maximal TLR5 induction being dependent on the K5 capsule, and that addition of purified K5 polysaccharide was able to induce TLR5 expression. In addition, the K5 capsule was necessary for both maximal induction of the adaptor proteins MyD88 and TRIF and induction of CD-14 expression by Nissle 1917. Further, we show that purified K5 polysaccharide could potentiate the activity of both TLR4 and TLR5 agonists to maximize the proinflammatory response. Analysis of the phosphorylation state of Jun N-terminal protein kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 in the presence of the K5 capsule demonstrated that the K5-mediated proinflammatory response is predominantly mediated via these mitogen-activated protein (MAP) kinase pathways. Overall, this paper provides the first data on the likely mechanism by which the K5 capsule mediates interactions between Nissle 1917 and host cells.
MATERIALS AND METHODS
Preparation of bacteria.
The Escherichia coli strain Nissle 1917 strain, the kfiC knockout mutant lacking a K5 capsule (EcNK5−), and plasmid pBkfiCD have been described previously (19). For all experiments, the bacteria were grown overnight in Luria-Bertani (LB) broth medium at 37°C on a shaker at 200 rpm. The cultures were then diluted 1:100 in fresh LB broth and reincubated under the same conditions until mid-log phase (optical density at 600 nm, 0.5). Where appropriate, the medium was supplemented with ampicillin (100 μg ml−1) or chloramphenicol (25 μg ml−1).
Purification of K5 polysaccharide.
The K5 polysaccharide was prepared from strain MS101 as described previously (9). Contaminating proteins were removed by phenol treatment, and the final polysaccharide preparation was analyzed for protein contamination by silver staining following SDS-PAGE. No detectable proteins were present in any polysaccharide preparations. LPS was removed by polymyxin B treatment, and the final polysaccharide preparation was assayed for LPS contamination using a Limulus amoebocyte lysate according to the manufacturer's instructions (AMS Biotechnology Ltd., Abingdon, United Kingdom). The K5 polysaccharide preparation was free of detectable contaminating LPS.
Cell culture condition.
The human colon adenocarcinoma cell line Caco-2 was maintained in Iscove's modified Dulbecco's medium (IMDM) for cell culture (Sigma Aldrich Company, Irvine, Ayrshire, United Kingdom) containing 10% fetal bovine serum (Invitrogen, Life Technologies, Paisley, United Kingdom) at 37°C in the presence of 5% CO2. The Caco-2 cells used for the experiments were at passages 10 to 20. The experiments were done using Caco-2 cell monolayers which were obtained by seeding the cells into six-well plates (Costar Corning) and growing them under the above-mentioned conditions for 4 days until they reached confluence.
Stimulation of Caco-2 cells.
Confluent Caco-2 cells in six-well plates (1 × 106 cells per well) were washed with phosphate-buffered saline (PBS) and incubated for 6 h at 37°C in 5% CO2 with bacterial suspension made in IMDM at a multiplicity of infection (MOI) of 1. Controls were carried out in similar way except that the bacterial suspension was replaced with IMDM. At the end of the stimulation period, RNA of the mammalian cells was extracted to be analyzed by quantitative reverse transcription-PCR (qRT-PCR).
RNA extraction and quantification.
RNA was extracted from cells by use of an RNeasy minikit (Qiagen, Hilden, Germany) as previously described (19). Genomic DNA contamination of the extracted RNA was removed by twice treating the purified RNA sample with RNase-free DNase (Qiagen, Hilden, Germany). The absence of DNA in isolated RNA was confirmed by performing PCR in the absence of a reverse transcriptase step. Two micrograms of each isolated RNA was then reverse transcribed for 60 min at 42°C in 25-μl assay mixtures containing 25 pM oligo(dT) primer, 12.5 mM deoxynucleoside triphosphates (dNTPs), 40 units avian myeloblastosis virus (AMV) reverse transcriptase (Roche Diagnostics, Mannheim, Germany) in addition to 20 units of RNase inhibitor (Roche Diagnostics, Mannheim, Germany). All samples were reverse transcribed under the same conditions and with a similar reverse transcription master mix to minimize differences in reverse transcription efficiency. The qPCR was performed with 50-μl reaction mixtures containing 2 μl cDNA and 300 nM each sense and antisense primer and with a SYBR green PCR kit from Eurogentec. The specific primers designed to amplify 90- to 250-bp fragments from the cDNA under investigation have previously been described (19). The PCRs were carried out with a thermocycler (ABI PRISM 7000 cycler; Applied Biosystems, Foster City, CA). The cycling conditions for PCR amplification were 95°C for 10 min and then 40 cycles including denaturation at 95°C for 15 s, annealing, and extension at 58°C for 1 min. The housekeeping gene used was the RPS9 (ribosomal protein S9) gene, which was assessed in parallel with each reaction as an internal standard. Relative mRNA levels were determined by using included standard curves for each individual gene, and further normalization to the levels for the housekeeping gene was carried out.
Flow cytometry.
Cell surface expression of TLR4 and -5 was determined by flow cytometry (cyan; Beckman-Coulter, High Wycombe, United Kingdom) with an excitation wavelength of 488 nm. The instrument was calibrated before each measurement with standardized fluorescent particles (SpectrAlign; Dako, Cambridgeshire, United Kingdom). Fluorescence signals of cells were measured using a 575-nm/25-nm band-pass filter. The flow cytometry was performed using monoclonal mouse anti-human antibodies (Imgenex, San Diego, CA) according to the manufacturer's instructions. Briefly, 5 × 105 Caco-2 cells were washed in PBS containing bovine serum albumin (BSA) and then incubated with the corresponding TLR antibodies (1/100 dilution) for 30 min at 4°C. After a wash, phycoerythrin (PE)-conjugated goat anti-mouse antibody was added and the suspension was incubated at 4°C for 30 min. Cells were then washed in PBS containing BSA. Negative controls were prepared by incubation with an isotype-matched control antibody. Samples were prepared and analyzed in duplicate, and a minimum of 5,000 cells were counted from each sample. Data are expressed as mean numbers of fluorescence units and percentages of cells staining positively.
Blocking TLR4 and TLR5.
To explore the role of TLR4 and TLR5 pathways in K5-mediated induction of a proinflammatory response, Caco-2 cells were treated with anti-human TLR4 or anti-human TLR5 blocking antibodies (Invivogen, San Diego, CA) prior to the infections. The blocking was done according to the protocol suggested by the manufacturer. Briefly, Caco-2 cells were incubated with 6 μg ml−1 of the neutralizing antibodies for 1 h min prior to the addition of bacteria. The blockade was confirmed by stimulation of the blocked Caco-2 cells with ultrapure E. coli LPS (a TLR4 agonist) or endotoxin-free Salmonella enterica serovar Typhimurium flagellin (a TLR5 agonist) at a concentration of 200 ng ml−1 or 20 ng ml−1, respectively. Both TLR agonists were purchased from Invivogen, San Diego, CA. In all cases, expression of IL-8 was used as a measure of TLR4 or TLR5 pathway activity.
Western blotting for MAP kinase pathways.
