

Abstract
Colistin is increasingly being utilized against Gram-negative pathogens, including Pseudomonas aeruginosa, resistant to all other antibiotics. Since limited data exist regarding killing by colistin at different initial inocula (CFUo), we evaluated killing of Pseudomonas aeruginosa by colistin at several CFUo and developed a mechanism-based mathematical model accommodating a range of CFUo. In vitro time-kill experiments were performed using ≥8 concentrations up to 64 × the MIC of colistin against P. aeruginosa PAO1 and two clinical P. aeruginosa isolates at CFUo of 106, 108, and 109 CFU/ml. Serial samples up to 24 h were simultaneously modeled in the NONMEM VI (results shown) and S-ADAPT software programs. The mathematical model was prospectively “validated” by additional time-kill studies assessing the effect of Ca2+ and Mg2+ on killing of PAO1 by colistin. Against PAO1, killing of the susceptible population was 23-fold slower at the 109 CFUo and 6-fold slower at the 108 CFUo than at the 106 CFUo. The model comprised three populations with different second-order killing rate constants (5.72, 0.369, and 0.00210 liters/h/mg). Bacteria were assumed to release signal molecules stimulating a phenotypic change that inhibits killing. The proposed mechanism-based model had a good predictive performance, could describe killing by colistin for all three studied strains and for two literature studies, and performed well in a prospective validation with various concentrations of Ca2+ and Mg2+. The extent and rate of killing of P. aeruginosa by colistin were markedly decreased at high CFUo compared to those at low CFUo. This was well described by a mechanism-based mathematical model, which should be further validated using dynamic in vitro models.
During the last decades, important strides have been made in our understanding of antimicrobial pharmacodynamics (PD) (16, 21). Identification of the pharmacokinetic/pharmacodynamic (PKPD) index that best predicts a successful outcome became a cornerstone of optimization of antimicrobial therapy (2, 16, 21). However, PKPD target values may vary between antibiotics and pathogens (2, 3) and may not be universal for all infections (27) and types of outcomes (44, 59). For example, severe infections, such as endocarditis, infections of prostheses, and ventilator-associated pneumonia are known to have a high density of bacteria, which is associated with a higher mortality (25, 47, 54, 60). Furthermore, an increased density of bacteria represents a greater challenge to the immune system and may result in higher bacterial toxin concentrations. In addition, bacterial killing by antibiotics has been shown to be attenuated at high initial inocula (CFUo) compared to low CFUo (9, 10, 47, 53, 67, 69).
Colistin, also known as polymyxin E, was introduced to the market nearly half a century ago (40). Due to the increasing prevalence of Pseudomonas aeruginosa strains resistant to virtually all other commercially available antibiotics, polymyxins are now being increasingly utilized as salvage therapy against infections due to multidrug-resistant Gram-negative pathogens, including P. aeruginosa (40, 42).
As the initial site of action, the polycationic polymyxin molecules displace Mg2+ and Ca2+ ions that cross-bridge between adjacent negatively charged phosphate groups of lipopolysaccharides (LPS) in the outer membrane of Gram-negative bacteria (19, 33, 39, 48, 61). High concentrations of Mg2+ and Ca2+ can competitively inhibit the effects of polymyxins (48, 55, 56). Changes in the composition of LPS reducing the net negative charge of the outer membrane and modifying the hydrophobic side chain have been proposed as a mechanism of resistance to polymyxins (14, 36). P. aeruginosa can sense certain environmental conditions that cause adaptive resistance to polymyxins (49, 51).
Infections by P. aeruginosa are especially difficult to treat if they are deep seated or sequestered or have a biofilm matrix at high bacterial densities (15, 28, 71), and infections with a high CFUo often carry a high number of resistant mutants. To limit the emergence of resistance, optimizing the PKPD of antimicrobials to combat infections with a high CFUo is therefore of the utmost importance. Adler and Finland (1) reported higher MICs for polymyxin B when tested at high compared CFUo to low CFUo, and Peyret et al. (52) and Tam et al. (65) found a lower rate of killing of P. aeruginosa by polymyxin B against high versus low CFUo. We are not aware of studies on the impact of CFUo on the rate and extent of killing by colistin. To our knowledge, there are no published mathematical models for the rate and extent of killing by polymyxins. We proposed and evaluated the first inoculum effect model studying ceftazidime against P. aeruginosa (10-11; J. B. Bulitta, J. C. Yang, B. T. Tsuji, W. J. Jusko, and A. Forrest, presented at the post-ICAAC symposium of the International Society of Anti-Infective Pharmacology, Chicago, IL, 20 September 2007), and subsequently, Udekwu et al. (70) proposed a simulation model with one subpopulation to describe the inoculum effect.
The objectives of the current study were to evaluate the impact of CFUo on the rate and extent of killing of P. aeruginosa by colistin and to develop and evaluate a mechanism-based mathematical PD model that can describe killing by colistin over a range of CFUo.
(This work was presented in part as a poster [no. A-6] at the 47th Interscience Conference of Antimicrobial Agents and Chemotherapy, Chicago, IL, 2007 [73], in part as an oral presentation at the post-ICAAC meeting of the International Society of Anti-Infective Pharmacology in Chicago, IL, 2007, in part as a poster [abstr. 1829] at the AAPS Annual Meeting, San Diego, CA, 2007 [11], and in part as a poster [no. A-119] at the 109th General Meeting of the American Society for Microbiology, Philadelphia, PA, 2009 (74)).
MATERIALS AND METHODS
Bacterial strains, media, and MICs.
Three isolates of P. aeruginosa were studied: a genetically characterized clinical isolate, PAO1, obtained from the R.E.W. Hancock Laboratory at the University of British Columbia, Vancouver, Canada (62), and two clinical isolates, URMC1 and URMC2, obtained from the University of Rochester Medical Center, Rochester, NY. Fresh solutions of colistin sulfate (Sigma-Aldrich, St. Louis, MO), dissolved in sterile water, were prepared prior to each experimental run. Luria-Bertani (LB) broth supplemented with Ca2+ (25 mg/liter) and Mg2+ (12.5 mg/liter) was utilized for all time-kill experiments. Bacterial counts were determined on LB agar. Luria-Bertani broth and agar were selected as rich media to facilitate growth at high CFUo in time-kill experiments (30). The MICs were determined by broth microdilution in quadruplicate using colistin sulfate according to Clinical Laboratory Standards Institute guidelines in cation-adjusted Mueller-Hinton broth (MHB) or cation-adjusted LB broth. The MICs in MHB (LB) broth were 2.0 (4.0) mg/liter for PAO1 and 0.5 (1.0) mg/liter for URMC1 and for URMC2.
Time-kill experiments.
Fresh bacterial colonies from an overnight growth were added to normal saline and adjusted spectrophotometrically to provide a standard suspension. This suspension was diluted with LB broth to achieve the desired CFUo. Each 20-ml culture was incubated in a water bath at 37°C with constant shaking. Initial inocula were 104, 106, 108, and 109 CFU/ml for PAO1 and 106, 108, and 109 CFU/ml for URMC1 and URMC2. Bacteria were allowed to grow for approximately 45 min in LB broth before colistin was added from a standard antibiotic stock solution to achieve 11 colistin concentrations up to 256 mg/liter colistin (64 × MIC in LB broth) for PAO1 and to achieve nine colistin concentrations up to 64 mg/liter colistin (64 × MIC in LB broth) for strains URMC1 and URMC2 at each CFUo.
Samples were collected for determination of bacterial counts at the following time points: 0, 0.25, 0.5, 1, 2, 3, 4, 8, 12, 16, and 24 h for PAO1 for initial characterization of colistin pharmacodynamics and 0, 0.5, 1, 2, 4, 6, 8, and 24 h for URMC1 and URMC2. Antimicrobial carryover was taken into account by serial dilution (10- to 100,000-fold) and centrifuging the bacterial samples for 5 min, after which they were reconstituted with sterile normal saline to their original volumes. Colony counts were determined as previously described (68) using an automated spiral dispenser. Plates were incubated for 18 to 24 h. To determine quantitative viable counts of less-susceptible cells for PAO1, bacterial suspensions at 24 h were centrifuged and resuspended in saline, and 200 μl of this suspension was manually plated on LB agar containing 0, 2, 4, 8, or 16 mg/liter colistin (MIC of PAO1 on LB agar, 2 mg/liter). Drug-containing plates were incubated for 48 h at 35°C. All time-kill experiments were performed in duplicate.
