

Abstract
We previously demonstrated that the biguanide-based compound NB325 inhibits human immunodeficiency virus type 1 (HIV-1) infection by interacting with the CXCR4 viral coreceptor. This interaction also appeared to be persistent, since HIV-1 infection was inhibited even when the virus was introduced subsequent to the removal of NB325 from the cell culture medium. The present studies were conducted to determine the extent and mechanism of this prolonged antiviral activity. Persistent inhibition of HIV-1 infection by NB325 was concentration dependent and was apparent up to 8 h after removal of the compound. Flow cytometric analyses of stimulated CD4+ T lymphocytes exposed to NB325 demonstrated concentration-dependent reductions in CXCR4 extracellular loop 2 epitope recognition that were maintained up to 24 h after removal of the compound. CXCL12-induced chemotaxis was also persistently inhibited following pre-exposure to NB325. These results demonstrate that persistent inhibition of X4 HIV-1 infection by NB325 involves extended perturbation of the viral coreceptor CXCR4.
The progression of disease associated with human immunodeficiency virus type 1 (HIV-1) infection can be successfully controlled in many individuals through the use of highly active antiretroviral therapy (HAART). Efficacious drugs that target specific elements of the viral replication cycle—reverse transcription, protease activity, integration, virus-cell fusion, and coreceptor usage—are the basis for the current chemotherapeutic approaches to HIV-1 infection. However, the expense associated with an effective course of treatment, the emergence of viral strains resistant to drugs currently in use, and slow progress in the field of vaccine development all emphasize the urgent need for the development of new anti-HIV-1 drugs that act through novel mechanisms of action and have unique properties that enhance their efficacy.
One such property has been referred to as a “chemical barrier” against HIV-1 infection (2), as “antiviral memory” (9), or as a “prolonged inhibitory effect” (8). Antiviral compounds that have this property can inhibit HIV-1 infection even after extracellular drug concentrations have decreased below effective levels. UC781 (1), which is a potent thiocarboxanilide nonnucleoside reverse transcriptase inhibitor (NNRTI), was shown to significantly delay X4 HIV-1 infection of MT2 cells after only a 10-min pre-exposure (and washout) (2). Similarly, pre-exposure of human cervical explants to UC781 prior to R5 HIV-1 infection resulted in reductions in HIV-1 release, proviral DNA copy number, and virus dissemination by migratory cells up to six days after drug exposure (9). The NNRTI TMC-120 (dapivirine), which can act as a potent inhibitor of cell-free virus infectivity (17), was also shown to provide a prolonged inhibitory effect against HIV-1 infection in human cervical explants (8). These unique activities have been attributed to tight binding interactions between these compounds and HIV-1 reverse transcriptase (15). However, persistent protection is not a general characteristic of reverse transcriptase inhibitors, since neither tenofovir nor zidovudine was able to provide antiviral activity subsequent to pretreatment (17). Persistent inhibition of infection is also not a trait exclusive to reverse transcriptase inhibitors, since the entry inhibitor PSC-RANTES is presumed to have persistent antiviral activity as a consequence of prolonged intracellular sequestration of CCR5 (11).
Our efforts to develop a safe and effective inhibitor of HIV-1 have focused on biguanide (BG)-based compounds, with particular emphasis on the compound polyethylene hexamethylene biguanide (PEHMB). PEHMB is a BG-based molecule that carries an overall positive charge and is composed of biguanide subunits flanked by alternating linkers containing two or six methylene groups (Fig. 1). PEHMB is characterized by low levels of in vitro and in vivo toxicity and considerable in vitro efficacy against both the X4 and R5 strains of HIV-1 (7, 14). PEHMB (herein referred to as NB325) interacts with extracellular loop 2 (ECL2) of CXCR4, resulting in effective inhibition of X4 HIV-1 infection and inhibition of chemotaxis induced by CXCL12 through CXCR4 (20). The mechanism by which NB325 inhibits R5 HIV-1 infection is currently under investigation.
FIG. 1.

Polyethylene hexamethylene biguanide (PEHMB) structure. The structural formula and space-filling model of PEHMB (also known as NB325) are shown. PEHMB consists of alternating ethylene and hexamethylene linkers connecting biguanide subunits. The compound is readily soluble in water and polydisperse, with molecular weights ranging from approximately 900 to 1,900 (median molecular weight, 1,400).