Western blot analysis was performed to measure MAP kinase (MAPK) protein activation within infected and control Caco-2 cells. Briefly, Caco-2 cell lysates were obtained by exposing cells to cold SDS loading buffer supplemented with NaF, Na3VO4, and α-glycerophosphate at 10 mM each. The samples were boiled for 10 min before gel loading. Proteins resolved on SDS-PAGE gel were then transferred to Western polyvinylidene difluoride (PVDF) membranes (Whatman) using an electrophoretic transfer system (Trans-blot semidry transfer cell; Bio-Rad) at 15 mA for 20 min. Membranes were then blocked overnight with T-PBS (PBS containing 0.1% [vol/vol] Tween) supplemented with 5% (vol/wt) BSA. After a wash with T-PBS, membranes were incubated at 4°C overnight with one of the primary antibodies (1:1,000 dilution). The primary antibodies used were p38 MAPK antibody, phospho-p38 MAPK (Thr180/Tyr182) antibody, ERK antibody, phospho-ERK (Thr202/Tyr204) antibody, JNK antibody, and phospho-JNK (Thr183/Tyr185) antibody (Cell Signaling Technology, Beverly, MA). Membranes were then washed with T-PBS and subsequently incubated with horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibody (1:2,000 dilution). The membranes were washed and developed with Western lightening chemiluminescence reagents (Perkin Elmer, Boston, MA).
MAP kinase inhibitors.
Confluent Caco-2 cells were incubated with SP600125 (JNK1, -2, and -3 inhibitor), SB203580 (p38 activity inhibitor), and U0126 (MEK1 and -2 inhibitor), which were prepared as stocks in dimethyl sulfoxide (DMSO) and then diluted to a final concentration of 20 μM in Dulbecco's modified Eagle's medium (DMEM). After incubation for 24 h at 37°C and 5% CO2, the bacteria were added at an MOI of 1. The plate was further incubated for 6 h, after which the mammalian cell RNA was extracted and reverse transcribed and the expression of IL-8 was assessed by qRT-PCR as mentioned earlier. It should be emphasized that the MAP kinase pathway inhibition was confirmed by the absence of phosphorylated JNK, p38, or ERK bands on the Western blot after stimulation of the cells with TNF-α (40 ng ml−1) for 1 h.
Statistical analysis.
Data are expressed as means ± standard deviations (SD). The significance of differences between means was tested using the two-tailed Student t test and analysis of variance. The differences between means were considered statistically significant when P was ≤0.01. The SPSS statistical package was used for analysis.
RESULTS
Effect of the K5 capsule on TLR expression of Caco-2 cells.
To determine the effect of the K5 capsular polysaccharide (CPS) on expression of TLRs, Caco-2 cells were infected with Nissle 1917 and the isogenic K5 minus mutant EcNK5− (19). The infection with Nissle 1917 resulted in significant rises in mRNA levels of TLR4 and -5, increasing 14- and 115-fold, respectively, compared to the level for uninfected Caco-2 cells (Fig. 1a). In contrast, infection with Nissle 1917 resulted in a more modest (1.7-fold) increase in TLR2 (Fig. 1a). Infection with EcNK5− resulted in levels of induction of TLR2 and -4 similar to those seen with Nissle 1917 but significantly (P ≤ 0.01) less induction of TLR5 than that seen with Nissle 1917 (Fig. 1a). The addition of exogenous K5 polysaccharide with EcNK5− during infection of Caco-2 cells was associated with a significant (P ≤ 0.01) (4-fold) rise in TLR5 mRNA levels compared to the level seen with Caco-2 cells infected with EcNK5− alone, with mRNA levels comparable to those induced by Nissle 1917 (Fig. 1a). Complementation of strain EcNK5− with plasmid pBkfiCD (19) resulted in a significant (P ≤ 0.01) increase in induction of TLR5 mRNA in infected Caco-2 cells compared to that seen with cells infected with EcNK5− (Fig. 1b).
FIG. 1.

Effect of infection by Nissle 1917 and EcNK5− strains on TLR expression of Caco-2 cells. Caco-2 cells were incubated with Nissle 1917 (EcN), EcNK5−, and EcNK5− plus 100 μg ml−1 of purified K5 polysaccharide (a) or incubated with EcN (pBlue Script II SK+), EcNK5− (pBlue Script II SK+), or EcNK5− (pBKfiCD) (b) for 6 h. After incubation, total RNA was extracted and reverse transcribed, and relative TLR mRNA was quantified by real-time RT-PCR and normalized to the level for the RPS-9 housekeeping gene. Relative messenger mRNA levels were expressed as fold changes compared to the level for untreated Caco-2 cells. The figure is representative of three independent experiments, each done at least in quadruplicate. Data are expressed as means + SD. In panel a, an asterisk indicates a significant difference in mRNA compared to the level for EcNK5−-infected cells (P < 0.01). In panel b, an asterisk indicates a significant difference compared to the level for EcNK5− (pSK+)-infected Caco-2 cells (P < 0.01).
When Caco-2 cells were incubated with increasing concentrations of purified K5 polysaccharide in the absence of bacteria, there was only a marginal increase in expression of TLR4 and the increase was not significant (data not shown). In contrast, under the same conditions, there was a significant (P ≤ 0.01) dose-dependent increase in expression of TLR5 (Fig. 2). To confirm that the changes in mRNA were reflected in increased TLR protein expression on the cell surface, flow cytometric analysis was used to determine TLR4 and -5 expression. There were low levels of expression of both TLR4 and -5 in the uninfected Caco-2 cells (Fig. 3). Infection with either Nissle 1917 or EcNK5− resulted in a significant increase in TLR4 expression (Fig. 3). While infection with EcNK5− increased TLR5 expression, this increase was significantly less than that seen with Nissle 1917-infected cells (Fig. 3). Purified K5 polysaccharide had a very modest effect on inducing TLR4 expression, in contrast to a significant increase in TLR5 expression (Fig. 3). Overall, these data indicate that Nissle 1917 induces both TLR4 and TLR5 expression in Caco-2 cells and that expression of a K5 capsule is required for maximal TLR5 expression. In addition, the data show that K5 polysaccharide alone is able to also stimulate TLR5 expression.
FIG. 2.

Effect of purified K5 capsular polysaccharide on the expression of TLR5 by Caco-2 cells. Caco-2 cells were incubated with increasing concentrations of purified K5 polysaccharide for 6 h, after which total RNA was isolated and reverse transcribed and the relative mRNA expression levels for the TLR5 gene and the RPS-9 housekeeping gene (as an internal control) were analyzed by real-time RT-PCR. Relative mRNA levels are expressed in fM. The figure is representative of three independent experiments, each done at least in quadruplicate. Data are expressed as means + SD.
FIG. 3.

Flow cytometric analysis of the expression of TLR4 and TLR5 on the surfaces of Caco-2 cells. Caco-2 cells were first infected with either Nissle 1917 (EcN) or EcNK5− or treated with 100 μg ml−1 purified K5 polysaccharide. Subsequently, following treatment of the cells with the appropriate antisera as described in the Materials and Methods, fluorescence signals of cells were measured using a 575-nm/25-nm band-pass filter. Shaded areas represent the background staining of the negative control, and the open areas show the fluorescence staining with TLR4- and TLR5-specific antibodies. Samples were prepared and analyzed in triplicate, and a minimum of 5,000 cells from each sample were counted. Data are expressed as mean fluorescence units (MFI) and percentages of cells staining positively.