Time-kill experiments to assess potential drug binding or degradation.
Additional experiments were performed to evaluate the potential effect of binding of colistin to LPS or other molecules in spent broth as a potential mechanism to account for the inoculum effect. A CFUo of PAO1 was grown from 106 to 109.4 CFU/ml over 24 h in cation-adjusted LB broth. Then, 16 or 64 mg/liter colistin or no drug was added to this suspension (without filtration of the suspension). After 0.75 or 24 h of coincubation at 37°C, the suspension was centrifuged and filter sterilized (0.22-μm filter). Fresh PAO1 cells in log-growth phase were added to the filtrate to achieve a CFUo of 106.2 CFU/ml. No additional colistin was added to the filtrate. Viable counts were recorded over an additional 24 h.
Time-kill experiments to support the target site model.
Time-kill experiments to assess the effect of Ca2+ and Mg2+ on the activity of colistin against PAO1 were performed as described above. For samples with low colony counts, 200 μl of the bacterial suspensions in saline were plated to increase the sensitivity of the viable counts. EDTA (pH adjusted to 7.0) as a chelating agent, Ca2+, or Mg2+ was added to LB broth to achieve the following concentrations of added cations or EDTA: (i) 20 mM EDTA, (ii) 2 mM EDTA, (iii) no cations added, (iv) 10 mg/liter (0.250 mM) Ca2+ and 5 mg/liter (0.206 mM) Mg2+, (v) 20 mg/liter Ca2+ and 10 mg/liter Mg2+, or (vi) 80 mg/liter Ca2+ and 40 mg/liter Mg2+. At these concentrations of EDTA, >95% of Ca2+ and Mg2+ should be bound.
Descriptive data analysis.
Initially, an integrated area approach was applied to describe the log ratio area by a Hill-type model as previously described (34).
Development and qualification of the population PD model. (i) Bacterial populations.
Models with two, three, or four preexisting bacterial populations were considered as proposed previously (24, 35, 45, 50). Since we had only time-kill data with a constant colistin concentration, the natural death rate constant (kd) from the work of Meagher et al. (45) of 0.3 h−1 was used, since their data were based on a one-compartment in vitro PD model with drug concentrations changing over time.
(ii) Growth model.
The growth model comprised a previously described, saturable growth function (45) with a maximal velocity of bacterial growth [VGmax, expressed in CFU/(ml·h)] and the bacterial density that results in 50% of the maximal rate of growth (CFUm, expressed in CFU/ml). We reparameterized this growth function (see the work of Harigaya et al. [34] for equations) and estimated the maximum population size (POPmax) and the fastest growth half-life at low bacterial densities [t1/2(kg,low CFU)], since these two parameters can be more readily interpreted than VGmax and CFUm. To incorporate a slower initial growth phase, a lag compartment that contained the susceptible population at time zero was included and linked by a first-order rate constant (klag) with the compartment of the susceptible population subject to growth (Fig. 1).
FIG. 1.

Structure of the pharmacodynamic model, including bacterial growth, killing, and the inoculum effect (A) and the receptor occupancy model (B). The bacterial model contains one compartment for the susceptible, intermediate, and least-susceptible (“resistant”) population and a lag compartment for the susceptible population. In the lag compartment, bacteria do not replicate and are not subject to natural death but are subject to drug-induced killing and the effect of signal molecules. For the other three bacterial compartments, loss of cells occurs via a first-order natural death and a second-order killing process by colistin. Signal molecules were assumed to inhibit bacterial killing by colistin and the rate of bacterial replication (only the arrow for the effect of signal molecules on the susceptible population is shown). Initial conditions (IC) were estimated (see Methods). The target site model comprised a competitive interaction of colistin with Mg2+ and Ca2+ at the LPS binding sites in the outer membrane. Binding sites occupied by Mg2+ and Ca2+ were assumed to decrease the colistin concentration that is available in the active conformation(s) at the site of action.
(iii) Receptor occupancy model.
We assumed that colistin has to competitively displace Mg2+ and Ca2+ from their binding sites on the outer membrane before it can cause killing at the target site. Schindler and Osborn (55) found a similar dissociation constant for Mg2+ and Ca2+ for the binding site on LPS extracted from Salmonella enterica serovar Typhimurium. The approximately four binding sites of LPS extracted from P. aeruginosa were shown to have equivalent affinities for polymyxin B (48). In the present modeling, the dissociation constants were fixed to 200 μmol/liter for Mg2+ and Ca2+ (KdCations) and to 0.3 μmol/liter for colistin (KdColistin) according to data on strain G30A of Salmonella serovar Typhimurium (55). This choice of values for KdColistin and KdCations was assessed in a systematic sensitivity analysis. Under these assumptions, the fractional occupancy (FrCations) of the receptor for these two cations can be expressed as a function of the sum of the molar Mg2+ and Ca2+ concentration (CCations) and the colistin concentration (CColistin in mg/liter):
 |
(1) |
The average molecular mass (Mm) of the two predominant components of colistin (colistin A and B) is 1,163 g/mol. The fraction of receptors not occupied by Mg2+ or Ca2+ was used in a Hill equation to calculate the effective colistin concentration at the target site (CColistin,eff) as a function of the colistin concentration in broth (CColistin). Binding was assumed to be rapid:
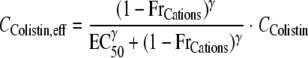 |
(2) |
(iv) Drug effect and inoculum effect.
Bacterial killing was specified as second-order processes with a different killing rate constant for each population. The data revealed a substantially slower killing at high CFUo than at low CFUo. To characterize this inoculum effect, all viable bacteria were assumed to synthesize and release freely diffusible signal molecules (57) that cause phenotypic changes inhibiting killing by polymyxins. The maximum inhibition of killing (ImaxKill) and of the replication rate (ImaxRep) and the signal molecule concentration associated with 50% of maximal inhibition (IC50) were estimated.
(v) Composite model.
The following equations describe the concentration of all viable bacteria (CFUALL) and inhibition of killing (INHKill) and of replication (INHRep) by signal molecules (concentration, CSign):
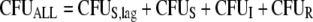 |
(3) |
 |
(4) |
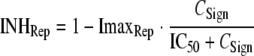 |
(5) |
The differential equations for susceptible cells in the lag compartment (CFUS,lag), replicating susceptible cells (CFUS), cells with intermediate susceptibility to colistin (CFUI), and least-susceptible cells (CFUR) are:
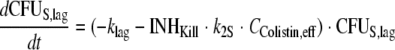 |
(6) |
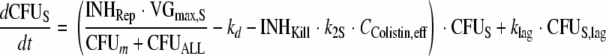 |
(7) |
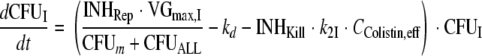 |
(8) |
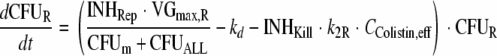 |
(9) |
The CFUo of all populations was estimated. Initial conditions of the intermediate and least-susceptible population were estimated as log10 of the fraction (FrI and FrR) of CFUo (see Table 1 for further explanation of parameters). We considered both models that assume the same FrI and FrR for all CFUo and models that allowed FrI and FrR to differ between CFUo. The initial condition for CFUS,lag was calculated as the difference between the CFUo and the initial condition of the intermediate and least-susceptible populations. The initial condition for CFUS was zero. To consider the potential absence of populations with a low mutation frequency at low CFUo, the initial condition of a bacterial population was set to zero if less than 1 cell would have been expected in 20 ml of broth.
TABLE 1.
