

Abstract
We report here that the enzymatic activity of phospholipase D2 (PLD2) is regulated by phosphorylation-dephosphorylation. Phosphatase treatment of PLD2-overexpressing cells showed a biphasic nature of changes in activity that indicated the existence of “activator” and “inhibitory” sites. We identified three kinases capable of phosphorylating PLD2 in vitro—epidermal growth factor receptor (EGFR), JAK3, and Src (with JAK3 reported for the first time in this study)—that phosphorylate an inhibitory, an activator, and an ambivalent (one that can yield either effect) site, respectively. Mass spectrometry analyses indicated the target of each of these kinases as Y296 for EGFR, Y415 for JAK3, and Y511 for Src. The extent to which each site is activated or inhibited depends on the cell type considered. In COS-7, cells that show the highest level of PLD2 activity, the Y415 is a prominent site, and JAK3 compensates the negative modulation by EGFR on Y296. In MCF-7, cells that show the lowest level of PLD2 activity, the converse is the case, with Y296 unable to compensate the positive modulation by Y415. MTLn3, with medium to low levels of lipase activity, show an intermediate pattern of regulation but closer to MCF-7 than to COS-7 cells. The negative effect of EGFR on the two cancer cell lines MTLn3 and MCF-7 is further proven by RNA silencing experiments that yield COS-7 showing lower PLD2 activity, and MTLn3 and MCF-7 cells showing an elevated activity. MCF-7 is a cancer cell line derived from a low-aggressive/invasive form of breast cancer that has relatively low levels of PLD activity. We propose that PLD2 activity is low in the breast cancer cell line MCF-7 because it is kept downregulated by tyrosyl phosphorylation of Y296 by EGFR kinase. Thus, phosphorylation of PLD2-Y296 could be the signal for lowering the level of PLD2 activity in transformed cells with low invasive capabilities.
Phospholipase D2 (PLD) catalyzes the hydrolysis of phosphatidylcholine to generate the lipid second messenger phosphatidic acid (PA) and choline. There are two mammalian isoforms: PLD1 (20) and PLD2 (12, 33). They share a highly conserved “HKD” domain necessary for catalysis (39). PLD has been implicated in a variety of physiological cellular functions, such as intracellular protein trafficking, cytoskeletal dynamics, membrane remodeling, and cell proliferation in mammalian cells and meiotic division and sporulation in yeast (6, 15, 16, 32, 42). PLD regulation in cells falls into two major signaling categories. One is by the small GTPase proteins Arf and Rho (3, 5, 11, 43), and the other is by growth factors/mitogens such as epidermal growth factor (EGF), platelet-derived growth factor, insulin, and serum that implicate tyrosine kinases (2, 12, 21, 22, 34). Several gaps in knowledge of the second arm remain in spite of its importance in cell signaling. The isoform PLD1 can be phosphorylated on tyrosine, but this does not lead to changes in activity (35, 36).
The isoform PLD2, which is expressed as a constitutively active enzyme in many cell types (47), can be detected as a phosphotyrosine protein both in vivo (25) and in vitro (17, 25, 41). PLD2 overexpression induces the transformation of cells (27, 34, 37, 48), and the activation of this enzyme enhances cellular processes favorable for the metastasis of EL4 lymphoma cells (30). PLD also activates STAT3 by interacting with the thyroid oncogenic kinase RET/PTC (29). PLD2 can form a complex with the EGF receptor (EGFR) (12, 46) or with Pyk2 and Src kinases (4). Hydrogen peroxide and EGF induce PLD phosphorylation and PKC-α activation (38), while activation of PLD by 8-Br-cAMP is accomplished through Src, Ras, and ERK (49).
Although all of these studies agree with respect to the presence of tyrosine kinases in PLD signaling, different conclusions were drawn regarding lipase activity. For example, whereas coexpression of PLD2 and Fyn or Fgr kinases lead to an enhancement of PLD activation and degranulation of mast cells (10), Ho et al. (23) noted that coexpression of Src and PLD1 results in an increased phosphorylation but no increase on its activity. Other authors have noted that the tyrosines Y11, Y14, Y165, and Y470 are important for PLD2 function (10) but, nevertheless, the overexpression of the phosphorylation-deficient mutant, PLD2-Y11F, increases PLD2 activity in resting or EGF-activated cells (12).
Closely related to the enhancing effect of phosphorylation-deficient mutants, a new line of research has also implicated tyrosine phosphatases in PLD, since PTP1B coimmunoprecipitates with PLD2 and Grb2 (24) through SH2 recognition sites on PLD2 which, in turn, bind to Sos (13, 14). To further connect PLD with the major pathways utilized by growth factors (Ras/MEK/ERK), Zhao et al. (51) demonstrated that PLD2-derived PA modulates Sos and Ras GTP/GDP exchange.
As just presented, fine work from leading laboratories over the past decade or so has provided a rich landscape of data for the molecular regulation of PLD2, clearly indicating that it is affected by tyrosyl phosphorylation and that the growth factor pathway is instrumental. However, the target(s) of phosphorylation within the PLD2 molecule that are key to its regulation have not been precisely mapped. We set out to define the parameters of tyrosyl regulation of PLD2 by identifying the targeted amino acids that could explain a putative increase in enzymatic activity. We have taken advantage of three different cell lines COS-7, MTLn3, and MCF-7 that express different levels of PLD activity, with the latter a breast cancer cell line, derived from a low-aggressive form of breast cancer, that has relatively low levels of PLD activity. We have identified three kinases capable of phosphorylating PLD2 (one newly discovered in the present study [JAK3]) and the phosphorylation sites as Y296, Y415, and Y511. We provide evidence that explains PLD2 activity regulation by phosphorylation-dephosphorylation, and we further show that PLD2 activity is low in MCF-7 cells because it is kept downregulated by tyrosyl phosphorylation on a specific residue by a specific kinase, which we have identified.
MATERIALS AND METHODS
Cells and plasmid transfections.
MCF-7 cells were cultured in Dulbecco modified Eagle medium (DMEM) plus 10% fetal calf serum (FCS), MTLn3 cells were cultured in α-MEM plus 5% FCS, and COS-7 cells were cultured in DMEM plus 10% newborn calf serum. When they reached ∼60% confluence, they were transfected with pcDNA3.1-mycPLD2 (0.3 ml of a lipid-DNA complex with 2 μg of pcDNA-mycPLD2 plasmid, 3 μl of Lipofectamine (Invitrogen, Carlsbad, CA), and 5 μl of Plus reagent (Invitrogen) in Opti-MEM (Invitrogen), previously mixed in sterile glass test tubes. After a 3-h incubation, the cells were washed and refed with the correct corresponding complete media. After 36 h, cells were harvested, and cell sonicates (∼0.8 mg of protein/ml) were immunoprecipitated with α-myc (9E-10) monoclonal antibody agarose matrix (Santa Cruz, Santa Cruz, CA).
Kinase reactions.