Effect of the K5 capsule on CD-14 gene expression in Caco-2 cells.
mCD-14 is a cell surface LPS binding protein that, following the binding of LPS, stimulates activation of downstream targets by interacting with TLR4 (24). Following infection with Nissle 1917, there was a significant (5-fold) increase in the level of CD-14-specific mRNA compared to the level for uninfected Caco-2 cells (Fig. 4). In contrast, there was no significant increase in CD-14 mRNA (P ≤ 0.01) following infection with EcNK5− (Fig. 4). Addition of purified K5 polysaccharide alone (100 μg ml−1) or in conjunction with EcNK5− induced a significant (P ≤ 0.01) increase in CD-14 mRNA expression (3- or 5-fold, respectively) compared to the level for uninfected Caco-2 cells (Fig. 4). Collectively, these data demonstrate (i) that infection with Nissle 1917 stimulates CD-14 expression, (ii) that this induction requires the presence of the K5 capsule, and (iii) that purified K5 CPS is able to induce CD-14 gene expression.
FIG. 4.

Effect of the K5 capsule on CD14 expression of Caco-2 cells. Caco-2 cells were infected with either Nissle 1917 (EcN), EcNK5−, or EcNK5− plus purified K5 polysaccharide (100 μg ml−1) or just treated with purified K5 polysaccharide (100 μg ml−1) for 6 h, after which total RNA was isolated and reverse transcribed and relative mRNA expression levels for selected genes and the RPS-9 housekeeping gene (as an internal control) were analyzed by real-time RT-PCR. Relative CD14 messenger mRNA levels are expressed in fM. The figure is representative of three independent experiments, each done at least in quadruplicate. Data are expressed as means + SD. An asterisk indicates a significant difference in mRNA compared to the level for the uninfected control (P < 0.01).
Blocking of either TLR4 or TLR5 diminishes the K5-mediated immunomodulatory effect.
To demonstrate that the immunomodulatory effects of the K5 capsule are mediated via TLR4 and -5, IL-8 induction in Caco-2 cells exposed to either K5 polysaccharide, Nissle 1917, or EcNK5− in the presence of TLR4 and TLR5 neutralizing polyclonal antibodies was assayed. In the presence of TLR4 neutralizing antibodies, LPS, a TLR4 agonist, was unable to stimulate a significant increase IL-8 expression (Fig. 5a), confirming that the neutralizing antibodies were blocking the TLR4 pathway. In contrast, flagellin, a TRL5 agonist, was able to induce a significant (P ≤ 0.01) increase in IL-8 expression (Fig. 5a). The addition of purified K5 polysaccharide to flagellin resulted in a significant (P ≤ 0.01) further induction in IL-8 expression compared to that seen with flagellin alone (Fig. 5a). As predicted from previous studies (19), K5 polysaccharide alone had a negligible effect on induction of IL-8 (Fig. 5a). These data confirm that K5 polysaccharide can act to potentiate the activity of a TLR5 agonist.
FIG. 5.

Effect of blocking TLR4 and TLR5 on the proinflammatory effect of Nissle 1917 and purified K5 polysaccharide. Caco-2 cells were treated with anti-human TLR4 neutralizing antibodies (a) or TLR5 neutralizing antibodies (b) before challenge with TLR4 and -5 agonists or were treated with TLR4 and/or TLR5 neutralizing antibodies before infection with Nissle 1917 (EcN) and EcNK5− (c). The preincubation with the antibodies was done for 1 h, after which bacteria or TLR agonists were added and incubated with cells for 6 h in the presence of the antibodies. After incubation, total RNA was isolated and reverse transcribed, and relative IL-8 mRNA was quantified by real-time RT-PCR and normalized to the level for the RPS-9 housekeeping gene. The figure is representative of three independent experiments, each done at least in quadruplicate. In panels a and b, an asterisk indicates a significant difference compared to the level for the uninfected control (P < 0.01). In panel c, an asterisk indicates a significant difference compared to the level for the sample lacking any neutralizing antibodies (P < 0.01).
In the presence of TLR5 neutralizing antibodies, flagellin was unable to induce a significant increase in IL-8 expression (Fig. 5b), confirming that the neutralizing antibodies were blocking the TLR5 pathway. In contrast, LPS, a TLR4 agonist, induced a significant (P ≤ 0.01) increase in IL-8 expression that was increased 10-fold by the addition of K5 polysaccharide to the LPS (Fig. 5b). These data confirm that the K5 polysaccharide can act to potentiate the activity of a TLR4 agonist to increase the proinflammatory response.
To confirm the roles of both TLR4 and TLR5 in stimulation of IL-8 expression by Nissle 1917, Caco-2 cells were infected with either Nissle 1917 or EcNK5− in the presence of either TLR4 or TLR5 neutralizing antibodies or with both antibodies being present. In the absence of either antibody, there was a 13-fold greater induction of IL-8 in cells infected by Nissle 1917 than in those infected by EcNK5− (Fig. 5c). In the presence of either TLR4 or TLR5 neutralizing antibodies, the ratio of IL-8 induction was significantly (P ≤ 0.01) reduced (2.8- or 2.6-fold, respectively) (Fig. 5c). When both neutralizing antibodies were present, the ratio of IL-8 induction dropped to 0.96 (Fig. 5c). These data confirm the essential role of the K5 capsule for maximal IL-8 induction and confirm that the effect of the K5 capsule is mediated via both TLR4 and TLR5.
Effect of K5 on expression of TLR4 and TLR5 adaptor molecules.
Signal transduction from TLR4 and TLR5 involves cytoplasmic adaptor proteins (1, 48). In the case of TLR4, there are two pathways, one via MyD88 and one MyD88-independent pathway via the adaptor proteins TRAM and TRIF (3, 48). In contrast, TLR5 uses only the MyD88 pathway (48). Infection with Nissle 1917 induced a 21-fold increase in MyD88 gene expression (Fig. 6a), which was significantly higher (P ≤ 0.01) than the 12-fold induction seen with infection with EcNK5− (Fig. 6a). Infection of Caco-2 cells with EcNK5− together with K5 polysaccharide (100 μg ml−1) doubled MyD88 expression, increasing it to a level comparable to that seen with cells infected with Nissle 1917 (Fig. 6a). In contrast, exposure of cells to the K5 CPS alone showed no significant upregulation of MyD88 expression (Fig. 6a).
FIG. 6.

Effect of K5 capsule on MyD88 (a) and TRIF (b) expression of Caco-2 cells. Caco-2 cells were infected with either Nissle 1917 (EcN), EcNK5−, or EcNK5− plus purified K5 polysaccharide (100 μg ml−1) or just treated with purified K5 polysaccharide (100 μg ml−1) for 6 h, after which total RNA was isolated, reverse transcribed, and quantified using real-time RT-PCR. Relative messenger mRNA levels are expressed as fold changes compared to the level for untreated Caco-2 cells. The figure is representative of three independent experiments, each done at least in quadruplicate. Data are expressed as means + SD. An asterisk indicates a significant difference compared to the level for uninfected cells (P < 0.01).
In the case of TRIF expression, infection with Nissle 1917 induced a 41-fold increase in expression compared to the level for uninfected cells (Fig. 6b). In contrast, infection with EcNK5− induced a modest (2-fold) increase in TRIF expression, similar to that seen when cells were exposed to purified K5 polysaccharide (100 μg ml−1) (Fig. 6b). Infection of Caco-2 cells with EcNK5− together with K5 polysaccharide (100 μg ml−1) induced a 28-fold increase in TRIF expression (Fig. 6b). These data establish that infection with Nissle 1917 induces expression of both MyD88 and TRIF, indicating that both pathways are induced by Nissle 1917. Further, for maximum induction of these two signaling pathways, expression of a K5 capsule is essential and that K5 polysaccharide can act in trans in the presence of EcNK5− to restore levels of both MyD88 and TRIF expression to that seen with cells infected with Nissle 1917.