Parameter estimates for population pharmacodynamic modelh
| Parameter | Symbol | Unit | Estimate (% variability) |
|||
|---|---|---|---|---|---|---|
|
P. aeruginosa PAO1 |
Strain URMC1 (S-ADAPT) | Strain URMC2 (S-ADAPT) | ||||
| NONMEM | S-ADAPT | |||||
| Parameters for bacterial growth | ||||||
| Shortest half-life of growth at low bacterial densities | ||||||
| Susceptible population | t1/2(kg,low CFU)S | min | 22.2 (24 [CV])b | 24.0 (12)b | 17.9 | 17.0 |
| Intermediate population | t1/2(kg,low CFU)I | min | 28.7 (24 [CV])b | 25.0 (12)b | 21.7 | 24.8 |
| Least-susceptible population | t1/2(kg,low CFU)R | min | 62.4 (24 [CV])b | 58.1 (12)b | 86.8 | 76.2 |
| Natural death rate constant | kd | h−1 | 0.3a | 0.3a | 0.3a | 0.3a |
| Half-life of growth lag process | t1/2(klag) | h | 1.31 | 1.10 (14) | 1.36 | 0.967 |
| Maximum population size | POPmax | CFU/ml | 109.59c | 109.53c | 109.68c | 109.16c |
| Parameters for bacterial inocula (initial conditions) | ||||||
| Initial inoculum size (sum of all populations) | ||||||
| 104 inoculum | Log10 CFUo4 | 4.34 | 4.34 ± 0.06 | |||
| 106 inoculum | Log10 CFUo6 | 6.17 | 6.31 ± 0.01 | 6.35 | 6.17 | |
| 108 inoculum | Log10 CFUo8 | 8.33 | 8.44 ± 0.25 | 7.82 | 8.07 | |
| 109 inoculum | Log10 CFUo9 | 8.96 | 9.07 ± 0.13 | 8.42 | 8.61 | |
| Intermediate population as fraction of initial inoculum | Log10 FrI | −3.43 | −3.53 | −3.97 | −4.93 | |
| Least-susceptible population as fraction of initial inoculum | Log10 FrR | −7.19 | −7.59 | −7.16 | −6.18 | |
| Parameters for the inoculum effect | ||||||
| Initial signal molecule conc relative to initial inoculum | FrSig | ml/CFU | 10−1.89d | 10−2.04d | 10−1.96d | 10−2.50d |
| Signal molecule concn resulting in 50% of max. effect | IC50 | 106.17± 0.79d | 106.51± 0.28d | 106.27d | 106.07d | |
| Degradation half-life of signal molecules | t1/2(kdeg) | h | 0.970 | 1.08 | 1.30 | 3.57 |
| Maximal inhibition of growth rate by signal molecules | ImaxRep | 0.422 | 0.407 | 0.569 | 0.111 | |
| Maximal inhibition of bacterial killing by signal molecules | ImaxKill | 0.992 | 0.999 | 0.999 | 0.999 | |
| Parameters for receptor occupancy and bacterial killing | ||||||
| Fraction of receptors not occupied by Mg2+ or Ca2+, resulting in an effective colistin concn of 50% relative to the colistin concn in broth | EC50 | 0.537e,f,g | 0.526 (9.2) | 0.316 | 0.123 | |
| Second-order killing rate constant | ||||||
| Susceptible population | k2S | liters/(mg·h) | 5.72 | 5.63 | 7.88 | 10.4 |
| Intermediate population | k2I | liters/(mg·h) | 0.369 | 0.353 | 0.627 | 0.522 |
| Least-susceptible population | k2R | liters/(mg·h) | 2.10·10−3 | 1.91·10−3 | 5.73·10−3 | 3.12·10−3 |
Fixed to the estimate reported by Meagher et al. (45).
Parameterized as difference in VGmax.
Due to the assumed inhibition of the growth rate by signal molecules, the maximum population size is approximately POPmax·(1 − ImaxRep).
This parameter is the signal molecule concentration at time zero divided by the sum of all bacteria at time zero. The signal molecule concentration has no units, since the concentration of signal molecules was not measured.
A Hill coefficient (γ) was estimated to fall between 10 and 20. The Hill coefficient was fixed to 10 in the final model to improve the stability of the estimation.
The supplemented concentration was 0.514 mmol/liter for Mg2+ and 0.624 mmol/liter for Ca2+. The sum of both molar concentrations (1.138 mmol/liter) was used in the model.
The dissociation constant was fixed to 200 μmol/liter for Mg2+ and Ca2+ and fixed to 0.3 μmol/liter for colistin as described in Materials and Methods. A sensitivity analysis with up to 50-fold different dissociation constants (in either direction) for colistin or for Mg2+ and Ca2+ showed that the numerical value of these constants only affected the EC50 notably.
The standard deviation of the additive error on a log10 scale (ɛCFU) was estimated as 0.474 in NONMEM and as 0.478 in S-ADAPT.
To account for a slower growth of the intermediate and least-susceptible populations than of the susceptible population, different VGmax terms were estimated for each population, and the associated t1/2(kg,low CFU) values were reported. The differential equation for hypothetical signal molecules is as follows (kdeg is the degradation rate constant):
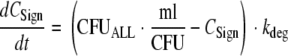 |
(10) |
The parameterization of equation 10 yields the same numerical value for the signal molecule concentration as for the bacterial concentration (CFUALL) at steady state.
(vi) Observation model.
The residual unidentified variability was described by an additive error model on a log10 scale, and data were fit as log10 (CFU/ml) if the observed bacterial density was 100 CFU/ml or more (equivalent to at least five colonies per plate). The CFUALL,obs is the observed CFU/ml, CFUALL,pred is the individual fitted CFU/ml (without error), and ɛCFU is a random variable with a mean of zero and standard deviation (SDCFU) describing the additive error on log10 scale:
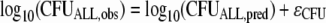 |
(11) |
For observations from undiluted samples with fewer than five colonies per plate, data were fit as actual colony counts, and the additive error on log scale was transformed to a proportional error on the scale of observed colony counts with the same variance as the additive error on log10 scale. At low colony counts, the random error due to observing only an integer number of colonies is most important compared to other sources of error. Therefore, a Poisson error (ɛPois) and an additive error (ɛAdd) were included if fewer than five colonies per plate were observed:
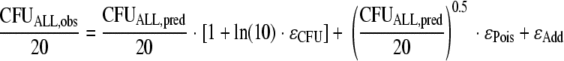 |
(12) |
Division by 20 converts the CFU/ml data into colony counts on the plate, since 50 μl of undiluted sample was plated for these low colony counts. The ɛPois and ɛAdd are random variables with a mean of 0 and fixed standard deviations of 1 for ɛPois and 0.25 for ɛAdd. The residual error model was evaluated in an exhaustive simulation-estimation study using a model with two subpopulations of different susceptibilities, first-order growth, logistic plateau function, and 2nd-order killing function.
(vii) Model qualification.
Models were assessed via standard diagnostic plots and a leave-one-inoculum-out internal cross-validation as described previously (10). For external model qualification involving two different clinical strains, selected model parameters for growth and killing of P. aeruginosa at low CFUo were reestimated based on data from the work of Gunderson et al. (31) and Li et al. (41). The Beal M3 method (6) was used for data from Gunderson et al. to appropriately handle data below the reported limit of detection.
(viii) Computation.
Nonlinear mixed-effects modeling in the NONMEM VI (7) and S-ADAPT (version 1.55 and 1.56) (5) software programs was applied as described previously (10). The first-order conditional estimation method with the interaction estimation option was used in NONMEM, and the importance sampling Monte-Carlo expectation-maximization method (pmethod = 4 or 8) was used in S-ADAPT. Variability of PD parameters between individual profiles was described by an exponential parameter variability model. An additive variability model was used for parameters estimated on log scale. A three-stage hierarchical analysis in S-ADAPT was performed to model the data of the time-kill experiments with various concentrations of Ca2+, Mg2+, or EDTA using methods described previously (37). The population PD results of the inoculum effect modeling in S-ADAPT were used as prior information. Plausible but uninformative priors were used for the maximum population size, initial inoculum, and half-life of growth and of the growth lag, since these parameters could be estimated well from the available data set. The sum of the free concentrations of Ca2+ and Mg2+ and the growth half-life in the presence of EDTA were estimated.