PLD2 was overexpressed in the three types of cells (MCF-7, MTLn3, or COS-7) for 36 h, and then lysates were prepared. The effect of kinases on PLD activity was measured according to two related experimental designs. In the first experimental design, PLD2 was immunoprecipitated with anti-myc antibodies and then the complex beads were mixed with the kinases. After the kinase reactions were completed, the products were taken for PLD activity assays. In the second experimental design, whole lysates were first mixed with kinases and, after the kinase reactions were completed, PLD was then immunoprecipitated with anti-myc antibodies; lastly, the complex beads were taken for PLD activity assays. In either case, before initiating the kinase reaction(s) each of the recombinant kinases was preincubated for 10 min at 30°C with cold ATP (100 μM) and 1 mM MgCl2, and the kinase reaction was allowed to proceed for 20 min at 30 to 37°C, in a rocking incubator. The three kinases (human sequences) were purchased from Millipore (Danvers, MA) or Active Motif (Carlsbad, CA) as truncated/active kinases (molecular masses were 86, 68.4, and 61.7 kDa for EGFR, Src, and JAK3, respectively). Kinase reactions (50 μl, containing each 500 pM concentrations of the active kinase) were performed according to manufacturers' protocols (Millipore, Danvers, MA, or Active Motif, Carlsbad, CA) and contained 25-μl aliquots of lysates from PLD2-overexpressing cells and reagents as listed in manufacturer's protocols (for instance, Src kinase buffer [20 mM morpholinepropanesulfonic acid-NaOH [pH 7.0], 0.1 mM EDTA, 0.01% Brij 35, 0.1% β-mercaptoethanol, 1 g of bovine serum albumin/ml, 5% glycerol]). For the immunoprecipitation step, α-myc antibodies bound to agarose were used (0.5 μg of antibody/reaction). Immunocomplexes were allowed to be formed at 4°C for 2 h. They were centrifuged and then washed twice with special lysis buffer (5 mM HEPES [pH 7.8], 100 μM sodium orthovanadate, 0.4% Triton X-100) and 5 μg of leupeptin and aprotinin/ml and then split into two 50-μl aliquots for duplicate determinations in the PLD2 assay.
PLD activity assay.
Immunocomplex samples were processed for PLD2 activity in PC8 liposomes and n-[3H]butanol, beginning with the addition of the following reagents (final concentrations): 3.5 mM PC8 phospholipid, 45 mM HEPES (pH 7.8), and 1.0 μCi of n-[3H]butanol in a liposome form (as indicated in reference 31). Samples were incubated for 20 min at 30°C with continuous shaking. The addition of 0.3 ml of ice-cold chloroform-methanol (1:2) stopped the reactions. Lipids were then isolated and resolved by thin-layer chromatography (TLC). TLC plates were developed in the upper phase of a ethyl acetate-iso-octane-acetic acid-water (4.3:0.6:1:3.3 [vol/vol/vol/vol]) mixture. The amount of [3H]PBut that comigrated with PBut standards was measured by scintillation spectrometry. Control reactions lacking PC8 were used to remove background counts.
Phosphate incorporation into kinases, autoradiography, and Western blotting.
PLD2-α-myc-agarose-bound immunocomplexes from cell lysates were used for measurement of PO4 incorporation into PLD2 as a substrate for the various kinases. After activation with [32P]ATP, 50-μl kinase reactions were split into two equal volumes, and 25-μl portions of each were spotted onto Whatman P81 filter paper for duplicate determinations. Filters were washed in running water to remove unincorporated substrate, dried, and liquid scintillation counted. Sodium dodecyl sulfate (SDS)-gel/autoradiography of EGFR phosphorylated PLD2 was performed. After kinase activations with or without [32P]ATP, the reactions were incubated at 30°C for 10 min with constant gentle shaking, and reactions were stopped by adding 12.5 μl of 4× SDS sample loading buffer. Proteins were electrophoresed and transferred onto polyvinylidene difluoride (PVDF) membrane. Radioactive, phosphorylated PLD2 was detected by autoradiography. For Western blotting of EGFR phosphorylated PLD2 and after kinase activations with or without 100 μM cold ATP, reactions were electrophoresed and transferred onto PVDF membrane. Nonradioactive, phosphorylated PLD2 was detected by using α-myc rabbit antibodies.
Detection of PLD2 phosphopeptides by mass spectrometry.
Lysates from COS-7 cells overexpressing PLD2 were immunoprecipitated with α-myc antibodies, phosphorylated, and run on SDS gels. Protein bands in the region of 105 ± 5 kDa were excised from the gels and trypsinized “in-gel.” Briefly, samples were heat denatured, followed by overnight trypsin digestion at a ratio (wt/wt) of 20 (sample) to 1 (trypsin). Samples were then acidified with trifluoroacetic acid to stop the digestion. The resulting tryptic peptides were separated by reversed-phase chromatography (C18 microbore column, 0.2 by 115 mm) with a gradient of 2 to 60% acetonitrile over 100 min. The eluted samples from the high-pressure liquid chromatography (HPLC) column were analyzed by a time-of-flight/mass spectrometry/electrospray approach (TOF-MS-ES) in a Micromass Q-TOF micro from Waters by Commonwealth Biotechnologies, Inc. (Richmond, VA). After this, an accurate mass analysis was performed to compare all parent ions to the theoretical tryptic digest of PLD2 with Biolynx software.
Generation and study of phosphorylation-deficient mutants.
YF point mutants were created from wt-PLD2 and transfected into MCF-7, MTLn3, or COS-7 cells. PLD2 α-myc-agarose immunocomplexes were used to measure the PLD activity after transfection. The construct pcDNA-mycPLD2 (33) was used as a template to create three independent YF substitutions (PLD2-Y415F, PLD2-Y296F, and PLD2-Y511F) according to the QuikChange XL site-directed mutagenesis protocol (Stratagene, La Jolla, CA), using the following mutagenesis primers: Y415F-forward, GGCATCAACAGTGGCTTTAGCAAGAGGGCG; Y415F-reverse, CGCCCTCTTGCTAAAGCCACTGTTGATGCC; Y296F-forward, CTCAAGTGCAGCAGCTTTCGGCAGGCACGGTGG; Y296F-reverse, CCACCGTGCCTGCCGAAAGCTGCTGCACTTGAG; Y511F-forward, GGCTGGGCAAGGACTTCAGCAATCTTATCACC; and Y511F-reverse, GGTGATAAGATTGCTGAAGTCCTTGCCCAGCC. All oligonucleotide primers were PAGE/HPLC purified (Integrated DNA Technologies, Coralville, IA). The molecular identity of all mutants was confirmed by direct sequence analysis (Agencourt Bioscience Corp., Beverly, MA).
Gene silencing.
For the EGFR gene, we used small interfering RNAs (siRNAs) synthesized with two deoxythymidine 3′ overhangs [d(TT)-3′], obtained from Ambion (Austin, TX) (“Select validated”), that targeted exon 10 [locus s563; sense, 5′-GAAUAGGUAUUGGUGAAUUd(TT)-3′]. For JAK3, we used siRNAs that targeted exon 19 [locus s7653; sense, 5′-GUAUCGUGGUGUCAGCUAUd(TT)-3′]. For Src, we used siRNAs that targeted exon 12 [locus s223346; sense, 5′-ACAAUUUCGUGCAUCGAGAd(TT)-3′]. The protocol for gene silencing involved transfection of 100 or 200 nM siRNAs into COS-7, MCF-7, or mTLN3 cells using Lipofectamine 2000 and the requisite cell culture media containing fetal calf serum. Transfection reactions were allowed to incubate at 37°C overnight, at which time the cells were washed and refed with complete medium minus antibiotics. Cells were allowed to be silenced for an additional 36 h, which then yielded 96 h of total gene silencing. Confirmation of gene silenced was achieved by Western blotting.
Statistical analysis.
Data are presented as means ± the standard error of the mean (SEM). The difference between means was assessed by the single-factor analysis of variance (ANOVA) test. A probability (P) of <0.05 was considered to indicate a significant difference.
RESULTS
Inhibition of tyrosine kinases lead to enhanced PLD2 activity in vivo.