K5-mediated activation of the MAP kinase pathway.
The transcription factor AP-1 is activated via the MAP kinase (MAPK) pathway (8, 29, 58). To determine the involvement of different components of the MAPK pathway in K5-mediated proinflammatory gene expression, Caco-2 cells were infected with either Nissle 1917 or EcNK5− in the presence or absence of K5 polysaccharide and the levels of total and phosphorylated JNK, ERK, and p38 assessed by Western blotting using specific antisera. The relative amount of the phosphorylated protein was taken as an indicator of their level of enzymatic activity. Infection of Caco-2 cells with Nissle 1917 resulted in increased levels of phosphorylated JNK1 and -2 compared to the level for noninfected cells or those infected with EcNK5−, where phosphorylated JNK1 and -2 were undetectable (Fig. 7). The addition of K5 polysaccharide during infection with EcNK5− resulted in detectable levels of phosphorylated JNK1 and -2 (Fig. 7). Interestingly, a similar pattern was observed in the cases of other MAPK pathways (ERK and p38), in which the levels of phosphorylation were much greater in cells infected with Nissle 1917 than in those infected with EcNK5− (Fig. 7).
FIG. 7.
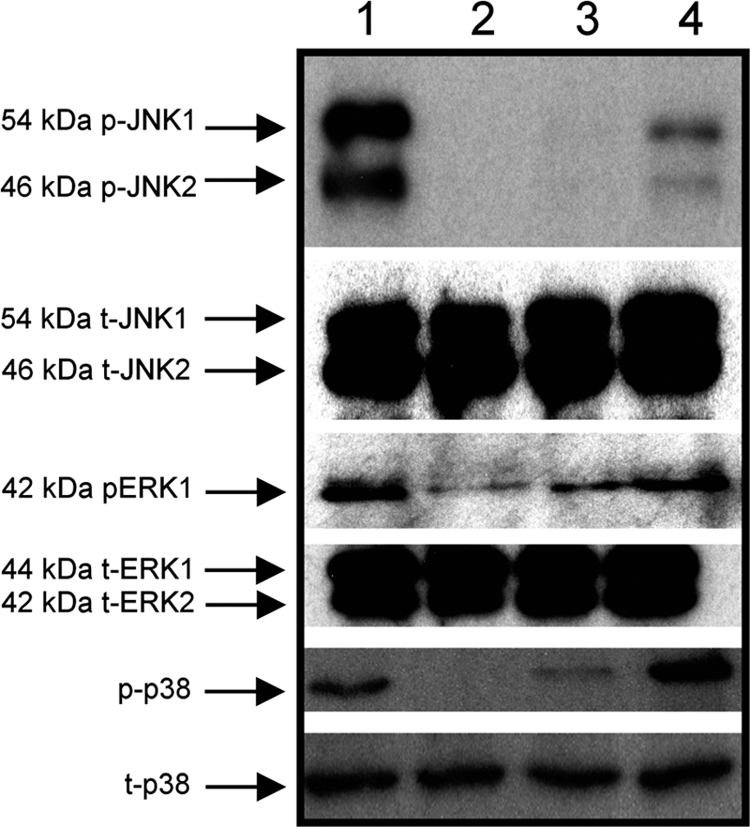
K5-mediated activation of the MAP kinase pathway. Caco-2 cells were incubated with Nissle 1917 (EcN) (lane 1), EcNK5− (lane 3), or EcNK5− plus purified K5 polysaccharide (100 μg ml−1) (lane 4) for 6 h, after which cells were lysed and the levels of total and phosphorylated JNK, ERK, and p38 were assessed by Western blotting using the specific antibodies to each kinase. Lane 2 represents a control lysate of uninfected Caco-2 cells.
To establish the contribution of different MAPK pathways to the K5-mediated proinflammatory response, the level of IL-8 expression induced by Nissle 1917 was compared to that induced by EcNK5− in the presence of specific MAPK inhibitors. The inhibitors used were SP600125 (JNK1, -2, and -3 pathway inhibitor), SB203580 (p38 pathway inhibitor), and U0126 (ERK pathway inhibitor). In the absence of the inhibitors, Nissle 1917 induced a 16.7-fold greater induction in IL-8 expression than EcNK5− (Table 1). Upon inhibition of JNK, ERK, and p38, the ratios were significantly (P ≤ 0.01) reduced, reaching 3.8, 6.6, and 6.9, respectively (Table 1). This observation clearly indicates that the K5-mediated proinflammatory effect is predominantly mediated via these MAPK pathways.
TABLE 1.
Effects of specific MAP kinase pathway inhibitors on induction of IL-8 expression by Nissle 1917 and EcNK5−
| Inhibition group | Ratioa of EcN/ECNK5−-induced IL-8 expression levels |
|---|---|
| Control | 16.7 |
| JNK1/2 inhibition (SP600125) | 3.8 |
| p38 inhibition (SB203580) | 6.6 |
| ERK inhibition (UO126) | 6.9 |
| JNK, ERK, and p38 inhibition | 2.4 |
Caco-2 cells were treated with specific inhibitors for 24 h prior to exposure to either Nissle 1917 (EcN) or EcNK5− as described in Materials and Methods and the ratios of IL-8 mRNA under the different conditions determined.
DISCUSSION
Previously, it had been shown that the K5 capsule is crucially important in mediating the immunomodulatory effects of Nissle 1917 (19). The data presented in this paper build on these earlier findings and add considerably to our understanding of the mechanisms by which the probiotic E. coli strain Nissle 1917 elicits a proinflammatory response and the role of the K5 capsule in this process. Infection of Caco-2 cells with Nissle 1917 induced cell surface expression of both TLR4 and TLR5 (Fig. 3). In the case of TLR5, the expression of a K5 capsule was essential for maximal TLR5 expression, with EcNK5− inducing significantly lower levels of TLR5 (Fig. 1 and 3). The observation that both the addition of exogenous K5 to strain EcNK5− and the complementation of the kfiC mutation increased TLR5 gene expression confirmed the role of the K5 capsule in this process. The finding that the addition of purified K5 polysaccharide also induced a dose-dependent increase in TLR5 gene expression (Fig. 2) would indicate that the K5 polysaccharide itself was able to induce TLR5 expression. This cannot be explained by contamination, since the K5 preparation was shown to be LPS free, with no detectable protein (data not shown). TLRs are key components in the innate immune response involved in the recognition of microbes, with particular TLRs recognizing different microbial products. In the case of Gram-negative bacteria, TLR4 is a receptor for LPS while TLR5 recognizes flagellin (1, 48). TLRs also play a crucial role in linking the innate and adaptive immune responses and instructing adaptive immune response to a particular infection (33, 37). It is known that TLR signals induced by commensal bacteria are needed to maintain gut homeostasis and defense against pathogenic bacteria and that intestinal flora upregulate TLRs (31). In addition, it has been shown that probiotic lactobacilli upregulate TLR2 expression (54, 55). The observation that Nissle 1917 increased TLR4 and TLR5 expression may in part explain the ability of Nissle 1917 to induce a chemokine response following exposure to the basolateral surfaces of polarized epithelial cells (19) and the finding that Nissle 1917 ameliorates experimental colitis via activation of the TLR4 and TLR2 signaling pathways (14, 47). The increase in signaling will also lead to induction of antimicrobial peptides that are induced following treatment with Nissle 1917 and will help to combat pathogenic bacteria (44).