RESULTS
The structural mathematical model (Fig. 1) described the rapid, concentration-dependent killing by colistin (Fig. 2) and the inhibition of killing at high CFUo compared to that at low CFUo. At the 106 CFUo, bacterial killing by colistin was rapid and concentration dependent against all P. aeruginosa isolates. For the 106 CFUo, bactericidal activity within 1 h was achieved at concentrations of 1 to 4 × the MIC, and concentrations of ≥16 × the MIC resulted in bacterial reductions to undetectable concentrations within 30 min. Against the 108 and 109 CFUo, colistin activity was markedly attenuated (Fig. 2). Up to 32-fold-higher concentrations were required at the 109 CFUo than at the 106 CFUo to achieve bactericidal activity. At the 109 CFUo, killing was less pronounced and much slower, with nadir CFU counts at 8 to 12 h for PAO1 (Fig. 2), and regrowth occurred for all three strains. Drug-containing plates (Fig. 3) showed an increased fraction of less-susceptible cells at 2 to 16 mg/liter for the 106 CFUo at 4 to 32 mg/liter for the 108 CFUo and at 4 to 128 mg/liter for the 109 CFUo for PAO1. The overall effect of colistin represented by the log ratio area approach was well described (r2 > 0.97) by Hill-type models (Fig. 4). The maximal effect (Emax) decreased systematically, and the colistin exposure (EC50) associated with 50% of Emax increased systematically with increasing CFUo for all three strains.
FIG. 2.

Population predicted versus observed log10 (CFU/ml) for three strains of P. aeruginosa at a low (top), intermediate (middle), or high (bottom) initial inoculum (see also Table 1 and Fig. 1; based on results from S-ADAPT). A log10 value of observed colony counts plotted as zero means no colony was on the plate. Given the volume of 50 μl plated, one colony is equivalent to 20 CFU/ml (approximately 1.3 on log10 scale) for an undiluted sample.
FIG. 3.

Viable counts on drug-free and drug-containing plates at 24 h for P. aeruginosa PAO1 for an initial inoculum of 106 (A), 108 (B), or 109 (C) CFU/ml (agar MIC of PAO1, 2 mg/liter). Colistin concentrations above 16 mg/liter were not studied at the 106-CFU/ml inoculum. Plates with 8 mg/liter colistin showed a log10 CFU/ml between 1.6 and 4.1 for 2 mg/liter at the 106 inoculum and for 2, 4, and 8 mg/liter colistin in broth at the 108-CFU/ml inoculum. The log10 CFU/ml was 2.75 for 16 mg/liter colistin at the 108-CFU/ml inoculum. All other plates with 8 or 16 mg/liter colistin showed no viable growth.
FIG. 4.

Pharmacodynamic relationship between colistin concentration/MIC and the log ratio area [log10 (area under the CFU curve from 0 to 24 h for colistin/area under the CFU curve from 0 to 24 h for growth control)] at each initial inoculum for three P. aeruginosa strains (all r2 values ranged between 0.978 and 0.999): PAO1 (A), URMC1 (B), and URMC2 (C).
We found the following extent of killing by colistin relative to baseline levels in the filtrate after coincubation for 0.75 or 24 h with 109.4 CFU/ml. For 0.75 h of coincubation, net killing was >5 log10 in the filtrate of the 16-mg/liter and 64-mg/liter suspensions. For 24 h of coincubation, net killing was 5 log10 (>5 log10) in the filtrate of the 16-mg/liter (64-mg/liter) suspension. The growth control in spent broth grew by 1.5 log10 at 4 h and 3.1 log10 at 12 h.
In the mathematical model (Fig. 1), the attenuated effect of colistin at a high CFUo was assumed to be triggered by high concentrations of hypothetical signal molecules. At a signal molecule concentration much larger than the concentration associated with 50% of maximal inhibition (IC50) of 106.17 (Table 1), bacterial killing of all populations by colistin could be almost completely inhibited (ImaxKill = 0.992). The parameter estimates from NONMEM (results shown in the text) and S-ADAPT were well comparable (Table 1) and precise. Relative standard errors of all fixed effects were below 20% in S-ADAPT (results not shown). Parameter estimates for our two clinical strains were comparable to those for PAO1 (Table 1). In agreement with the lower MIC for the clinical isolates, these strains had a faster 2nd-order killing rate constant for the intermediate population (k2I) than PAO1 (Table 1).
Population predictions were reasonably precise and unbiased (Fig. 2 and 5B). The individual fits were more precise (Fig. 5A for PAO1). Slopes (correlation coefficients) of observed versus individual fitted log10 CFU/ml were 0.986 (r = 0.989) for URMC1 and 0.982 (r = 0.991) for URMC2. At the 109 CFUo, the predicted viable population counts showed more killing than observed for the 4 and 8 × MIC profiles. These two profiles primarily caused the mispredictions in panels B and C of Fig. 5. Figure 5C shows predictions for the observations that were not used during the four estimation runs of the cross-validation. Predictions were reasonably precise and unbiased during cross-validation of the three lower inocula. For the 109 CFUo, mispredictions were only slightly more pronounced during cross-validation (Fig. 5C) than during estimation of all data (panel B). The model described the time course of killing for our two clinical strains at three CFUo and the two literature studies (Fig. 6) well. Slopes (correlation coefficients) of observed versus fitted log10 CFU/ml were 1.023 (r = 0.927) for the data from the work of Gunderson et al. (31) and 1.002 (r = 0.985) for the data from the work of Li et al. (41).
FIG. 5.

Observed versus predicted bacterial counts on log10 scale for P. aeruginosa PAO1 (based on results from NONMEM). (A) Observed versus individual fitted CFU/ml. (B) Observations versus population predictions when all data were used for estimation. (C) Observations versus population predictions from the internal cross-validation. Importantly, none of the observations at the respective inocula were used during cross-validation when data at this inoculum were predicted. A log10 value of observed colony counts plotted as zero means no colony was on the plate.
FIG. 6.

Fitted and observed bacterial counts versus time for one-compartment in vitro model data from the work of Gunderson et al. (32) (A) (lower limit of detection: log10 CFU/ml of 2.48; observations below this reported lower limit are plotted as 0 log10 [CFU/ml]) and time-kill data from Li et al. (42) (B) (plates with zero colonies are plotted as zero).
The most important model assumptions are discussed in Table 2. Models with a first-order conversion between the susceptible, intermediate, and least-susceptible populations were estimated, but neither significantly improved either the objective function or the curve fits of our static time-kill data (results not shown). Models with four instead of three populations provided no significant benefit, and models with two populations were inadequate to describe the observations (results not shown). Estimating different FrI and FrR values at each CFUo yielded only a small improvement in the objective function and well-comparable estimates for FrI and FrR for each CFUo (results not shown). Therefore, FrI and FrR were assumed to be the same for all CFUo. Models with different relative affinities of colistin relative to Mg2+ and Ca2+ for the susceptible, intermediate, and least-susceptible populations in addition to a different killing rate constant for each population achieved only a slightly lower objective function in S-ADAPT but did not notably improve the diagnostic plots. Thus, we did not implement different relative affinities of colistin and Mg2+ and Ca2+ for each population following the rule of parsimony.
TABLE 2.