Transfection of COS-7 cells with a pcDNA-mycPLD2a-WT construct led to the expression of a recombinant PLD2 protein and concomitant activity that was measurable starting at ∼12 h (Fig. 1A). As cells were allowed longer times for the expression of PLD2, the activity gradually increased. This is in agreement with early observations that PLD2 is expressed in cells with a constitutively high enzymatic activity (47). To ascertain whether this effect was specific to COS-7 or more universal, we extended the experiment to two other cell lines: the lung cancer cell line MTLn3 and the breast cancer cell line MCF-7 which, like COS-7, are of epithelioid origin. As seen in Fig. 1B and C, the activity of overexpressing cells also increased with the time after transfection, but the maximum activity for MTLn3 was ∼5,000 dpm/mg of protein and for MCF-7 was only ∼3,000 dpm/mg of protein (compared to ∼9,000 dpm/mg of protein for COS-7 cells, Fig. 1A). PLD activity is frequently elevated in breast and other cancer cell lines, such as MDA-MB-231 (8). However, the MCF-7 cell line, derived from a less aggressive form of breast cancer, has relatively low levels of PLD activity (52). The data presented in Fig. 1B indicate that MTLn3 cells also display lower basal activity than COS-7 cells. Consequently, and in addition to the low endogenous or basal level, we were also able to document here the consistently lower level of PLD activity of the MCF-7 and MTLn3 cell lines overexpressing mycPLD2WT protein. This is lower relative to the noncancerous COS-7 cells that exhibit high PLD activity when transfected with the same construct.
FIG. 1.

Expression of a PLD2 constitutively active in COS-7, MTLn3, and MCF-7 cells. Either COS-7 (A), MTLn3 (B), or MCF-7 (C) cells were transfected with pcDNA-mycPLD2-WT and divided into several sets. Each set was incubated with 0.1 μg/ml of genistein for the indicated times. Regardless of those variable lengths of time, all cells were allowed a full 36-h time for overexpression of PLD2. Overexpressed mycPLD2 was immunoprecipitated with 9E10 monoclonal antibody agarose matrix and assayed for lipase activity as indicated above. Enzyme reaction units in the y axes was calculated in relation to PLD2 protein, so comparison between the cell lines is possible. The results represent means ± the SEM of three independent experiments in duplicate. ANOVA tests were performed on the means and SEM of the different points and, when statistically significant differences (P < 0.05) were found, the points are denoted with an asterisk.
Regardless of the basal level of PLD2 activity in overexpressing cells, when the tyrosine kinase inhibitor genistein was included in the culture medium continuously since the moment of transfection, the basal level of PLD2 increased (Fig. 1). This was somewhat surprising, since earlier reports have indicated that PLD2 is upregulated by tyrosine phosphorylation. Since this experiment was conducted in a living cell, genistein could have affected a kinase that phosphorylates PLD2, or it could have affected globally the biosynthesis of this or other related proteins. Regardless, the results shown in Fig. 1 warranted further investigation as PLD2 could be regulated by dephosphorylation.
Protein tyrosine phosphatase CD45 and PTP1B dually modulate PLD2 activity in vitro.
We reasoned that if PLD2 was upregulated by inhibition of tyrosine kinases, then phosphatases would also elevate its activity. Treatment of PLD2 from COS-7, MTLn3, and MCF-7 cells with the phosphatases CD45 or PTP1B resulted in a complex pattern of activity regulation (Fig. 2). Whereas concentrations of PTP of 0.3 to 1 μg/ml in COS-7 cells or 5 to 10 μg/ml in MTLn3 or MCF-7 cells indeed enhanced PLD activity, concentrations of >10 μg/ml of either tyrosine phosphatase (but particularly PTP1B) were inhibitory. This indicates a dual regulation of PLD2 by phosphorylation-dephosphorylation, and upon close inspection of the biphasic patterns of cells of Fig. 2, a different behavior for COS-7 on one hand and for MTLn3 and MCF-7 on the other. The biphasic nature of the changes in Fig. 2 can be explained by the existence of sites on PLD2 that, once phosphorylated, can exert positive or negative functional effects on the lipase activity. We call these “activator” or “inhibitory” sites, respectively. It is possible that with low concentrations of phosphatases the inhibitory site(s) is dephosphorylated and the enzyme gains activity. With higher concentrations of phosphatases, both the inhibitory and the activator sites are dephosphorylated, and the enzyme loses activity completely. The difference in the concentration of phosphatases for COS-7 and MTLn3/MCF-7 is due to the way the two sites are regulated in these cells, as will be made apparent below. To further clarify the biphasic nature of the PLD regulation, a model for this is presented in Fig. 9.
FIG. 2.

Biphasic regulation of PLD2 activity by tyrosine phosphatases in vitro. Either MCF-7, MTLn3, or COS-7 cells were transfected with pcDNA3.1-mycPLD2 for 36 h. Cell lysates were treated with the indicated concentrations of CD45 (A) or PTP1B (B) and incubated for 30 min at 30°C. Samples were then processed for PLD2 activity in PC8 liposomes and n-[3H]butanol. The results represent means ± the SEM of four independent experiments in duplicate. ANOVA tests were performed on the means and SEM of the different points comparing to controls and, when statistically significant differences (P < 0.05) were found, these points were denoted by “*” or “#”, respectively, for values above or below the basal level.
FIG. 9.

Model highlighting the biological significance of the various findings from the present study. (A) Kinase action on PLD2. EGFR, JAK3, and Src are capable of phosphorylating PLD2 in vitro, and the targets are Y296 for EGFR, Y415 for JAK3, and Y511 for Src. EGFR phosphorylates an “inhibitory site,” JAK3 phosphorylates an “activator site,” and Src phosphorylates an “ambivalent site” (one that can yield either effect). The extent to which each site is activated or inhibited depends on the cell type considered. In COS-7 cells, which bear the highest level of PLD2 activity, the Y415 is a prominent site that, by being phosphorylated by JAK3, compensates for the negative modulation by EGFR on Y296. In MCF-7 cells, which show the lowest level of PLD2 activity, the converse is true, with Y296 being unable to compensate for the positive modulation by Y415. MTLn3 cells, with medium or low levels of lipase activity, show an intermediate pattern of regulation but one that is closer to that of MCF-7 cells than that of COS-7 cells. (B) Phosphatase action on PLD2. Low concentrations of phosphatases or phosphatases targeting Y511 or Y296 leave the enzyme regulated positively by the “activator” sites and thus with high levels of lipase activity. Conversely, with higher concentrations of phosphatases or with phosphatases targeting Y415 (the activator site), the enzyme loses activity to different extents based on how heavily the cell relies on activator or inhibitory sites. We propose that Y296, an inhibitory site in MCF-7 cancer cells, is the reason for the observed low lipase activity in them, and this underscores the importance of this site for regulation of PLD2.
Effect of EGFR, Src, and JAK3 kinases on PLD2 activity.
In the next series of experiments we sought to investigate the effect of specific tyrosine kinases on PLD2 activity in vitro, with the first challenge being which tyrosine kinases to choose among the many known to regulate the pathways utilized by growth factors. Based on the current literature, the likely tyrosine kinase candidates regarding PLD2 phosphorylation, activation, and mast cell degranulation include EGFR, which is known to phosphorylate PLD2 (12), or the Src family of kinases Fyn and Fgr, as demonstrated by Choi et al. (10). Moreover, Ahn et al. (2) have observed a physical interaction between the pleckstrin homology domain of PLD2 and c-Src, which provides a molecular basis for the interaction of PLD2 and its kinase. Our own tyrosine kinase consensus sites search based on PhosphoBase and Swiss-Prot (through KinasePhos [http://kinasephos.mbc.nctu.edu.tw/index.html]) yielded four potential phosphorylation sites for Src/Syk and five possible sites for EGFR. In addition to this, the search also yielded six possible sites for JAK3 kinase phosphorylation with E-values of >100. Thus, we tested the hypothesis that EGFR, Src, and JAK3 would phosphorylate PLD2 and modulate its activity.