The studies presented here demonstrated that NB325 is also characterized by persistent antiviral activity against HIV-1 infection. The persistent activity of NB325 against HIV-1 IIIB (X4) infection, which was evident in experiments up to 8 h after exposure to and removal of the compound, was hypothesized to involve the same CXCR4-dependent mechanism previously shown to inhibit infection in studies of immediate antiviral activity (20). Persistent interactions between NB325 and ECL2 of CXCR4 were demonstrated as prolonged reductions in CXCR4 epitope recognition and persistent inhibition of CXCL12-induced chemotaxis. The results of binding experiments using fluorescently labeled NB325 suggested that persistent virus inhibition (PVI) is a consequence of NB325 retention on the cell surface in association with CXCR4.
MATERIALS AND METHODS
Cells and viruses.
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood (Biological Specialty Corporation, Colmar, PA) using Ficoll-Paque Plus density gradient centrifugation as described by the manufacturer (GE HealthCare). Cells were cultured in RPMI medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin-streptomycin, and 0.05% sodium bicarbonate (RPMI complete medium). PBMC preparations were stimulated with 5 μg/ml phytohemagglutinin (PHA-P, catalog number L-1668; Sigma-Aldrich, St. Louis, MO) and 20 U/ml interleukin-2 (IL-2, catalog number 136; NIH AIDS Research and Reference Reagent Program, Germantown, MD) for 48 h and then IL-2 (20 U/ml) alone for 24 h. Subsequent to stimulation, CD4+ T cells were isolated by magnetic separation using CD4+ T cell enrichment technology and Auto MACS Pro (Miltenyi Biotec, Auburn, CA). HIV-1 strain IIIB was obtained from Advanced Biotechnologies, Inc. (Columbia, MD).
Compounds.
Dextran sulfate (DS) (Dextralip 50, catalog number D8787, lot 71K1278) was purchased from Sigma-Aldrich. NB325 synthesis was performed using procedures similar to those described for PEHMB (14) and polyhexamethylene biguanide (PHMB) (18) synthesis. During the production of NB325, the synthesis reaction was performed in a closed vessel under ambient atmosphere for 24 h. NB325 was water soluble and polydisperse, with molecular masses ranging from approximately 900 to 1,900 Da (at 1,400 Da median molecular mass, 0.02% NB325 is equivalent to approximately 143 μM). Fluorescently labeled NB325 was produced using an Alexa Fluor 647 protein labeling kit (catalog number A20173; Molecular Probes, Invitrogen, Eugene, OR). Briefly, after labeling and purification, the degree of labeling was determined to be approximately 95% (data not shown).
Viral infection inhibition assay.
Primary CD4+ T lymphocytes, stimulated as described above, were seeded at a density of 1 × 105 cells/well in a 96-well plate. Cells were then infected with HIV-1 IIIB for 1 h in the absence or presence of NB325 or DS. After 1 h, cells were washed and subsequently cultured for 3 days, at which time the cells were washed and supplied with new medium supplemented with IL-2. After an additional 3 days, HIV-1 production was assayed by determining the level of p24 core antigen in the supernatant using an HIV-1 p24 antigen enzyme-linked immunosorbent assay (ELISA) (Zeptometrix/VWR). The remaining infectivity is expressed relative to that of mock-treated HIV-1-infected cells. The IC50 and IC90 (concentrations at which exposure to the compound resulted in 50% or 90% decreases, respectively, in infectivity relative to that of mock-treated HIV-1-infected cells) were calculated using the Forecast function of Microsoft Excel. Data points are shown as mean values and calculated standard deviations.
Persistent virus inhibition assay.
Primary CD4+ T lymphocytes, stimulated as described above, were incubated with the IC50 or IC90 of NB325 or DS for 1 h. Cells were then challenged with HIV-1 IIIB for 1 h at 37°C either immediately after removal of the compound (by extensive cell washes), up to 24 h after compound washout and incubation in new medium without the compound, or concurrently with incubation with the compound. Following infection, cells were washed and then assayed for HIV-1 production as described above.
Cytotoxicity assay.
Primary CD4+ T lymphocytes, stimulated as described above, were incubated with the NB325 compound or mock exposed for 1 h at 37°C. Following thorough washing, cells were incubated with fluorochrome-conjugated annexin V (BD Biosciences, San Jose, CA), propidium iodide (BD Biosciences), and antibodies specific for CD4 and CD45RO either immediately following exposure or at the time points after exposure indicated below. Annexin V indicates early apoptosis (23), while reaction with propidium iodide is indicative of necrotic cell death (13). For the delayed toxicity component of the experiments, cells were resuspended in RPMI complete medium and incubated at 37°C until they were assayed for cytotoxicity at the time points indicated below.