Overview of assumptions and supporting data for proposed model features (see Methods and Discussion for further details)
| Assumption | Justification and supporting data |
|---|---|
| 1. There are three preexisting subpopulations (two subpopulations at low inocula) with different susceptibilities to colistin. | Killing by colistin is extremely rapid. Periplasmic proteins are released within 2 min of polymyxin B treatment. After longer incubation (up to 60 min), 100% of cytoplasmic proteins are released (12, 61). This suggests that there were preexisting less-susceptible cells. |
| 2. P. aeruginosa displays a first-order growth at low bacterial densities, and the rate of growth becomes saturable at high bacterial concentrations. The plateau is achieved due to an equilibrium between the saturable rate of growth and a first-order natural death rate constant. | At high bacterial concentrations, bacterial growth is slower due to a shortage of nutrients. |
| The same saturable growth function was employed by Meagher et al. (45). | |
| 3. Bacteria reside in a lag compartment to describe the short lag time of growth. | A short lag time of bacteria that have not yet achieved log growth phase has been previously described and implemented (66). A growth lag time of the intermediate and resistant subpopulations was ignored to keep the model simpler, since these subpopulations would not grow much during the 1- to 2-h lag phase. |
| 4. Divalent cations such as Ca2+ and Mg2+ can competitively inhibit killing by colistin. | This competitive interaction has been previously described for polymyxins by several groups (48, 55, 56). We used dissociation constants for Ca2+, Mg2+, and colistin reported by Schindler and Osborn (55) for LPS extracted from S. Typhimurium. |
| A sensitivity analysis of our model showed that the exact ratio of dissociation constants for Ca2+ and Mg2+ compared to colistin had a minimal impact on parameter estimates and curve fits (see Table 1). The target site model was also supported by the prospective “validation” shown in Fig. 7. | |
| 5. The rate of killing of P. aeruginosa by colistin can be described by a 2nd-order process. | The rate of killing by colistin showed essentially no saturation at high colistin concentrations. A 2nd-order killing process described the observed viable counts well. |
| 6. All viable P. aeruginosa cells synthesize and release freely diffusible signal molecules. | Freely diffusible signal molecules that are associated with quorum sensing have been previously described for P. aeruginosa (57). |
| 7. High signal molecule concentrations were assumed to inhibit the rate of killing by colistin and the rate of growth. | Changes in the outer membrane of P. aeruginosa in response to signal molecule concentrations were described previously (36, 49, 51) |
| Signal molecules were shown to inhibit the rate of growth of P. aeruginosa (18), and this model feature was previously incorporated in a mathematical model (9). |
It was estimated that 53.7% (EC50) of the binding sites for Mg2+ and Ca2+ were required to be unoccupied by Mg2+ or Ca2+ to achieve an effective colistin concentration of 50% of the colistin concentration in broth (Fig. 1). The Hill coefficient of this relationship was initially estimated to fall between 10 and 20, indicating cooperativity, and subsequently fixed to 10 to improve model stability. This choice did not affect curve fits.
Our three-stage hierarchical analysis showed excellent individual curve fits at various concentrations of Ca2+, Mg2+, and EDTA (Fig. 7). All ratios of the population mean parameter estimates from the 3-stage hierarchical analysis divided by their prior estimates from the population PD analysis of the inoculum effect data set in S-ADAPT were within 0.90 to 1.13 (average ± SD: 1.00 ± 0.056). This supported the proposed target site model and indicated consistency between both series of experiments. Compared to LB broth without addition of cations or EDTA (Fig. 7B), addition of cations inhibited killing by colistin (Fig. 7C to E). Addition of 2 mM EDTA to chelate Ca2+ and Mg2+ enhanced killing by colistin (Fig. 7A) but increased the estimated growth half-life of the susceptible population to 48.5 min. The rate and extent of killing were very similar for 2 mM EDTA and 20 mM EDTA (data not shown). However, 20 mM EDTA inhibited net growth of PAO1 substantially, and therefore, these data were not included in the modeling analysis. The sum of the free concentrations of Ca2+ and Mg2+ in LB broth was estimated to be small (0.06 mM).
FIG. 7.

Observed and individual fitted viable counts of P. aeruginosa PAO1 for various concentrations of Ca2+, Mg2+, or EDTA added to LB broth: 2.0 mM EDTA (A), no cations and no EDTA added (B), 10 mg/liter Ca2+ and 5 mg/liter Mg2+ (C), 20 mg/liter Ca2+ and 10 mg/liter Mg2+ (D), or 80 mg/liter Ca2+ and 40 mg/liter Mg2+ (E). Observed viable counts of zero mean no colony on the plate (using a volume of 200 μl plated manually for samples with low viable counts).
Our simulation-estimation study assessing the proposed residual error model showed that this model was robust and curve fits were unbiased and precise using the cutoff value of 5 colonies per plate applied in this colistin study (equivalent to 100 CFU/ml for a 50-μl plating volume). For fixed-effect parameters, bias was ≤0.07 for estimates on log10 scale and ≤4% for all other estimates. Precision of fixed effects had a standard deviation (SD) of <0.09 for estimates on log10 scale and a coefficient of variation (CV) of <10% for all other estimates. This new residual error model was more robust in NONMEM than the Beal M3 method (6).
DISCUSSION
The increasing prevalence of Gram-negative bacteria, including P. aeruginosa, that are resistant to almost all commercially available antibiotics is a cause for significant concern (22, 40). The lack of alternative therapeutic options has forced clinicians to use polymyxins due to their activity against these multidrug-resistant pathogens (40, 42). Although colistin has displayed rapid bactericidal activity against low CFUo of P. aeruginosa, it has not yet been determined whether this profile would also be seen against high CFUo. We therefore studied the impact of CFUo on colistin killing activity in vitro for three isolates of P. aeruginosa. In our study, killing by colistin was drastically attenuated at high CFUo, and substantially higher colistin concentrations were necessary to achieve bactericidal activity at high CFUo (Fig. 2). Consistent with the inverted-U concept (64), intermediate concentrations caused resistance at 24 h, whereas low or (very) high concentrations did not increase the fraction of less-susceptible cells (Fig. 3).
Hancock et al. (32, 33, 63) highlighted that it is critical to understand the mechanisms of antibiotic killing to prevent emergence of bacterial resistance. Consequently, we developed a mathematical model that incorporates known features of the mechanism of action of colistin (39). After displacing Mg2+ and Ca2+ from their LPS binding sites, colistin disrupts the outer and cytoplasmic membrane, and leakage of small intracellular molecules and bacterial killing are observed within less than 2 min (39, 48, 56, 61). The proposed model (Fig. 1 and Table 2) incorporates the competitive interaction of colistin with Mg2+ and Ca2+ as a first step of colistin action. After colistin sufficiently destabilized the outer membrane by increasing the fraction of receptors not occupied by Mg2+ or Ca2+ to 53.7% (EC50), colistin reached its target site and caused killing. The assumed relative affinity for LPS binding sites was 670-fold higher for colistin than for Mg2+ and Ca2+ (55), which is consistent with the literature as it relates to P. aeruginosa (13, 43, 48). All model parameters except EC50 were essentially unaffected in a sensitivity analysis (in S-ADAPT) that varied the dissociation constants for colistin and Mg2+ and Ca2+ up to 50-fold in either direction. In agreement with data in the literature (43, 48), the (estimated) Hill coefficient of 10 suggests cooperativity in loss of membrane integrity after sufficient displacement of Ca2+ and Mg2+.
A prospective experimental validation (Fig. 7) coupled with a three-stage hierarchical analysis supported the proposed target site model (Fig. 1B). To our knowledge, this analysis for a mechanism-based model is the first application of a three-stage hierarchical population PD analysis in antibacterial PD. This method is very helpful for borrowing results from other studies in the literature and using these results as prior information with uncertainty.
The mathematical model developed in this study incorporates a second-order killing function and three preexisting populations with different susceptibilities to colistin. This characterization of colistin PD is in agreement with the rapid, concentration-dependent killing of P. aeruginosa by colistin and with the regrowth of resistant cells (4, 8, 31, 41). At high colistin concentrations against low CFUo, 99.9% killing was achieved within 15 min and undetectable levels were maintained until 24 h. The descriptive log ratio area method yielded a robust measure for the average drug effect and indicated a systematic trend toward less killing at high CFUo (Fig. 4).
Our mathematical model could describe the time course of killing and regrowth for a range of CFUo (Fig. 2 and Fig. 5). Initially, the slower killing at high CFUo compared to low CFUo was modeled as different second-order killing rate constants for each population at each CFUo. In this descriptive analysis for strain PAO1, the killing rate constant of the susceptible population was approximately 23-fold (6-fold) smaller for the 109 (108) CFUo than for the 106 CFUo.
Subsequently, the inoculum effect was described as a phenotypic change of bacterial cells. Potential reasons for the inhibition of killing by colistin are a decreased negative charge of LPS and changes in the hydrophobic side chain in the outer membrane of P. aeruginosa in response to cells sensing their environment (14, 36, 49, 51). Parameter estimates from NONMEM and S-ADAPT using notably different estimation algorithms were well comparable (Table 1). The model described the killing by colistin well for clinical strains URMC1 and URMC2 (Fig. 2B and 2C) and for two literature data sets (Fig. 6). The proposed mechanism-based model incorporates certain features of the mode of action of colistin, can predict the time course of bacterial killing by colistin over a wide range of CFUo, and can be extended to describe killing by combinations of antibiotics. The new proposed residual error model was very robust, yielded unbiased and precise estimates in a simulation-estimation study with a simplified PD model, and could account for plates with zero colonies during estimation and for the Poisson error at low viable counts.