To prove this, we set up two interrelated experimental designs for the study of in vitro phosphorylation of PLD2. In the first experimental design, PLD2 was immunoprecipitated with anti-myc antibodies, and then the complex beads were mixed with the kinases. After the kinase reactions were completed, the products were taken for PLD activity assays. Figure 3A to C shows that EGFR and Src kinases are inhibitory of PLD2 in all three cell types, whereas JAK3 causes no effect. The inhibitory effects of these kinases confirm the activating effects seen with the phosphatase treatments of Fig. 2. However, Fig. 2 shows a biphasic pattern, indicating also the presence of activator sites on PLD2, which cannot be seen in panels A to C. We have reasoned that this is because positive effectors for these sites are lost during immunoprecipitation of intact cells. To gain a better knowledge of this, a second experimental approach was necessary. In this one, whole lysates were first mixed with kinases and after the kinase reactions were completed, PLD was then immunoprecipitated with anti-myc antibodies; lastly, the complex beads were taken for PLD activity assays.
FIG. 3.

Differential effect of EGFR, Src, and JAK3 kinases on PLD2 activity. COS-7 (A and D), MTLn3 (B and E), or MCF-7 (C and F) cells (transfected with myc-PLD2 or mock transfected) were used for in vitro kinase reactions with EGFR, JAK3, or Src. In panels A to C, PLD2 was immunoprecipitated with anti-myc antibodies, and then the complex beads were mixed with the kinases. After the kinase reactions were completed, the products were taken for PLD activity assays. In panels D to F, whole lysates were first mixed with kinases and, after the kinase reactions were completed, PLD was immunoprecipitated with anti-myc antibodies. Finally, the complex beads were taken for PLD activity assays. In either case, the samples were split into two 50-μl aliquots for duplicate determinations in the PLD2 assay. The results represent means ± the SEM of four independent experiments in duplicate. *, differences between means (no-kinase versus kinase treatments) that were statistically significant (P < 0.05) as determined by ANOVA.
Figure 3D to F shows that EGFR kinase continues being inhibitory for the cancer cell lines MTLn3 and MCF-7, but JAK3 and Src have now an activating effect on PLD2. Thus, EGFR phosphorylates an inhibitory site, JAK3 phosphorylates an activator site, and Src phosphorylates an ambivalent site (one that can yield either effect). A model for this is presented in Fig. 9. We can only speculate at this point on the nature of the conditions that exist in lysates that enable the full effect of JAK3 and Src, but it could be because transfection of PLD2 causes an elevation of the expression of JAK3 itself (data not shown). In summary, JAK3 is an overall activating kinase for PLD2 inasmuch as EGFR is an inhibitory kinase for PLD2 of cancer cells.
Identification of specific amino acid residues on PLD2, targeted by the kinases.
To precisely decipher the detailed mechanism, down to the molecular level, of how PLD2 is regulated by phosphorylation, we needed precise identification of the phosphorylation sites with their corresponding kinases. For this, we used proteomics-based techniques that required phosphate incorporation on PLD2 by the chosen kinases. We demonstrated this by either direct radioactivity liquid scintillation counting (Fig. 4A) or by autoradiography (Fig. 4B). The latter figure indicates that a phospho-PLD2 band, the result of EGFR action, was present at the expected molecular mass of PLD2, which was confirmed in Western blots with α-myc antibodies (Fig. 4C). Figure 4D also shows that the other two kinases, Src and JAK3, were also effective in producing a phospho-PLD2 band. Thus, EGFR, Src, and JAK3 resulted in specific phosphorylation of PLD2. This begged the question as to where on PLD2 the phosphate was incorporated.
FIG. 4.

Investigation of the targets of phosphorylation. (A) Phosphate was incorporated into COS-7 PLD2 by the action of EGFR, Src, or JAK3 kinases in vitro. PLD2 α-myc-agarose-bound immunocomplexes were used for measurement of PO4 incorporation into PLD2. The results represent means ± the SEM of three independent experiments in duplicate. *, differences between means that were statistically significant (P < 0.05) as determined by ANOVA. (B) SDS-gel/autoradiograph of EGFR phosphorylated PLD2. Following kinase activations with or without [32P]ATP, proteins were electrophoresed and transferred onto PVDF membranes. Radioactive, phosphorylated PLD2 was detected by autoradiography. (C) Western blotting of EGFR phosphorylated PLD2. Nonradioactive, phosphorylated PLD2 was detected by using α-myc rabbit antibodies. (D) Representative autoradiograph of Src (first panel) or JAK3 (second panel). A representative Western blot of mycPLD2 (third panel) is also shown to indicate equal loading of the gels.
To shed light onto this subject, we used LC-MS analyses to prove the presence of PLD2-peptides. After in vitro phosphorylation of immunoprecipitated PLD2, gel-size selection and tryptic digestion, peptides were subjected first to liquid chromatography by reverse phase HPLC (Fig. 5A) and then analyzed by TOF-MS-ES (Fig. 5B). The MS spectrum recorded several peptide peaks and ions of interest for the present study at m/z = 900 to 1,300 were marked as T40 to T42 for samples that had been previously phosphorylated by EGFR; T61-62 and T61 for samples that had been previously phosphorylated by Src; and T52-53, T53, T53-55, and T51-53 for samples that had been previously phosphorylated by JAK3. The masses were compared to proteins in the Swiss-Prot databank with Biolynx software. The actual phosphopeptides had molecular masses within ±0.03% of the calculated masses of the corresponding theoretically predicted PLD2 tryptic peptides (Fig. 5C). Of the potential 15 possible sites that were theoretically predicted to be phosphorylated by KinasePhos, we found by MS analysis that Y296, Y415, and Y511 were targets of phosphorylation. This does not necessarily indicate that EGFR, Src, or JAK3 kinases specifically target the newly found sites exclusively.
FIG. 5.

Detection of PLD2 phosphopeptides by MS. (A) HPLC. Lysates from COS-7 cells overexpressing PLD2 were immunoprecipitated with α-myc antibodies, phosphorylated, and run on SDS gels. Protein bands in the region of 105 ± 5 kDa were excised from the gels and trypsinized “in-gel.” The resulting tryptic peptides were separated by reversed-phase chromatography as shown. (B) The eluted samples from the HPLC column were analyzed by TOF-MS-ES. After this, an accurate mass analysis was performed to compare all parent ions to the theoretical tryptic digest of PLD2 using Biolynx software. A section of the spectrogram with an m/z of ∼1,100 for samples phosphorylated by Src is shown. (C) Identification of phosphorylated peptides. The compilation indicates m/z of samples that were positive for ions corresponding to phosphate adducts with double or triple charges and compared to the parent ions with the sequences of PLD2 (NCBI accession no. O14939). Also indicated are the sites most likely recognized for each of the kinases used in the present study (EGFR, Src, and JAK3).
Investigation of the role of each site with phosphorylation-deficient mutants.