Although there was no evidence for an effect of the K5 capsule on TLR4 expression, we were able to show that expression of CD14 was induced 5-fold by Nissle 1917 and that this induction was dependent on the expression of a K5 capsule (Fig. 4). In addition, purified K5 polysaccharide alone could induce a 3-fold increase in CD14 expression (Fig. 4). CD14 acts as an opsonic receptor for LPS-dependent TLR4 signaling (24), such that an increase in CD14 may sensitize cells to the presence of LPS (24). The purified type 2 capsule of Streptococcus suis has been shown to increase expression of both CD14 and TLR2 (15). However, the effects on CD14 expression are much more modest than those seen here with the K5 capsule, with less-than-2-fold increases in both TLR2 and CD14 expression (15). Collectively, these data indicate that Nissle 1917 increases the expression of TLR4, TLR5, and CD14, with the induction of TLR5 and CD14 expression being dependent on the expression of a K5 capsule and with purified polysaccharide alone being able to induce the expression of these two molecules. To our knowledge, this is the first example of a capsular polysaccharide inducing increased expression of TLR5.
The purified K5 polysaccharide also potentiated the proinflammatory effects of flagellin (a TLR5 agonist) and LPS (a TLR4 agonist), with no evidence of a direct proinflammatory effect (Fig. 5). Such potentiation was completely abolished upon blocking of TLR4 and -5, respectively. Taken together, these results confirm the earlier reports indicating that the K5 polysaccharide has no direct proinflammatory effect (19) but that, rather, it potentiates the effect of TLRs agonists. In part, this potentiation might be explained by the observed upregulation of both TLR5 and the TLR4-associated molecule, CD14. The overall net effect is that the K5 polysaccharide on the surface of Nissle 1917 potentiates the activity of both TLR4 and -5 agonists to promote a proinflammatory response.
The role of TLR4 and TLR5 in mediating the K5 polysaccharide proinflammatory response of Nissle 1917 was demonstrated by blocking either TLR4 or TLR5 (Fig. 5). In the absence of neutralizing antibodies, Nissle 1917 was able to induce 13-fold more IL-8 expression than EcNK5−, the mutant lacking the K5 capsule, clearly demonstrating the role of the K5 capsule in this process. However, when either TLR4 or TLR5 was blocked, the ratio of IL-8 induction dropped to approximately 2.5-fold, and when both TLR4 and TLR5 were blocked at the same time, there was no difference in the abilities of Nissle 1917 and EcNK5− to induce IL-8. This clearly confirms that the ability of the K5 polysaccharide to potentiate the proinflammatory effect of Nissle 1917 is predominantly mediated via TLR4 and TLR5.
It is known that in mice, TLR4 plays an important role in the host defense against DSS-induced colitis and that hyaluronic acid (HA) can help preserve the epithelia through TLR4 activation (61). The observation that dimethylsulfoxide (DSS) increased endogenous HA synthesis is in keeping with a role for endogenous HA synthesis in protection from colitis (61). At this stage, we cannot state whether such a mechanism involving HA and TLR4 could be important in the protective and restorative effects of Nissle 1917 in treating colitis. However, our data showing the induction of TLR4 by Nissle 1917 and its ability to potentiate the TLR4 agonist LPS might suggest that this could be a possible mechanism by which Nissle 1917 ameliorates colitis.
TLR5-expressing lamina propria dendritic cells (LPDCs) have been identified as playing key roles in the regulation of humoral and cellular gut immunity (50, 51). Stimulation of LPDCs with flagellin induced differentiation of naïve B cells into IgA-producing plasma cells, as well as stimulating the differentiation of IL-17-producing T helper cells and type 1 T helper cells (50). On the basis of these findings and the data presented here, indicating that Nissle 1917 can stimulate expression of TLR5 on epithelial cells and potentiate the activity of TLR5 agonists, it is possible that Nissle 1917 may interact with LPDCs to stimulate TLR5 expression and potentiate TLR5 activation, thereby limiting bacterial infections by inducing a local IgA secretion. Likewise, the induction of TH-17 and TH-1 cells following TLR stimulation may modulate the pathogenesis of inflammatory bowel disease (49). Studies are currently under way in our laboratory to study possible interactions between Nissle 1917 and dendritic cells (DCs).
Infection with either Nissle 1917 or EcNK5− resulted in a significant induction of MyD88 gene expression, with a K5 capsule being required for maximal induction of MyD88 gene expression (Fig. 6). In the case of TRIF expression, infection with Nissle 1917 induced a large increase in expression and this induction was K5 capsule dependent (Fig. 6), with only a modest induction of TRIF gene expression detectable in cells infected with EcNK5−. These data indicate that Nissle 1917 stimulates expression of genes encoding adaptor proteins involved in the TLR4 and TLR5 signal transduction pathway and that TRIF expression requires a K5 capsule.
The signal transduction events following the internalization of the bound TLR complex lead to the activation of the IκB kinase α/β (IKKα/β) and/or MAPK pathways, leading to the activation of transcription factors NF-κΒ and AP-1, respectively (8, 21, 48). It is well known that transcriptional factor NF-κΒ plays a pivotal role in expression of LPS-induced inflammatory factors, leading to enhanced expression of proinflammatory cytokines, chemokines, and inflammatory enzymes (38). In unstimulated cells, NF-κΒ is sequestered in the cytoplasm through interaction with the inhibitory protein Iκβα. Following IKKβ kinase phosphorylation of Iκβα, NF-κβ is released and translocates into the nuclei, where it initiates gene transcription (38). It has previously been shown that infection of Caco-2 cells by Nissle 1917 increased expression of a number of inhibitors of NF-κΒ activation, such as PRDX4 and NF-κΒIA, suggesting that Nissle 1917 does not stimulate chemokine expression via the NF-κΒ pathway (52). Indeed, preliminary results from our laboratories show that infection of Caco-2 cells with Nissle 1917 leads to a 40-fold increase in the expression of NF-κΒIA and a 1,000-fold increase in TNF-αIP3 (M. Hafez and I. S. Roberts, unpublished results), both of which will inhibit activation of NF-κΒ (38). Overall, this would confirm that infection of Caco-2 cells with Nissle 1917 and chemokine induction are unlikely to proceed via the NF-κΒ-dependent pathway. This is in agreement with other published data that show that other probiotic bacteria are capable of attenuating the activation of the NF-κΒ pathway (23, 42, 59). In addition, it has been suggested that commensal bacteria may specifically prevent NF-κB activation by blocking ubiquitination of its inhibitor (Iκβα) (36). However, induction of expression of the human β-defensin gene by Nissle 1917 has shown to involve both JNK and NF-κΒ (56). It is possible that the induction of the NF-κΒ pathway in these experiments reflects differences in the experimental protocols used; in particular, in these experiments Nissle 1917 was heat killed and left for 24 h in contact with the Caco-2 cells (56). However, at this stage it is reasonable to conclude that interaction between Nissle 1917 and epithelial cells may induce a complex pattern of signal transduction.