Our model is largely simplified, since it contains only one compartment for hypothetical, unmeasured signal molecules that were assumed to cause the phenotypic changes of cells leading to a slower killing by colistin (Table 2). Several signal molecules are known to be involved in cell-to-cell communication (quorum sensing) of P. aeruginosa (20, 57) and to cause a delayed onset of expression of quorum-induced genes (57, 58). The proposed model can account for this delay and had a good predictive performance for interpolation (when leaving out data of the 106 or 108 CFUo) and extrapolation (when leaving out data of the 104 or 109 CFUo) between CFUo (Fig. 5C). Further studies are needed to assess the influence of antibiotics on bacterial signaling, gene expression, proteomics, and bacterial physiology at high and low CFUo in vitro and in vivo.
A potential reason for the inoculum effect of colistin might arise from its antiendotoxin activity in neutralizing bacterial LPS (26, 29, 72). Colistin forms mixed monolayers with phospholipids and is incorporated in micelles in vitro (46). “Binding” of colistin to LPS fragments of killed bacteria might decrease the free colistin concentration in vitro and contribute to the inoculum effect. Our coincubation experiments with colistin added to a CFUo of 109.4 CFU/ml for 0.75 or 24 h suggest that binding to LPS or other molecules in spent broth was not the major reason for the inoculum effect in our study. This coincubation experiment and the slow killing with late nadir CFU counts at 8 to 12 h for PAO1 suggested that a phenotypic change of bacterial cells is the most likely reason for the inoculum effect in our in vitro experiments.
Davis (17) found no inoculum effect for colistin at the two lower dose levels in mouse protection tests with P. aeruginosa, whereas the mean effective dose increased approximately 16-fold for gentamicin or tobramycin with a 100-fold increase in CFUo. Since there was no washout of signal molecules or other substances, a time-kill experiment as in our study may overestimate the inoculum effect in vivo. However, several animal infection model studies showed the significance of the inoculum effect in vivo (25, 38, 47, 60). It is important to note that the clinical relevance of the inoculum effect has rarely been demonstrated, and numerous factors, such as host defense and severity of infection, greatly influence antimicrobial effect and treatment success in patients.
Based on our in vitro time-kill data, our mathematical model predicted (results not shown) that colistin regimens with a large colistin exposure during the first approximately 12 h may be able to achieve enough net killing that the immune system can eradicate the remaining colistin-resistant cells. Further in vitro and animal studies are required to substantiate this model prediction.
Our study has potential limitations. First, the mathematical model of the present study was developed based on data from static time-kill experiments. Our in vitro model also does not account for the complex protein binding of colistin in vivo (23). Second, our study was not designed to assess the mechanism(s) of colistin resistance. To build mathematical models for the time course of colistin resistance, hollow-fiber infection models determining the viable counts of the total and resistant subpopulation(s) and resistance mechanisms will need to be studied for clinically relevant colistin dosage regimens over approximately 10 to 14 days. Finally, other potential limitations of our study are that the resistant subpopulation(s) might have been growing faster in LB broth, which is richer in nutrients than many infection sites in vivo, and that colistin concentrations were not directly measured. However, time-kill experiments in spent broth after coincubating colistin and a high CFUo of PAO1 suggested that drug degradation was not the primary reason for the inoculum effect of colistin in our study.
In summary, the extent and rate of killing of PAO1 and two clinical P. aeruginosa isolates by colistin were markedly inhibited at high CFUo compared to low CFUo. Killing of PAO1 was approximately 23-fold (6-fold) slower for the 109 (108) CFUo than for the 106 CFUo. The proposed mechanism-based mathematical model described bacterial growth and killing of three P. aeruginosa strains across a range of CFUo well, had a good predictive performances for all studied CFUo, and could well describe data from two additional clinical strains from the literature. A prospective experimental “validation” using different concentrations of Ca2+ and Mg2+ in combination with a three-stage hierarchical population PD analysis supported the proposed target site model. The proposed mathematical model should be further validated using dynamic in vitro models with colistin in mono- and combination therapy. Overall, this study emphasizes the potential need for higher colistin exposure or combination regimens to treat deep-seated, difficult-to-treat infections with high CFUo. Further animal studies are warranted to assess the toxicity and inoculum effect of colistin in vivo.
Acknowledgments
We thank Roger Nation, Jian Li, and Cornelia Landersdorfer for very fruitful discussion and comments on the manuscript. We thank Robert E. Hancock, Joseph McPhee, and Manjeet Bains for their insights and for the gift of PAO1. We thank Dwight Hardy for the gift of URMC1 and URMC2 and Curtis Hass and Ashley-Dodds for their contribution to this project.
This work was supported in part by Public Health Service grant R01AI079330 from the National Institute of Allergy and Infectious Diseases and in part by the University at Buffalo, State University of New York. J.B.B. was supported by a postdoctoral fellowship from Johnson & Johnson, and W.J.J. was supported by NIH grant GM 57980.
We have no conflicts of interest to report.
Footnotes
Published ahead of print on 8 March 2010.
REFERENCES
- 1.Adler, J. L., and M. Finland. 1971. Susceptibility of recent isolates of Pseudomonas aeruginosa to gentamicin, polymyxin, and five penicillins, with observations on the pyocin and immunotypes of the strains. Appl. Microbiol. 22:870-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambrose, P. G., S. M. Bhavnani, C. M. Rubino, A. Louie, T. Gumbo, A. Forrest, and G. L. Drusano. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin. Infect. Dis. 44:79-86. [DOI] [PubMed] [Google Scholar]
- 3.Andes, D., and W. A. Craig. 2002. Animal model pharmacokinetics and pharmacodynamics: a critical review. Int. J. Antimicrob. Agents 19:261-268. [DOI] [PubMed] [Google Scholar]
- 4.Aoki, N., K. Tateda, Y. Kikuchi, S. Kimura, C. Miyazaki, Y. Ishii, Y. Tanabe, F. Gejyo, and K. Yamaguchi. 2009. Efficacy of colistin combination therapy in a mouse model of pneumonia caused by multidrug-resistant Pseudomonas aeruginosa. J. Antimicrob. Chemother. 63:534-542. [DOI] [PubMed] [Google Scholar]
- 5.Bauer, R. J. 2008. S-ADAPT/MCPEM user's guide (version 1.56). Software for pharmacokinetic, pharmacodynamic and population data analysis. Biomedical Simulations Resource, Berkeley, CA.
- 6.Beal, S. L. 2001. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 28:481-504. [DOI] [PubMed] [Google Scholar]
- 7.Beal, S. L., L. B. Sheiner, and A. J. Boeckmann. 2006. NONMEM users guides (1989-2006). Icon Development Solutions, Ellicott City, MD.
- 8.Bergen, P. J., J. Li, R. L. Nation, J. D. Turnidge, K. Coulthard, and R. W. Milne. 2008. Comparison of once-, twice- and thrice-daily dosing of colistin on antibacterial effect and emergence of resistance: studies with Pseudomonas aeruginosa in an in vitro pharmacodynamic model. J. Antimicrob. Chemother. 61:636-642. [DOI] [PubMed] [Google Scholar]
- 9.Brook, I. 1989. Inoculum effect. Rev. Infect. Dis. 11:361-368. [DOI] [PubMed] [Google Scholar]
- 10.Bulitta, J. B., N. S. Ly, J. C. Yang, A. Forrest, W. J. Jusko, and B. T. Tsuji. 2009. Development and qualification of a pharmacodynamic model for the pronounced inoculum effect of ceftazidime against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53:46-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bulitta, J. B., J. C. Yang, B. T. Tsuji, W. J. Jusko, and A. Forrest. 2007. Mechanistic PK/PD models for the inoculum effect (over 5 orders of magnitude) of colistin and ceftazidime against Pseudomonas aeruginosa (PA), poster W4553 (abstr. 1829). AAPS Annual Meeting, San Diego, CA, 11 to 15 November 2007.