To investigate what specific role each of the three newly identified sites would play in the mechanism of PLD2 phosphorylation and activation, we generated PLD2 mutants with Y→F point substitutions on the Y296, Y415, and Y511 sites (Fig. 6). The conclusions of the data presented in Fig. 6 are as follows. First, residues Y415 and Y511 are essential to PLD2 activity, regardless of the cell line used, since these two Y→F substitutions rendered PLD2 inactive (Fig. 6A to C). Second, Y296 regulates COS-7 and MTLn3 and MCF-7 oppositely: prevention of phosphorylation decreases PLD2 activity in COS-7, whereas it increases PLD2 activity in the cancer cells. This is the first detection of an inhibitory site for PLD2 phosphorylation: Y296. Based on this, we hypothesized that phosphorylation of Y296 is what keeps MTLn3 and MCF-7 at a low basal level of activity (as found in Fig. 1 and 3).
FIG. 6.
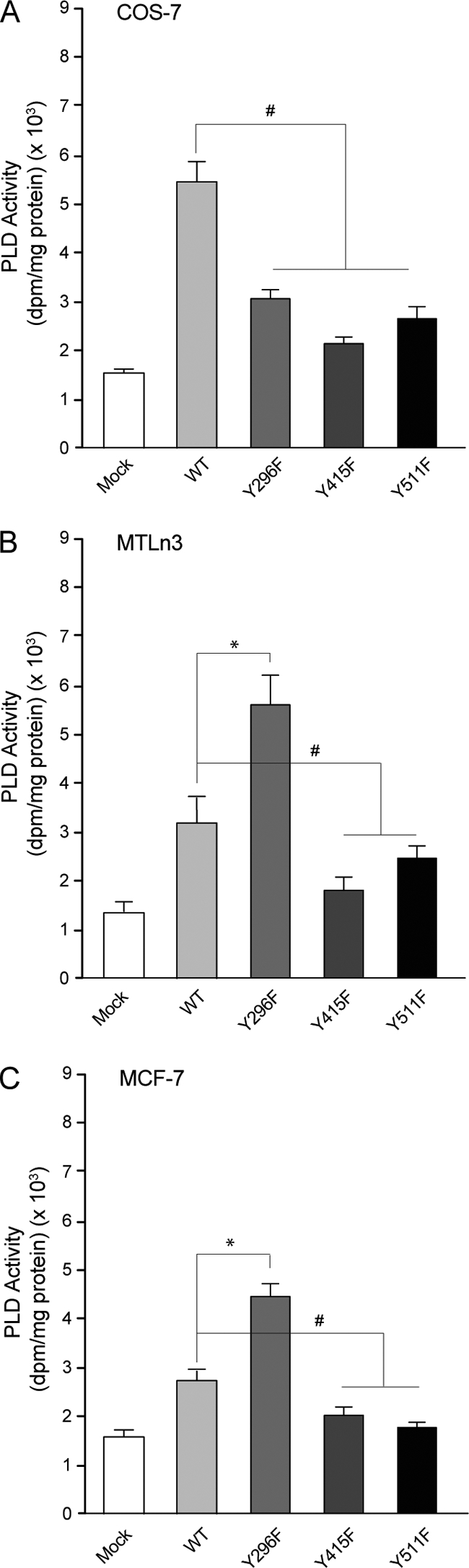
Study of lipase activity for the three phosphorylation-incapable mutants. Phosphorylation-incapable PLD2-Y296F, PLD2-Y415F, and PLD2-Y511F, Y→F point mutants were generated by site-directed mutagenesis by using myc-pcDNA-PLD2-WT as a template. Each one of these mutants alone or the wild-type PLD2 (“WT”) or mock reagents was transfected into COS-7 (A), MTLn3 (B), or MCF-7 (C) cells. After 36 h, cells were harvested, and whole lysates were prepared from overexpressing cells and mixed in kinase buffer with either EGFR, JAK3, or Src kinases. After this, PLD2's were immunoprecipitated with α-myc antibodies. The PLD2-α-myc-agarose immunocomplexes were used for measurement of PLD activity in PC8 liposomes and n-[3H]butanol. * and #, differences between means, as determined by ANOVA, above and below, respectively, the wild-type levels that were statistically significant (P < 0.05).
The next series of experiments addressed which residue each of the kinases would phosphorylate. As indicated in Fig. 7, the addition of either EGFR, JAK3, or Src to any of the cells, which specifically overexpressed either PLD2-Y296F (Fig. 7A, D, and G), PLD2-Y511F (Fig. 7C, F, and I), or PLD2-Y415F (Fig. 7B, E, and H), respectively, had no effect on PLD2 activity. This is to be expected, since according to Fig. 5C, EGFR's consensus site recognizes Y296, JAK3's consensus site recognizes Y415, and JAK3's consensus site recognizes Y511. These sites theoretically cannot be phosphorylated in these Y→F mutants. Surprisingly, JAK3 or Src caused a substantial elevation of PLD2 activity in cells transfected with PLD2-Y296F (Fig. 7A, D, and G), indicating that when Y296 can no longer be utilized by EGFR, other kinases are then able to use PLD2 more efficiently as a substrate, which yields greater PLD activity. A similar, albeit less robust, situation for JAK3 and Src was observed with the other mutants, PLD2-Y415F and PLD2-511F; it was clear that JAK3 and Src worked at different levels than EGFR and can phosphorylate Y415 and Y511, respectively, although probably not exclusively.
FIG. 7.

Effect of exogenous kinases on the PLD activity of phosphorylation-deficient mutants. PLD2-Y296F (A, D, and G), PLD2-Y415F (B, E, and H), and PLD2-Y511F (C, F, and I) PLD2 mutants were transfected into COS-7 (A to C), MTLn3 (D to F), or MCF-7 (G to I) cells. After 36 h of expression, lysates were used for in vitro kinase reactions with EGFR, JAK3, or Src. After this, PLD2 was immunoprecipitated with α-myc antibodies, and samples were split into two 50-μl aliquots for duplicate determinations in the PLD2 assay. The results represent means ± the SEM of four independent experiments in duplicate. *, differences between means (no-kinase versus kinase treatments) that were statistically significant (P < 0.05) as determined by ANOVA.
The last series of experiments involved gene expression silencing for the three kinases at hand. As presented in Fig. 8, silencing of EGFR resulted in decreased PLD2 activity in COS-7 but was increased in MTLn3 cells and, to an even greater extent, in MCF-7 cells. This reinforces the kinase data presented in Fig. 7. Silencing Jak3 and Src results in decreased PLD activity in all cell lines tested. The extent of silencing varied with the cell line used, as indicated in the companion Western blots Fig. 8, but was in general 60 to 80% at concentrations of 200 nM siRNAs.
FIG. 8.

Silencing endogenous kinases and status of PLD activity. EGFR, JAK3, or Src -siRNAs were transfected into COS-7 (A-C), MTLn3 (D-F) or MCF-7 (G-I) for 2 days. After this, duplicate cell samples were used to generate cell lysates for PLD activity assays and for the detection of protein expression by Western blotting. For the latter, blots were cut in half; the upper part (containing an ∼175-kDa region, an ∼115-kDA region, or an ∼60-kDA region) was probed with the appropriate anti-kinase antibodies, while the lower part (containing an ∼50-kDa region) was probed with anti-β-actin to ascertain equivalent protein loading.
In conclusion, the present study shows that phosphorylation of Y415 and Y511 residues are essential for activity in COS-7, MTLn3, and MCF-7 cells. JAK3 and Src phosphorylate these sites, although other kinases might also phosphorylate them. PLD2-Y296 is a specific target of EGFR kinase. Y296 is an activator site in COS-7, whereas it is an inhibitory site in the cancer cells MTLn3 and MCF-7. We propose that PLD2-Y296 is an inhibitory site in these cancer cells and responsible for the observed low PLD activity in them, particularly in the low-aggressive/invasive form of breast cancer cell line MCF-7.