As an alternative to the NF-κB pathway, chemokine induction may take place via MAPK-dependent pathways (29, 43, 58, 60). Three main families of MAPK exist in mammalian species: the extracellular signal-regulated protein kinases (ERKs), the p38 MAP kinases, and the c-Jun NH2-terminal kinases (JNKs) (58). The present study showed that Nissle 1917 clearly activated the three MAPK pathways. These results are in agreement with recent data showing that probiotic lactobacilli activate the MAPK pathway (4, 23). The increased MAPK activation by the capsulated Nissle 1917 strain compared to the level for its noncapsulated mutant EcNK5−, coupled with the observation that the addition of purified K5 polysaccharide to strain EcNK5− increased activation of all three MAPKs, clearly establishes a pivotal role for the K5 polysaccharide in mediating the activation of the MAPKs by Nissle 1917. The Y4 capsular polysaccharide of the dental pathogen Actinobacillus actinomycetemcomitans has also been shown to induce IL-1 expression via activation of the JNK pathway in human gingival fibroblasts (22).
To establish whether K5-mediated MAPK activation is linked to the proinflammatory effect of the capsule, chemokine induction in Caco-2 cells infected with either Nissle 1917 or EcNK5− after specific MAPK pathway inhibition was assessed (Table 1). In the absence of MAPK inhibitors, the Nissle/EcNK5−-mediated IL-8 induction ratio was 16.7. However, this ratio was significantly reduced (to between 3.8- and 6.9-fold) when specific MAPK inhibitors were included and reduced (to 2.4-fold) when all three MAPK pathways were inhibited simultaneously (Table 1). These results clearly indicate that all the three MAPK pathways are effectively contributing to the K5-mediated proinflammatory effect. It is noteworthy that the complete inhibition of the three MAPK pathways still allowed significantly higher levels of IL-8 expression with Nissle 1917 than with EcNK5−, implying that other MAPK-independent pathways may have minimal but significant contribution to the capsule-mediated proinflammatory effect. One possibility is that inhibition of the MAPK pathways results in some activation of the NF-κΒ pathway. The activation of the MAPK pathways by Nissle 1917 will contribute to its observed probiotic properties. It is known that Nissle 1917 inhibits gut leakage and promotes barrier function by enhancing tight junctions through increased ZO-1 expression (52) and that ZO-1 expression is ERK dependent (26). Likewise, among probiotic Lactobacillus species, MAPK-dependent signaling has been shown to be important in stimulating epithelial cell tight junctions and heat shock protein production (45, 49). As such, activation of the MAPK pathways by Nissle 1917 will have a pleiotropic effect on epithelial cell function.
The effects of the purified K5 polysaccharide on epithelial cells described in this paper add to the growing literature indicating that bacterial capsular polysaccharides are capable of interacting directly with host cells and moderating host inflammatory responses. Capsular polysaccharides have been shown to induce responses from host cells at either end of the inflammatory response. The Vi antigen of Salmonella enterica serotype Typhi reduces TLR-dependent IL-8 production and IL-17 secretion (40, 41), thereby reducing inflammation. Likewise, the PSA polysaccharide of Bacteroides fragilis has been shown to have potent anti-inflammatory effects (32). In contrast, capsular polysaccharides from Streptococcus pneumoniae, Staphylococcus aureus, and Porphyromonas gingivalis have all been shown to elicit inflammatory cytokines. (5, 12, 46). The observation that capsular polysaccharides can be released from the surfaces of bacteria raises the possibility that capsular polysaccharides could interact with host cells at sites distal to the site at which the encapsulated bacterium is in intimate contact with the host cell. In the host gut, with its large microbiome, it is possible that the capsular polysaccharide of one bacterium could influence the response of a host cell to another bacterium. It will be interesting to determine the effects that purified K5 polysaccharide has in mediating interactions between host cells and bacteria other than Nissle 1917.
In conclusion, in this paper we demonstrate that Nissle 1917 induces TLR4 and TLR5 expression in epithelial cells, with induction of TLR5 being potentiated by the K5 capsule, which alone was capable of increasing TLR5 expression. In addition, we show that Nissle 1917 upregulates TLR adaptor proteins and stimulates the three MAPK pathways in a K5-dependent fashion. These data add considerably to our understanding of the mechanism by which Nissle 1917 interacts with epithelial cells and reinforce the pivotal role played by the K5 capsule in mediating these interactions.
Acknowledgments
Work in the laboratory of Ian Roberts is supported by the BBSRC and MRC of the United Kingdom. The laboratory of Richard Grencis is supported by the Wellcome Trust. Mohamed Hafez gratefully acknowledges the award of a postdoctoral scholarship from the Ministry of Higher Education Egypt.
We thank U. Dobrindt for the generous gift of E. coli Nissle 1917 and David Corbett for help with the figures.
Editor: A. J. Bäumler
Footnotes
Published ahead of print on 9 February 2010.
REFERENCES
- 1.Akira, S., K. Takeda, and T. Kaisho. 2001. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2:675-680. [DOI] [PubMed] [Google Scholar]
- 2.Bambou, J. C., A. Giraud, S. Menard, B. Begue, S. Rakotobe, M. Heyman, F. Taddei, N. Cerf-Bensussan, and V. Gaboriau-Routhiau. 2004. In Vitro and ex Vivo Activation of the TLR5 signaling pathway in intestinal epithelial cells by a commensal Escherichia coli strain. J. Biol. Chem. 279(41):42984-42992. [DOI] [PubMed] [Google Scholar]
- 3.Biswas, S. K., P. Bist, M. K. Dhillon,. T. Kajiji, C. del Fresno, M. Yamamoto, E. Lopez-Collazo, S. Akira, and V. Tergaonkar. 2007. Role for MyD88-independent, TRIF pathway in lipid A/TLR4-induced endotoxin tolerance. J. Immunol. 179:4083-4092. [DOI] [PubMed] [Google Scholar]
- 4.Bleau, C., R. Savard, and R. Lamontagne. 2007. Murine immunomodulation of IL-10 and IL-12 induced by new isolates from avian type 2 Lactobacillus acidophilus. Can. J. Microbiol. 53:944-956. [DOI] [PubMed] [Google Scholar]
- 5.Bootsma, H. J., M. Egmont-Petersen, and P. W. Hermans. 2007. Analysis of the in vitro transcriptional response of human pharyngeal epithelial cells to adherent Streptococcus pneumoniae: evidence for a distinct response to encapsulated strains. Infect. Immun. 75:5489-5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudeau, J., A. L. Glasser, S. Julien, J. F. Colombel, and A. Darfeuille-Michaud. 2003. Inhibitory effect of probiotic Escherichia coli strain Nissle 1917 on adhesion to and invasion of intestinal epithelial cells by adherent-invasive E. coli strains isolated from patients with Crohn's disease. Aliment. Pharmacol. Ther. 18:45-56. [DOI] [PubMed] [Google Scholar]
- 7.Reference deleted.