- 12.Cerny, G., and M. Teuber. 1972. Comparative polyacrylamide electrophoresis of periplasmic proteins released from gram-negative bacteria by polymyxin B. Arch. Mikrobiol. 82:361-370. [DOI] [PubMed] [Google Scholar]
- 13.Chen, C. C., and D. S. Feingold. 1972. Locus of divalent cation inhibition of the bactericidal action of polymyxin B. Antimicrob. Agents Chemother. 2:331-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conrad, R. S., and C. Galanos. 1989. Fatty acid alterations and polymyxin B binding by lipopolysaccharides from Pseudomonas aeruginosa adapted to polymyxin B resistance. Antimicrob. Agents Chemother. 33:1724-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costerton, J. W., P. S. Stewart, and E. P. Greenberg. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318-1322. [DOI] [PubMed] [Google Scholar]
- 16.Craig, W. A. 1998. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin. Infect. Dis. 26:1-12. [DOI] [PubMed] [Google Scholar]
- 17.Davis, S. D. 1975. Activity of gentamicin, tobramycin, polymyxin B, and colistimethate in mouse protection tests with Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 8:50-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diggle, S. P., K. Winzer, S. R. Chhabra, K. E. Worrall, M. Camara, and P. Williams. 2003. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol. Microbiol. 50:29-43. [DOI] [PubMed] [Google Scholar]
- 19.Dixon, R. A., and I. Chopra. 1986. Leakage of periplasmic proteins from Escherichia coli mediated by polymyxin B nonapeptide. Antimicrob. Agents Chemother. 29:781-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donabedian, H. 2003. Quorum sensing and its relevance to infectious diseases. J. Infect. 46:207-214. [DOI] [PubMed] [Google Scholar]
- 21.Drusano, G. L. 2004. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug.’ Nat. Rev. Microbiol. 2:289-300. [DOI] [PubMed] [Google Scholar]
- 22.Drusano, G. L. 2007. Pharmacokinetics and pharmacodynamics of antimicrobials. Clin. Infect. Dis. 45(Suppl. 1):S89-S95. [DOI] [PubMed] [Google Scholar]
- 23.Dudhani, R. V., J. Li, and R. L. Nation. 2009. Plasma binding (PB) of colistin (C) involves multiple proteins and is concentration dependent: potential clinical implications, abstr. A1-576. Abstr. 49th Annu. Intersci. Conf. Antimicrob. Agents Chemotherapy. American Society for Microbiology, Washington, DC.
- 24.Dudley, M. N., H. D. Mandler, D. Gilbert, J. Ericson, K. H. Mayer, and S. H. Zinner. 1987. Pharmacokinetics and pharmacodynamics of intravenous ciprofloxacin. Studies in vivo and in an in vitro dynamic model. Am. J. Med. 82:363-368. [PubMed] [Google Scholar]
- 25.Eagle, H. 1949. The effect of the size of the inoculum and the age of the infection on the curative dose of penicillin in experimental infections with streptococci, pneumococci, and Treponema pallidum. J. Exp. Med. 90:595-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Escartin, P., J. A. Rodriguez-Montes, V. Cuervas-Mons, I. Rossi, J. Alvarez-Cienfuegos, P. Maganto, and J. L. Castillo-Olivares. 1982. Effect of colistin on reduction of biliary flow induced by endotoxin in E. coli. Dig. Dis. Sci. 27:875-879. [DOI] [PubMed] [Google Scholar]
- 27.Forrest, A., D. E. Nix, C. H. Ballow, T. F. Goss, M. C. Birmingham, and J. J. Schentag. 1993. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob. Agents Chemother. 37:1073-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garcia-Medina, R., W. M. Dunne, P. K. Singh, and S. L. Brody. 2005. Pseudomonas aeruginosa acquires biofilm-like properties within airway epithelial cells. Infect. Immun. 73:8298-8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gardiner, K. R., P. J. Erwin, N. H. Anderson, M. D. McCaigue, M. I. Halliday, and B. J. Rowlands. 1995. Lactulose as an antiendotoxin in experimental colitis. Br. J. Surg. 82:469-472. [DOI] [PubMed] [Google Scholar]
- 30.Gooderham, W. J., M. Bains, J. B. McPhee, I. Wiegand, and R. E. Hancock. 2008. Induction by cationic antimicrobial peptides and involvement in intrinsic polymyxin and antimicrobial peptide resistance, biofilm formation, and swarming motility of PsrA in Pseudomonas aeruginosa. J. Bacteriol. 190:5624-5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gunderson, B. W., K. H. Ibrahim, L. B. Hovde, T. L. Fromm, M. D. Reed, and J. C. Rotschafer. 2003. Synergistic activity of colistin and ceftazidime against multiantibiotic-resistant Pseudomonas aeruginosa in an in vitro pharmacodynamic model. Antimicrob. Agents Chemother. 47:905-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hancock, R. E. 2005. Mechanisms of action of newer antibiotics for Gram-positive pathogens. Lancet Infect. Dis. 5:209-218. [DOI] [PubMed] [Google Scholar]
- 33.Hancock, R. E., and D. S. Chapple. 1999. Peptide antibiotics. Antimicrob. Agents Chemother. 43:1317-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harigaya, Y., J. B. Bulitta, A. Forrest, G. Sakoulas, A. J. Lesse, J. M. Mylotte, and B. T. Tsuji. 2009. Pharmacodynamics of vancomycin at simulated epithelial lining fluid concentrations against methicillin-resistant Staphylococcus aureus (MRSA): implications for dosing in MRSA pneumonia. Antimicrob. Agents Chemother. 53:3894-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jumbe, N., A. Louie, R. Leary, W. Liu, M. R. Deziel, V. H. Tam, R. Bachhawat, C. Freeman, J. B. Kahn, K. Bush, M. N. Dudley, M. H. Miller, and G. L. Drusano. 2003. Application of a mathematical model to prevent in vivo amplification of antibiotic-resistant bacterial populations during therapy. J. Clin. Invest. 112:275-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon, D. H., and C. D. Lu. 2006. Polyamines induce resistance to cationic peptide, aminoglycoside, and quinolone antibiotics in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 50:1615-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landersdorfer, C. B., M. Kinzig, J. B. Bulitta, F. F. Hennig, U. Holzgrabe, F. Sorgel, and J. Gusinde. 2009. Bone penetration of amoxicillin and clavulanic acid evaluated by population pharmacokinetics and Monte Carlo simulation. Antimicrob. Agents Chemother. 53:2569-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee, D., Y. Murakami, T. Stamstad, K. Marchillo, J. Ashbeck, D. R. Andes, and W. A. Craig. 2007. Inoculum effect of daptomycin (D), linezolid (L), vancomycin (V), and ceftobiprole (C) with Staphylococcus aureus (SA) and Streptococcus pneumoniae (SP) in neutropenic mice at 105 and 107 CFU in opposite thighs. Abstr. 47th Annu. Intersci. Conf. Antimicrob. Agents Chemother. (ICAAC), abstr. A-37. American Society for Microbiology, Washington, DC.