DISCUSSION
We have established that PLD2 activity is regulated by phosphorylation-dephosphorylation and have identified three of the kinases involved, EGFR and Src (already known to play a role in PLD2 modulation) and JAK3 (newly discovered here), as well as the specific sites of action: Y296, Y415, and Y511. Also, and thanks to the use of three cell lines and their comparison, we learned the reason why PLD2 activity is kept low in cancer cells. Experiments with phosphatases (Fig. 2) indicated the existence of “activator” and “inhibitory” sites that would be confirmed later on. Low concentrations of phosphatases or phosphatases targeting Y511 or Y296 leave the enzyme regulated positively by the “activator” sites and thus with high levels of lipase activity. Conversely, with higher concentrations of phosphatases or with phosphatases targeting Y415, the activator sites, the enzyme loses activity at different extents based on how heavily the cell relies on activator or inhibitory sites.
The dephosphorylation obtained in vitro could be extrapolated, at least theoretically, to an in vivo situation. Dephosphorylation is normally accomplished in vivo as EGFR, platelet-derived growth factor receptor (PDG-R), and insulin receptor are all known to associate with tyrosine phosphatases. A well-studied example is the cytoplasmic protein tyrosine phosphatase Shp-2 that serves to propagate signal transduction proximal to EGFR and enhance information flow from the receptor leading to cell growth and development (44). Another example is protein tyrosyl phosphatase 1B (PTP1B), which is able to dephosphorylate substrates of the epidermal growth factor receptor and can regulate its functions in vivo (18). Our laboratory has shown earlier that phosphorylated PLD2 complexes with Grb2 (14) and with PTP1B (24) through two newly defined SH2 recognition sites on PLD2 that in turn bind to Sos (13). Sos binds to PA and, through the Ras GTP/GDP exchange, the PLD system is tied to the Ras/MEK/ERK pathway (19, 51).
PLD2 is expressed with a high enzymatic activity that cannot be significantly activated above those levels either in vitro or in vivo. This suggests that PLD2 is regulated by inhibitory mechanisms, and the prime example for that is provided in the present study. It will be beneficial for a normal cell to have a controlling mechanism to shut down or slow down the production of PA, a pleiotropic second messenger with potentially harmful effects to the cell if unchecked. We know that inhibition of PLD-derived PA production inhibits migration of adenocarcinoma cells (1, 8). In this line of reasoning, we present here for the first time three phosphorylation-incapable mutants that can be an index of a new PLD2 regulatory mechanism. Phosphorylation and dephosphorylation of Y296 could be the signal for altering PLD activity and concomitant proliferation and transformation levels.
The use of cancer cells, particularly MCF-7, has been instrumental in deciphering the complex regulation of PLD2 phosphorylation in the present study, since it offered results in contraposition to COS-7 cells. The use of MCF-7 has also led to an understanding of why these cells bear a low PLD2 activity. High levels of PLD are found in many human cancers (26, 40, 50). Cell proliferation by Ras oncogenes is functionally linked to PLD activation (7), and PLD2 is the target of aberrant transcription of the Ewing's sarcoma fusion protein (28). PLD activity is frequently elevated in breast and other cancer cell lines, such as MDA-MB-231 (8). However, the MCF-7 cell line, derived from a less aggressive form of breast cancer, has relatively low levels of PLD activity (52). MCF-7 and MDA-MB-231 are estrogen receptor-positive (ER+) and -negative (ER−) cells, respectively.
The Foster group has proposed that PLD induces the phenotype change from ER+ to ER− that coincides with greater invasiveness of tumors and that PLD is a signal for survival in serum-deprived cells (8, 9, 45, 52). In addition to this, we show the existence of a low-activity PLD2 due to tyrosine phosphorylation of Y296 by EGFR kinase. It is possible that a transition to a higher PLD activity would involve the removal of the inhibition caused on Y296. The work presented here is consistent with Zheng et al. (52), whereby elevated PLD2 expression in MCF-7 cells made them somewhat more mobile (migrating and invasive), but was still much less invasive than MDA-MB-231 breast cancer cells. Our results with PLD2-Y296F are also consistent with another phosphorylation-deficient mutant (mouse PLD2-Y11F), which was found to cause a robust increase in PLD2 activity in the presence or absence of EGF (12).
In summary, the results on the present study indicate that there are at least three kinases capable of phosphorylating PLD2 in vitro: EGFR, JAK3, and Src. The model presented in Fig. 9 attempts to pull together the results of the present study in a comprehensive picture that explains the complex interplay between kinases, phosphatases and the levels of enzymatic activity of PLD2. Figure 9A indicates that the extent to which each site is activated or inhibited depends on the cell type considered. In COS-7, cells that show the highest level of PLD2 activity, the Y415 is a prominent site that by being phosphorylated by JAK3 compensates the negative modulation by EGFR on Y296. In MCF-7, cells that show the lowest level of PLD2 activity, the converse is true, with Y296 unable to compensate the positive modulation by Y415. MTLn3, with medium/low levels of lipase activity, show an intermediate pattern of regulation but closer to MCF-7 than to COS-7 cells. Figure 9B indicates that the enzymatic activity of PLD2 is regulated by phosphorylation-dephosphorylation in a complex or biphasic way. We are proposing the existence of sites on PLD2 that once phosphorylated can exert positive or negative functional effects on the lipase activity. We called these “activator” or “inhibitory” sites, respectively. EGFR phosphorylates an inhibitory site (Y296), JAK3 phosphorylates an activator site (Y415), and Src phosphorylates an ambivalent site (one that can yield either effect) (Y511).
Even though we initiated the present study to explain the relationship between phosphorylation and activity of PLD2 and discover the complex pattern of phosphorylation-dephosphorylation, the use of the three cell lines (two of which are the cancer cell lines MTLn3 and MCF-3) has provided additional and important insights. MCF-7 is a cancer cell line derived from a low-aggressive/invasive form of breast cancer that has relatively low levels of PLD activity. This low level of activity is increased by JAK3 and Src treatment also in vitro. We propose that PLD2 activity is low in the breast cancer cell line MCF-7 and in MTLn3 cells because it is kept downregulated by tyrosyl phosphorylation of Y296 by EGFR kinase. Thus, phosphorylation of PLD2-Y296 could be the signal for lowering the level of PLD2 activity, which is associated with a delayed onset of cell transformation.
Acknowledgments
We thank Steven J. Berberich (Wright State University School of Medicine, Dayton, OH) for providing the MCF-7 cell line and Jeff Segall (Albert Einstein College of Medicine, Yeshiva University, NY) for providing the MTLn3 cell line. We also thank Steven J. Berberich and David Foster (Hunter College, CUNY) for helpful suggestions.
This study has been supported by the National Institutes of Health grant HL056653 (J.G.-C.).
Footnotes
Published ahead of print on 22 February 2010.