- 8.Chang, L., and M. Karin. 2001. Mammalian MAP kinase signaling cascades. Nature 410:37-40. [DOI] [PubMed] [Google Scholar]
- 9.Clarke, B. R., F. Esumeh, and I. S. Roberts. 2000. Cloning, expression, and purification of the K5 capsular polysaccharide lyase (KflA) from coliphage K5A: evidence for two distinct K5 lyase enzymes. J. Bacteriol. 182:3761-3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cross, M. L., A. Ganner, D. Teilab, and L. M. Fray. 2004. Patterns of cytokine induction by gram-positive and gram-negative probiotic bacteria. FEMS Immunol. Med. Microbiol. 42:173-180. [DOI] [PubMed] [Google Scholar]
- 11.Cukrowska, B., R. Lodinova-Zadnikova, C. Enders, U. Sonnenborn, J. Schulze, and H. Tlaskalova-Hogenova. 2002. Specific proliferative and antibody responses of premature infants to intestinal colonization with non-pathogenic probiotic E. coli strain Nissle 1917. Scand. J. Immunol. 55:204-209. [DOI] [PubMed] [Google Scholar]
- 12.d'Empaire, G., M. T. Baer, and F. C. Gibson III. 2006. The K1 serotype capsular polysaccharide of Porphyromonas gingivalis elicits chemokine production from murine macrophages that facilitates cell migration. Infect. Immun. 74:6236-6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Furrie, E., S. Macfarlane, G. Thomson, and G. T. Macfarlane. 2005. Toll-like receptors-2, -3, and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology 115:565-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grabig, A., D. Paclik, C. Guzy, A. Dankof, D. C. Baumgart, J. Erckenbrecht, B. Raupach, U. Sonnenborn, J. Eckert, R. R. Schumann, B. Wiedenmann, A. U. Dignass, and A. Sturm. 2006. Escherichia coli strain Nissle 1917 ameliorates experimental colitis via Toll-like receptor 2- and Toll-like receptor 4-dependent pathways. Infect. Immun. 74:4075-4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graveline, R., M. Segura, D. Radzioch, and M. Gottschalk. 2007. TLR2-dependent recognition of Streptococcus suis is modulated by the presence of capsular polysaccharide which modifies macrophage responsiveness. Int. Immunol. 19(4):375-389. [DOI] [PubMed] [Google Scholar]
- 16.Gribar, S. C., R. J. Anand, C. P. Sodhi, and D. J. Hackam. 2008. The role of epithelial Toll-like receptor signaling in the pathogenesis of intestinal inflammation. J. Leukoc. Biol. 83(3):493-498. [DOI] [PubMed] [Google Scholar]
- 17.Grozdanov, L., C. Raasch, J. Schulze, U. Sonnenborn, G. Gottschalk, H. Hacker, and U. Dobrindt. 2004. Analysis of the genome structure of the nonpathogenic probiotic Escherichia coli strain Nissle 1917. J. Bacteriol. 186:5432-5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guzy, C., D. Paclik, A. Shirbel, U. Sonnenborn, B. Wiedenmann, and A. Sturm. 2008. The probiotic Escherichia coli strain Nissle 1917 induces γδ T cell apoptosis via caspase and FasL dependent pathways. Int. Immunol. 20:829-840. [DOI] [PubMed] [Google Scholar]
- 19.Hafez, M., K. Hayes, M. Goldrick, G. Warhurst, R. Grencis, and I. S. Roberts. 2009. The K5 capsule of Escherichia coli strain Nissle 1917 is important in mediating interactions between intestinal epithelial cells and chemokine induction. Infect. Immun. 77:2995-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henker, J., M. Laass, B. M. Blokhin, Y. K. Bolbot, V. G. Maydannik, M. Elze, C. Wolff, and J. Schulze. 2007. The probiotic Escherichia coli strain Nissle 1917 (EcN) stops acute diarrhoea in infants and toddlers. Eur. J. Pediatr. 166:311-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holtmann, H., R. Winzen, P. Holland, S. Eickemeier, E. Hoffmann, D. Wallach, N. L. Malinin, J. A. Cooper, K. Resch, and M. Kracht. 1999. Induction of interleukin-8 synthesis integrates effects of transcription and mRNA degradation from at least three different cytokine- or stress-activated signal transduction pathways. Mol. Cell. Biol. 19:6742-6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwata, T., A. Mitani, Y. Ishihara, S. Tanaka, G. Yamamoto, T. Kikuchi, T. Naganawa, Y. Matsumura, T. Suga, M. Koide, T. Sobue, T. Suzuki, and T. Noguchi. 2005. Actinobacillus actinomycetemcomitans Y4 capsular polysaccharide induces IL-1β mRNA expression through the JNK pathway in differentiated THP-1cells. Clin. Exp. Immunol. 141:261-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iyer, C., A. Kosters, G. Sethi, A. B. Kunnumakkara, B. B. Aggarwal, and J. Versalovic. 2008. Probiotic Lactobacillus reuteri promotes TNF-induced apoptosis in human myeloid leukemia-derived cells by modulation of NF-kappaB and MAPK signalling. Cell. Microbiol. 10:1442-1452. [DOI] [PubMed] [Google Scholar]
- 24.Jerala, R. 2007. Structural biology of LPS recognition. Int. J. Med. Microbiol. 297:353-363. [DOI] [PubMed] [Google Scholar]
- 25.Kelly, D., S. Conway, and R. Aminov. 2005. Commensal gut bacteria: mechanisms of immune modulation. Trends Immunol. 26:326-333. [DOI] [PubMed] [Google Scholar]
- 26.Ko, J. A., S. Murata, and T. Nishida. 2009. Up-regulation of the tight junction protein ZO-1 by substance P and IGF-1 in A431 cells. Cell Biochem. Funct. 27:388-394. [DOI] [PubMed] [Google Scholar]
- 27.Kruis, W., E. Schutz, P. Fric, B. Fixa, G. Judmaier, and M. Stolte. 1997. Double-blind comparison of an oral Escherichia coli preparation and mesalazine in maintaining remission of ulcerative colitis. Aliment. Pharmacol. Ther. 11:853-858. [DOI] [PubMed] [Google Scholar]
- 28.Kruis, W., P. Fric, J. Pokrotnieks, M. Lukas, B. Fixa, M. Kascak, M. A. Kamm, J. Weismueller, C. Beglinger, M. Stolte, C. Wolff, and J. Schulze. 2004. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 53:1617-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kyosseva, S. V. 2004. Mitogen-activated protein kinase signaling. Int. Rev. Neurobiol. 59:201-220. [DOI] [PubMed] [Google Scholar]
- 30.Lodinova-Zadnikova, R., and U. Sonnenborn. 1997. Effect of preventive administration of a nonpathogenic Escherichia coli strain on the colonization of the intestine with microbial pathogens in newborn infants. Biol. Neonate 71:224-232. [DOI] [PubMed] [Google Scholar]
- 31.Lundin, A., C. M. Bok, L. Aronsson, B. Björkholm, J. A. Gustafsson, S. Pott, V. Arulampalam, M. Hibberd, J. Rafter, and S. Pettersson. 2008. Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell. Microbiol. 10:1093-1103. [DOI] [PubMed] [Google Scholar]
- 32.Mazmanian, S. K., J. L. Round, and D. Kasper. 2008. A microbial symbiosis factor prevents inflammatory disease. Nature 453:620-625. [DOI] [PubMed] [Google Scholar]
- 33.Medzhitov, R. 2001. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1:135-145. [DOI] [PubMed] [Google Scholar]
- 34.Mollenbrink, M., and E. Bruckschen. 1994. Treatment of chronic constipation with physiologic Escherichia coli bacteria. Results of a clinical study of the effectiveness and tolerance of microbiological therapy with the E. coli Nissle 1917 strain (Mutaflor). Med. Klin. (Munich) 89:587-593. (In German.) [PubMed] [Google Scholar]
- 35.Reference deleted.