- 39.Li, J., R. L. Nation, R. W. Milne, J. D. Turnidge, and K. Coulthard. 2005. Evaluation of colistin as an agent against multi-resistant Gram-negative bacteria. Int. J. Antimicrob. Agents 25:11-25. [DOI] [PubMed] [Google Scholar]
- 40.Li, J., R. L. Nation, J. D. Turnidge, R. W. Milne, K. Coulthard, C. R. Rayner, and D. L. Paterson. 2006. Colistin: the emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 6:589-601. [DOI] [PubMed] [Google Scholar]
- 41.Li, J., J. Turnidge, R. Milne, R. L. Nation, and K. Coulthard. 2001. In vitro pharmacodynamic properties of colistin and colistin methanesulfonate against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob. Agents Chemother. 45:781-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linden, P. K., S. Kusne, K. Coley, P. Fontes, D. J. Kramer, and D. Paterson. 2003. Use of parenteral colistin for the treatment of serious infection due to antimicrobial-resistant Pseudomonas aeruginosa. Clin. Infect. Dis. 37:e154-e160. [DOI] [PubMed] [Google Scholar]
- 43.Loh, B., C. Grant, and R. E. Hancock. 1984. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 26:546-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Louie, A., H. S. Heine, K. Kim, D. L. Brown, B. VanScoy, W. Liu, M. Kinzig-Schippers, F. Sorgel, and G. L. Drusano. 2008. Use of an in vitro pharmacodynamic model to derive a linezolid regimen that optimizes bacterial kill and prevents emergence of resistance in Bacillus anthracis. Antimicrob. Agents Chemother. 52:2486-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meagher, A. K., A. Forrest, A. Dalhoff, H. Stass, and J. J. Schentag. 2004. Novel pharmacokinetic-pharmacodynamic model for prediction of outcomes with an extended-release formulation of ciprofloxacin. Antimicrob. Agents Chemother. 48:2061-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mestres, C., M. A. Alsina, M. A. Busquets, I. Muranyi, and F. Reig. 1998. Interaction of colistin with lipids in liposomes and monolayers. Int. J. Pharm. 160:99-107. [Google Scholar]
- 47.Mizunaga, S., T. Kamiyama, Y. Fukuda, M. Takahata, and J. Mitsuyama. 2005. Influence of inoculum size of Staphylococcus aureus and Pseudomonas aeruginosa on in vitro activities and in vivo efficacy of fluoroquinolones and carbapenems. J. Antimicrob. Chemother. 56:91-96. [DOI] [PubMed] [Google Scholar]
- 48.Moore, R. A., N. C. Bates, and R. E. Hancock. 1986. Interaction of polycationic antibiotics with Pseudomonas aeruginosa lipopolysaccharide and lipid A studied by using dansyl-polymyxin. Antimicrob. Agents Chemother. 29:496-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moskowitz, S. M., R. K. Ernst, and S. I. Miller. 2004. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J. Bacteriol. 186:575-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mouton, J. W., A. A. Vinks, and N. C. Punt. 1997. Pharmacokinetic-pharmacodynamic modeling of activity of ceftazidime during continuous and intermittent infusion. Antimicrob. Agents Chemother. 41:733-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pamp, S. J., M. Gjermansen, H. K. Johansen, and T. Tolker-Nielsen. 2008. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 68:223-240. [DOI] [PubMed] [Google Scholar]
- 52.Peyret, M., J. P. Flandrois, and G. Fardel. 1991. Time-killing curves of Pseudomonas aeruginosa strains exposed to polymyxin B. Pathol. Biol. (Paris) 39:446-450. [PubMed] [Google Scholar]
- 53.Piantieri, G., E. Magliulo, A. Cafarelli, G. Bossi, and M. Cruciani. 1981. Antibacterial activity in vitro of several aminoglycosides and cephalosporins. G. Batteriol. Virol. Immunol. 74:77-100. (In Italian.) [PubMed] [Google Scholar]
- 54.Roosendaal, R., I. A. Bakker-Woudenberg, M. van den Berghe-van Raffe, J. C. Vink-van den Berg, and M. F. Michel. 1991. Impact of the duration of infection on the activity of ceftazidime, gentamicin and ciprofloxacin in Klebsiella pneumoniae pneumonia and septicemia in leukopenic rats. Eur. J. Clin. Microbiol. Infect. Dis. 10:1019-1025. [DOI] [PubMed] [Google Scholar]
- 55.Schindler, M., and M. J. Osborn. 1979. Interaction of divalent cations and polymyxin B with lipopolysaccharide. Biochemistry 18:4425-4430. [DOI] [PubMed] [Google Scholar]
- 56.Schindler, P. R., and M. Teuber. 1975. Action of polymyxin B on bacterial membranes: morphological changes in the cytoplasm and in the outer membrane of Salmonella typhimurium and Escherichia coli B. Antimicrob. Agents Chemother. 8:95-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schuster, M., and E. P. Greenberg. 2006. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int. J. Med. Microbiol. 296:73-81. [DOI] [PubMed] [Google Scholar]
- 58.Schuster, M., C. P. Lostroh, T. Ogi, and E. P. Greenberg. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 185:2066-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stearne, L. E., W. H. Goessens, J. W. Mouton, and I. C. Gyssens. 2007. Effect of dosing and dosing frequency on the efficacy of ceftizoxime and the emergence of ceftizoxime resistance during the early development of murine abscesses caused by Bacteroides fragilis and Enterobacter cloacae mixed infection. Antimicrob. Agents Chemother. 51:3605-3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stevens, D. L., S. Yan, and A. E. Bryant. 1993. Penicillin-binding protein expression at different growth stages determines penicillin efficacy in vitro and in vivo: an explanation for the inoculum effect. J. Infect. Dis. 167:1401-1405. [DOI] [PubMed] [Google Scholar]
- 61.Storm, D. R., K. S. Rosenthal, and P. E. Swanson. 1977. Polymyxin and related peptide antibiotics. Annu. Rev. Biochem. 46:723-763. [DOI] [PubMed] [Google Scholar]
- 62.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 63.Straus, S. K., and R. E. Hancock. 2006. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta 1758:1215-1223. [DOI] [PubMed] [Google Scholar]
- 64.Tam, V. H., A. Louie, M. R. Deziel, W. Liu, and G. L. Drusano. 2007. The relationship between quinolone exposures and resistance amplification is characterized by an inverted U: a new paradigm for optimizing pharmacodynamics to counterselect resistance. Antimicrob. Agents Chemother. 51:744-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tam, V. H., A. N. Schilling, G. Vo, S. Kabbara, A. L. Kwa, N. P. Wiederhold, and R. E. Lewis. 2005. Pharmacodynamics of polymyxin B against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49:3624-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Treyaprasert, W., S. Schmidt, K. H. Rand, U. Suvanakoot, and H. Derendorf. 2007. Pharmacokinetic/pharmacodynamic modeling of in vitro activity of azithromycin against four different bacterial strains. Int. J. Antimicrob. Agents 29:263-270. [DOI] [PubMed] [Google Scholar]
- 67.Tsuji, B. T., and M. J. Rybak. 2005. Short-course gentamicin in combination with daptomycin or vancomycin against Staphylococcus aureus in an in vitro pharmacodynamic model with simulated endocardial vegetations. Antimicrob. Agents Chemother. 49:2735-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tsuji, B. T., C. von Eiff, P. A. Kelchlin, A. Forrest, and P. F. Smith. 2008. Attenuated vancomycin bactericidal activity against Staphylococcus aureus hemB mutants expressing the small-Colony-variant phenotype. Antimicrob. Agents Chemother. 52:1533-1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tuomanen, E., and A. Tomasz. 1990. Mechanism of phenotypic tolerance of nongrowing pneumococci to beta-lactam antibiotics. Scand. J. Infect. Dis. Suppl. 74:102-112. [PubMed] [Google Scholar]
- 70.Udekwu, K. I., N. Parrish, P. Ankomah, F. Baquero, and B. R. Levin. 2009. Functional relationship between bacterial cell density and the efficacy of antibiotics. J. Antimicrob. Chemother. 63:745-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Venkatesan, A., C. Spalding, A. Speedie, G. Sinha, and J. A. Rumbaugh. 2005. Pseudomonas aeruginosa infective endocarditis presenting as bacterial meningitis. J. Infect. 51:e199-e202. [DOI] [PubMed] [Google Scholar]
- 72.Warren, H. S., S. A. Kania, and G. R. Siber. 1985. Binding and neutralization of bacterial lipopolysaccharide by colistin nonapeptide. Antimicrob. Agents Chemother. 28:107-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang, J. C., J. B. Bulitta, A. Forrest, and B. T. Tsuji. 2007. High inocula Pseudomonas aeruginosa (PA) attenuates colistin bactericidal activity and alters pharmacodynamics (PD). Abstr. 47th Annu. Intersci. Conf. Antimicrob. Agents Chemother. (ICAAC), poster A-6. American Society for Microbiology, Washington, DC.
- 74.Yohonn, L., S. V. Brown, J. B. Bulitta, A. Forrest, H. Sun, C. McPhee, P. N. Holden, P. A. Kelchlin, E. Dodds Ashley, C. E. Haas, D. Hardy, and B. T. Tsuji. 2007. Bactericidal activity of colistin against clinical isolates of Pseudomonas aeruginosa at high and low bacterial density, abstr. A-119. Abstr. 109th Gen. Meet. Am. Soc. Microbiol. American Society for Microbiology, Washington, DC.