REFERENCES
- 1.Aguirre Ghiso, J. A., E. F. Farias, D. F. Alonso, C. Arregui, and E. Bal de Kier Joffe. 1997. A phospholipase D and protein kinase C inhibitor blocks the spreading of murine mammary adenocarcinoma cells altering f-actin and beta1-integrin point contact distribution. Int. J. Cancer 71:881-890. [DOI] [PubMed] [Google Scholar]
- 2.Ahn, B. H., S. Y. Kim, E. H. Kim, K. S. Choi, T. K. Kwon, Y. H. Lee, J. S. Chang, M. S. Kim, Y.-H. Jo, and D. S. Min. 2003. Transmodulation between phospholipase D and c-Src enhances cell proliferation. Mol. Cell. Biol. 23:3103-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bae, C. D., D. S. Min, I. N. Fleming, and J. H. Exton. 1998. Determination of interaction sites on the small G protein RhoA for phospholipase D. J. Biol. Chem. 273:11596-11604. [DOI] [PubMed] [Google Scholar]
- 4.Banno, Y., K. Ohguchi, N. Matsumoto, M. Koda, M. Ueda, A. Hara, I. Dikic, and Y. Nozawa. 2005. Implication of phospholipase D2 in oxidant-induced phosphoinositide 3-kinase signaling via Pyk2 activation in PC12 cells. J. Biol. Chem. 280:16319-16324. [DOI] [PubMed] [Google Scholar]
- 5.Brown, H. A., S. Gutowski, C. R. Moomaw, C. Slaughter, and P. C. Sternweis. 1993. ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell 75:1137-1144. [DOI] [PubMed] [Google Scholar]
- 6.Brown, H. A., L. G. Henage, A. M. Preininger, Y. Xiang, and J. H. Exton. 2007. Biochemical analysis of phospholipase D. Methods Enzymol. 434:49-87. [DOI] [PubMed] [Google Scholar]
- 7.Carnero, A., A. Cuadrado, L. del Peso, and J. C. Lacal. 1994. Activation of type D phospholipase by serum stimulation and ras-induced transformation in NIH3T3 cells. Oncogene 9:1387-1395. [PubMed] [Google Scholar]
- 8.Chen, Y., Y. Zheng, and D. A. Foster. 2003. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 22:3937-3942. [DOI] [PubMed] [Google Scholar]
- 9.Chen, Y., V. Rodrik, and D. A. Foster. 2005. Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene 24:672-679. [DOI] [PubMed] [Google Scholar]
- 10.Choi, W. S., T. Hiragun, J. H. Lee, Y. M. Kim, H. P. Kim, A. Chahdi, E. Her, J. W. Han, and M. A. Beaven. 2004. Activation of RBL-2H3 mast cells is dependent on tyrosine phosphorylation of phospholipase D2 by Fyn and Fgr. Mol. Cell. Biol. 24:6980-6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cockcroft, S., G. M. Thomas, A. Fensome, B. Geny, E. Cunningham, I. Gout, I. Hiles, N. F. Totty, O. Truong, and J. J. Hsuan. 1994. Phospholipase D: a downstream effector of ARF in granulocytes. Science 263:523-526. [DOI] [PubMed] [Google Scholar]
- 12.Colley, W. C., T. C. Sung, R. Roll, J. Jenco, S. M. Hammond, Y. Altshuller, D. Bar-Sagi, A. J. Morris, and M. A. Frohman. 1997. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr. Biol. 7:191-201. [DOI] [PubMed] [Google Scholar]
- 13.Di Fulvio, M., N. Lehman, X. Lin, I. Lopez, and J. Gomez-Cambronero. 2006. The elucidation of novel SH2 binding sites on PLD2. Oncogene 25:3032-3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Fulvio, M., K. Frondorf, K. M. Henkels, N. Lehman, and J. Gomez-Cambronero. 2007. The Grb2/PLD2 interaction is essential for lipase activity, intracellular localization and signaling in response to EGF. J. Mol. Biol. 367:814-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Exton, J. H. 1999. Regulation of phospholipase D. Biochim. Biophys. Acta 1439:121-133. [DOI] [PubMed] [Google Scholar]
- 16.Gomez-Cambronero, J. 1995. Immunoprecipitation of a phospholipase D activity with antiphosphotyrosine antibodies. J. Interferon Cytokine Res. 15:877-885. [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Cambronero, J., J. Horn, C. C. Paul, and M. A. Baumann. 2003. Granulocyte-macrophage colony stimulating factor is a chemoattractant cytokine for human neutrophils: involvement of the ribosomal p70 S6 kinase signaling pathway. J. Immunol. 171:6846-6855. [DOI] [PubMed] [Google Scholar]
- 18.Gunaratne, P., C. Stoscheck, R. E. Gates, L. Li, L. B. Nanney, and L. E. King. 1994. Protein tyrosyl phosphatase-1B is expressed by normal human epidermis, keratinocytes, and A-431 cells and dephosphorylates substrates of the epidermal growth factor receptor. J. Invest. Dermatol. 103:701-706. [DOI] [PubMed] [Google Scholar]
- 19.Hammond, S. M., J. M. Jenco, S. Nakashima, K. Cadwallader, Q. Gu, S. Cook, Y. Nozawa, G. D. Prestwich, M. A. Frohman, and A. J. Morris. 1997. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J. Biol. Chem. 272:3860-3868. [DOI] [PubMed] [Google Scholar]
- 20.Han, J. M., Y. Kim, J. S. Lee, C. S. Lee, B. D. Lee, M. Ohba, T. Kuroki, P. G. Suh, and S. H. Ryu. 2002. Localization of phospholipase D1 to caveolin-enriched membrane via palmitoylation: implications for epidermal growth factor signaling. Mol. Biol. Cell 13:3976-3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hess, J. A., A. H. Ross, R. G. Qiu, M. Symons, and J. H. Exton. 1997. Role of Rho family proteins in phospholipase D activation by growth factors. J. Biol. Chem. 272:1615-1620. [DOI] [PubMed] [Google Scholar]
- 22.Ho, W. T., Z. Xie, Z. J. Zhao, and J. H. Exton. 2005. Tyrosine phosphorylation of phospholipase D1 by v-Src does not per se result in activation. Cell. Signal. 17:691-699. [DOI] [PubMed] [Google Scholar]
- 23.Horn, J., I. Lopez, M. W. Miller, and J. Gomez-Cambronero. 2005. The uncovering of a novel regulatory mechanism for PLD2: formation of a ternary complex with protein tyrosine phosphatase PTP1B and growth factor receptor-bound protein GRB2. Biochem. Biophys. Res. Commun. 332:58-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houle, M. G., and S. Bourgoin. 1999. Regulation of phospholipase D by phosphorylation-dependent mechanisms. Biochim. Biophys. Acta 1439:135-149. [DOI] [PubMed] [Google Scholar]
- 25.Hui, L., Y. Zheng, Y. Yan, J. Bargonetti, and D. A. Foster. 2006. (Mutant p53 in MDA-MB-231 breast cancer cells is stabilized by elevated phospholipase D activity and contributes to survival signals generated by phospholipase D. Oncogene 25:7305-7310. [DOI] [PubMed] [Google Scholar]
- 26.Joseph, T., R. Wooden, A. Bryant, M. Zhong, Z. Lu, and D. A. Foster. 2001. Transformation of cells overexpressing a tyrosine kinase by phospholipase D1 and D2. Biochem. Biophys. Res. Commun. 289:1019-1024. [DOI] [PubMed] [Google Scholar]
- 27.Kikuchi, R., M. Murakami, S. Sobue, T. Iwasaki, K. Hagiwara, A. Takagi, T. Kojima, H. Asano, M. Suzuki, Y. Banno, Y. Nozawa, and T. Murate. 2007. Ewing's sarcoma fusion protein, EWS/Fli-1 and Fli1 protein induce PLD2 but not PLD1 gene expression by binding to an ETS domain of 5′ promoter. Oncogene 26:1802-1810. [DOI] [PubMed] [Google Scholar]