- 36.Neish, A. S., A. T. Gewirtz, H. Zeng, A. N. Young, M. E. Hobert, V. Karmali, A. S. Rao, and J. L. Madara. 2000. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science 289:1560-1563. [DOI] [PubMed] [Google Scholar]
- 37.Palm, N. W., and R. Medzhitov. 2009. Pattern recognition receptors and control of adaptive immunity. Immunol. Rev. 227:221-233. [DOI] [PubMed] [Google Scholar]
- 38.Pasparakis, M. 2008. IKK/NF-κΒ signaling in intestinal epithelial cells controls immune homeostasis in the gut. Mucosal Immunol. 1(Suppl. 1):S54-S57. [DOI] [PubMed] [Google Scholar]
- 39.Patzer, S. I., M. R. Baquero, D. Bravo, F. Moreno, and K. Hantke. 2003. The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors. Microbiology 149:2557-2570. [DOI] [PubMed] [Google Scholar]
- 40.Raffatellu, M., D. Chessa, R. P. Wilson, R. Dusold, S. Rubino, and A. J. Baumler. 2005. The Vi capsular antigen of Salmonella enterica serotype Typhi reduces Toll-like receptor-dependent interleukin-8 expression in the intestinal mucosa. Infect. Immun. 73:3367-3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raffatellu, M., R. L. Santos, D. Chessa, R. P. Wilson, S. E. Winter, C. A. Rossetti, S. D. Lawhon, H. Chu, T. Lau, C. L. Bevins, L. G. Bevins, and A. J. Baumler. 2007. The capsule encoding the viaB locus reduces interleukin-17 expression and mucosal innate responses in the bovine intestinal mucosa during infection with Salmonella enterica serotype Typhi. Infect. Immun. 75:4342-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riedel, C. U., F. Foata, D. Philippe, O. Adolfsson, B. J. Eikmanns, and S. Blum. 2006. Anti-inflammatory effects of bifidobacteria by inhibition of LPS-induced NF-κB activation. World J. Gastroenterol. 12(23):3729-3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaeffer, H. J., and M. J. Weber. 1999. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol. Cell. Biol. 19:2435-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schlee, M., J. Wehkamp, A. Altenhoefer, T. A. Oelschlaeger, E. F. Stange, and K. Fellermann. 2007. Induction of human B-defensin 2 by probiotoc Escherichia coli Nissle 1917 is mediated through flagellin. Infect. Immun. 75:2399-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seth, A., F. Yan, D. B. Polk, and R. K. Rao. 2008. Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a PKC- and MAP kinase-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 294:G1060-G1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soell, M., M. Diab, G. Haan-Archipoff, A. Beretz, C. Herbelin, B. Poutrel, and J. P. Klein. 1995. Capsular polysaccharide types 5 and 8 of Staphylococcus aureus bind specifically to human epithelial (KB) cells, endothelial cells, and monocytes and induce release of cytokines. Infect. Immun. 63:1380-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sturm, A., K. Rilling, D. C. Baumgart, K. Gargas, T. Abou-Ghazale, B. Raupach, J. Eckert, R. R. Schumann, C. Enders, U. Sonnenborn, B. Wiedenmann, and A. U. Dignass. 2005. Escherichia coli Nissle 1917 distinctively modulates T-cell cycling and expansion via Toll-like receptor 2 signaling. Infect. Immun. 73:1452-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takeda, K., T. Kaisho, and S. Akira. 2003. Toll-like receptors. Annu. Rev. Immunol. 21:335-376. [DOI] [PubMed] [Google Scholar]
- 49.Tao, Y., K. Drabik, T. S. Waypa, M. W. Musch, J. C. Alverdy, O. Schneewind, E. B. Chang, and E. O. Petrof. 2006. Soluble factors of Lactobacillus GG activate MAPKs and induce cytoprotective heat shock proteins in intestinal epithelial cells. Am. J. Physiol. Cell Physiol. 290:C1018-C1030. [DOI] [PubMed] [Google Scholar]
- 50.Uematsu, S., K. Fujimoto, M. H. Jang, B. G. Yang, Y. J. Jung, M. Nishiyama, S. Sato, T. Tsujimura, M. Yamamoto, Y. Yokota, H. Kiyono, M. Miyasaka, K. J. Ishii, and S. Akira. 2008. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 9:769-776. [DOI] [PubMed] [Google Scholar]
- 51.Uematsu, S., and S. Akira. 2009. Immune responses of TLR5+ lamina propria dendritic cells in enterobacterial infection. J. Gastroenterol. 44:803-811. [DOI] [PubMed] [Google Scholar]
- 52.Ukena, S. N., A. M. Westendorf, W. Hansen, M. Rohde, R. Geffers, S. Coldewey, S. Suerbaum, J. Buer, and F. Gunzer. 2005. The host response to the probiotic Escherichia coli strain Nissle 1917: specific up-regulation of the proinflammatory chemokine MCP-1. BMC Med. Genet. 6:43-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Underhill, D. M., and A. Ozinsky. 2002. Toll-like receptors: key mediators of microbe detection. Curr. Opin. Immunol. 14:103-110. [DOI] [PubMed] [Google Scholar]
- 54.Vizoso-Pinto, M. G., M. R. Gomez, S. Seifert, B. Watzl, W. H. Holzapfel, and C. M. A. P. Franz. 2009. Lactobacilli stimulate the innate immune response and modulate the TLR expression of HT29 intestinal epithelial cells in vitro. Int. J. Food Microbiol. 133:86-93. [DOI] [PubMed] [Google Scholar]
- 55.Voltan, S., I. Castagliuolo, M. Elli, S. Longo, P. Brun, R. D'Incà, A. Porzionato, V. Macchi, G. Palù, G. C. Sturniolo, L. Morelli, and D. Martines. 2007. Aggregating phenotype in Lactobacillus crispatus determines intestinal colonization and TLR2 and TLR4 modulation in murine colonic mucosa. Clin. Vaccine Immunol. 14:1138-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wehkamp, B. J., J. Harder, K. Wehkamp, B. W. von Meissner, M. Schlee, C. Enders, U. Sonnenborn, S. Nuding, S. Bengmark, K. Fellermann, J. M. Schroder, and E. F. Stang. 2004. NFB and AP-1-mediated induction of human beta defensin-2 in intestinal epithelial cells by Escherichia coli Nissle 1917: a novel effect of a probiotic. Infect. Immun. 72:5750-5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Westendorf, A. M., F. Gunzer, S. Deppenmeier, J. K. Hunger, M. A. Schmidt, and J. Buer. 2005. Intestinal immunity of E. coli Nissle 1917: a safe carrier for therapeutic molecules. FEMS Immunol. Med. Microbiol. 43:373-384. [DOI] [PubMed] [Google Scholar]
- 58.Whitmarsh, A. J., and R. J. Davis. 1996. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 74:589-607. [DOI] [PubMed] [Google Scholar]
- 59.Zhang, L., N. Li, R. Caicedo, and J. Neu. 2005. Alive and dead Lactobacillus rhamnosus GG decrease tumor necrosis factor-alpha-induced interleukin-8 production in Caco-2 cells. J. Nutr. 135:1752-1756. [DOI] [PubMed] [Google Scholar]
- 60.Zhang, Y., and C. Dong. 2005. MAP kinases in immune responses. Cell. Mol. Immunol. 2(1):20-27. [PubMed] [Google Scholar]
- 61.Zheng, L., T. E. Riehl, and W. F. Stenson. 2009. Regulation of colonic epithelial repair in mice by toll-like receptors and hyaluronic acid. Gastroenterology 137:2041-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zyrek, A. A., C. Cichon, S. Helms, C. Enders, U. Sonnenborn, and M. A. Schmidt. 2007. Molecular mechanisms underlying the probiotic effects of Escherichia coli Nissle 1917 involve ZO-2 and PKCz redistribution resulting in tight junction and epithelial barrier repair. Cell. Microbiol. 9:804-816. [DOI] [PubMed] [Google Scholar]