- 28.Kim, Y. R., H. S. Byun, M. Won, K. A. Park, J. M. Kim, B. L. Choi, H. Lee, J. H. Hong, J. Park, J. H. Seok, D. W. Kim, M. Shong, S. K. Park, and G. M. Hur. 2008. Modulatory role of phospholipase D in the activation of signal transducer and activator of transcription (STAT)-3 by thyroid oncogenic kinase RET/PTC. BMC Cancer 8:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knoepp, S. M., M. S. Chahal, Y. Xie, Z. Zhang, D. J. Brauner, M. A. Hallman, S. A. Robinson, S. Han, M. Imai, S. Tomlinson, and K. E. Meier. 2008. Effects of active and inactive phospholipase D2 on signal transduction, adhesion, migration, invasion, and metastasis in EL4 lymphoma cells. Mol. Pharmacol. 74:574-584. [DOI] [PubMed] [Google Scholar]
- 30.Lehman, N., M. Di Fulvio, N. McCray, I. Campos, F. Tabatabaian, and J. Gomez-Cambronero. 2006. Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. Blood 108:3564-3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liscovitch, M., M. Czarny, G. Fiucci, and X. Tang. 2000. Phospholipase D: molecular and cell biology of a novel gene family. Biochem. J. 345:401-415. [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez, I., R. S. Arnold, and J. D. Lambeth. 1998. Cloning and initial characterization of a human phospholipase D2 (hPLD2): ADP-ribosylation factor regulates hPLD2. J. Biol. Chem. 273:1284612852. [DOI] [PubMed] [Google Scholar]
- 33.Lu, Z., A. Hornia, T. Joseph, T. Sukezane, P. Frankel, M. Zhong, S. Bychenok, L. Xu, L. A. Feig, and D. A. Foster. 2000. Phospholipase D and RalA cooperate with the epidermal growth factor receptor to transform 3Y1 rat fibroblasts. Mol. Cell. Biol. 20:462-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcil, J., D. Harbour, P. H. Naccache, and S. Burgoin. 1997. Human phospholipase D1 can be tyrosine phosphorylated in HL-60 granulocytes. J. Biol. Chem. 272:20660-20664. [DOI] [PubMed] [Google Scholar]
- 35.Min, D. S., E.-G. Kim, and J. H. Exton. 1998. Involvement of tyrosine phosphorylation and protein kinase C in the activation of phospholipase D by H2O2 in Swiss 3T3 fibroblasts. J. Biol. Chem. 273:29986-29994. [DOI] [PubMed] [Google Scholar]
- 36.Min, D. S., B. H. Ahn, and Y. H. Jo. 2001. Differential tyrosine phosphorylation of phospholipase D isozymes by hydrogen peroxide and the epidermal growth factor in A431 epidermoid carcinoma cells. Mol. Cells 11:369-378. [PubMed] [Google Scholar]
- 37.Min, D. S., T. K. Kwon, W. S. Park, J. S. Chang, S. K. Park, B. H. Ahn, Z. Y. Ryoo, Y. H. Lee, Y. S. Lee, D. J. Rhie, S. H. Yoon, S. J. Hahn, M. S. Kim, and Y. H. Jo. 2001. Neoplastic transformation and tumorigenesis associated with overexpression of phospholipase D isozymes in cultured murine fibroblasts. Carcinogenesis 22:1641-1647. [DOI] [PubMed] [Google Scholar]
- 38.Morris, A., J. Engebrecht, and M. A. Frohman. 1996. Structure and regulation of phospholipase D. Trends Pharmacol. Sci. 17:182-185. [DOI] [PubMed] [Google Scholar]
- 39.Pannequin, J., N. Delaunay, C. Darido, T. Maurice, P. Crespy, M. A. Frohman, M. S. Balda, K. Matter, D. Joubert, J. F. Bourgaux, J. P. Bali, and F. Hollande. 2007. Phosphatidyl ethanol accumulation promotes intestinal hyperplasia by inducing ZONAB-mediated cell density increase in response to chronic ethanol exposure. Mol. Cancer Res. 5:1147-1157. [DOI] [PubMed] [Google Scholar]
- 40.Pochet, S., M. Mètioui, K. Grosfils, A. Góm̂ez-Muñoz, A. Marino, and J. P. Dehaye. 2003. Regulation of phospholipase D by muscarinic receptors in rat submandibular ductal cells. Cell. Signal. 15:103-113. [DOI] [PubMed] [Google Scholar]
- 41.Powner, D. J., T. R. Pettitt, and M. J. Wakelam. 2005. Assays to study phospholipase D regulation by inositol phospholipids and ADP-ribosylation factor 6. Methods Enzymol. 404:398-410. [DOI] [PubMed] [Google Scholar]
- 42.Preininger, A. M., L. G. Henage, W. M. Oldham, E. J. Yoon, H. E. Hamm, and H. A. Brown. 2006. Direct modulation of phospholipase D activity by Gβγ. Mol. Pharmacol. 70:311-318. [DOI] [PubMed] [Google Scholar]
- 43.Prelich, G., and B. Stillman. 1998. Coordinated leading and lagging strand synthesis during SV40 DNA replication in vitro requires PCNA. Cell 53:117-126. [DOI] [PubMed] [Google Scholar]
- 44.Qu, C. K., W. M. Yu, B. Azzarelli, and G. S. Feng. 1999. Genetic evidence that Shp-2 tyrosine phosphatase is a signal enhancer of the epidermal growth factor receptor in mammals. Proc. Natl. Acad. Sci. U. S. A. 96:8528-8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodrik, V., Y. Zheng, F. Harrow, Y. Chen, and D. A. Foster. 2005. Survival signals generated by estrogen and phospholipase D in MCF-7 breast cancer cells are dependent on Myc. Mol. Cell. Biol. 25:7917-7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Slaaby, R., T. Jensen, H. S. Hansen, M. A. Frohman, and K. Seedorf. 1998. PLD2 complexes with the EGF receptor and undergoes tyrosine phosphorylation at a single site upon agonist stimulation. J. Biol. Chem. 273:33722-33727. [DOI] [PubMed] [Google Scholar]
- 47.Xu, L., Y. Shen, T. Joseph, A. Bryant, J. Q. Luo, P. Frankel, T. Rotunda, and D. A. Foster. 2000. Mitogenic phospholipase D activity is restricted to caveolin-enriched membrane microdomains. Biochem. Biophys. Res. Commun. 273:77-83. [DOI] [PubMed] [Google Scholar]
- 48.Yoon, M. S., J. B. Koo, J. H. Hwang, K. S. Lee, and J. S. Han. 2005. Activation of phospholipase D by 8-BrcAMP occurs through novel pathway involving Src, Ras, and ERK in human endometrial stromal cells. FEBS Lett. 579:5635-5642. [DOI] [PubMed] [Google Scholar]
- 49.Zhao, Y., H. Ehara, Y. Akao, M. Shamoto, Y. Nakagawa, Y. Banno, T. Deguchi, N. Ohishi, K. Yagi, and Y. Nozawa. 2000. Increased activity and intranuclear expression of phospholipase D2 in human renal cancer. Biochem. Biophys. Res. Commun. 278:140-143. [DOI] [PubMed] [Google Scholar]
- 50.Zhao, C., G. Du, K. Skowronek, M. A. Frohman, and D. Bar-Sagi. 2007. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 9:706-712. [DOI] [PubMed] [Google Scholar]
- 51.Zheng, Y., V. Rodrik, A. Toschi, M. Shi, L. Hui, Y. Shen, and D. A. Foster. 2006. Phospholipase D couples survival and migration signals in stress response of human cancer cells. J. Biol. Chem. 281:15862-15868. [DOI] [PubMed] [Google Scholar]
- 52.Zhong, M., Y. Shen, Y. Zheng, T. Joseph, D. Jackson, S. Beychenok, and D. A. Foster. 2003. Phospholipase D prevents apoptosis in v-Src-transformed rat fibroblasts and MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 302:615-619. [DOI] [PubMed] [Google Scholar]