Flow cytometry.
Primary CD4+ T lymphocytes, stimulated as described above, were incubated with NB325 or mock exposed for 1 h at 37°C. Following incubation, cells were washed three times with Hank's balanced salt solution (HBSS; Cellgro, Manassas, VA) supplemented with 3% FBS and 0.02% NaN3 and then incubated with fluorochrome-conjugated antibodies or their corresponding isotype controls for 45 min at 4°C either immediately following exposure or up to 24 h after exposure. Assays included the following antibodies: anti-CD4 (clone RPA-T4; BD Biosciences), anti-CD45RO (clone UCHL1; BD Biosciences), and anti-CXCR4 (clones 12G5 and 1D9 [BD Biosciences] and clone 173 [R&D Systems, Minneapolis, MN]). The isotype control antibodies used in these studies included mouse IgG1 (eBioscience), mouse IgG2a (eBioscience), mouse IgG2b (eBioscience), and rat IgG2a (BD Biosciences). Antibodies (and their corresponding isotype controls) were used in the following quantities per assay: anti-CD4 and anti-CXCR4, 1 μg; anti-CD45RO, 0.06 μg; and annexin V, 0.01 μg (as described by the manufacturer). Subsequently, cells were washed with supplemented HBSS and then fixed with 1% formaldehyde at 4°C. Samples were examined on a FACSCaliber (Becton Dickinson, Franklin Lakes, NJ) and analyzed using Flow Jo software, version 8.5.3 (Tree Star, Ashland, OR). Annexin V was also examined in every sample, and annexin V-positive cells were eliminated from all analyses.
The presence of NB325 on the surface of CD4+ T lymphocytes was also quantified using flow cytometry. Briefly, cells were incubated with unlabeled or labeled NB325 for 1 h at 37°C. Following exposure, cells were left unwashed or washed three times with supplemented HBSS. Cells were then incubated with specific antibodies or reagents, fixed, and collected for flow cytometry as described above.
Chemotaxis.
CD4+ T lymphocytes, stimulated as described above, were incubated with the NB325 compound or mock exposed for 1 h or 2 h (as indicated) at 37°C. Following exposure, mock-exposed and NB325-pretreated cells were washed and then seeded at 1 × 106 cells in 0.1 ml in the absence or presence of NB325 in the top well of a 24-well transmigration chamber (5-μm pore size, polycarbonate filter Transwell; Costar, Cambridge, MA). The lower chamber was filled with 0.6 ml of RPMI complete medium alone or supplemented with 250 ng/ml CXCL12, the compound at the concentration indicated below, or a combination of CXCL12 and the compound. Plates were incubated for 2 h at 37°C. Following the incubation, cells that had migrated to the lower chamber were quantitated using Trypan blue dye exclusion on a hemacytometer. Chemotaxis inhibition is expressed relative to the CXCL12-induced chemotaxis in the absence of NB325. For pre-exposure inhibition of CXCL12-induced chemotaxis, the background migration toward medium alone was subtracted from the migration in response to CXCL12 for the corresponding samples.
RESULTS
NB325 provides persistent protection from infection by X4 HIV-1 strain IIIB.
Because previous studies demonstrated that inhibition of X4 HIV-1 infection by NB325 was attributable to CXCR4 antagonism (20), it was hypothesized that PVI was the result of persistent interactions between NB325 and CXCR4 that provide a window of antiviral activity after the removal of NB325 from the medium. To test this hypothesis, experiments were performed to document the level and duration of PVI provided by NB325 and to determine the underlying mechanism of this extended inhibition of HIV-1 infection.
Primary human CD4+ T lymphocytes were first incubated with HIV-1 IIIB and NB325 or dextran sulfate (DS) for 1 h to demonstrate concentration-dependent inhibition of HIV-1 infection in the presence of NB325 in the medium. In these experiments, NB325 inhibited HIV-1 IIIB infection with an IC50 of 0.018% and an IC90 of 0.05% (Fig. 2A). In subsequent assays, cells were incubated with the IC50 or IC90 of NB325 or DS for 1 h and then washed extensively. Cells were then immediately challenged with cell-free HIV-1 for 1 h or incubated in NB325-free medium for up to 7 h prior to HIV-1 infection.
FIG. 2.
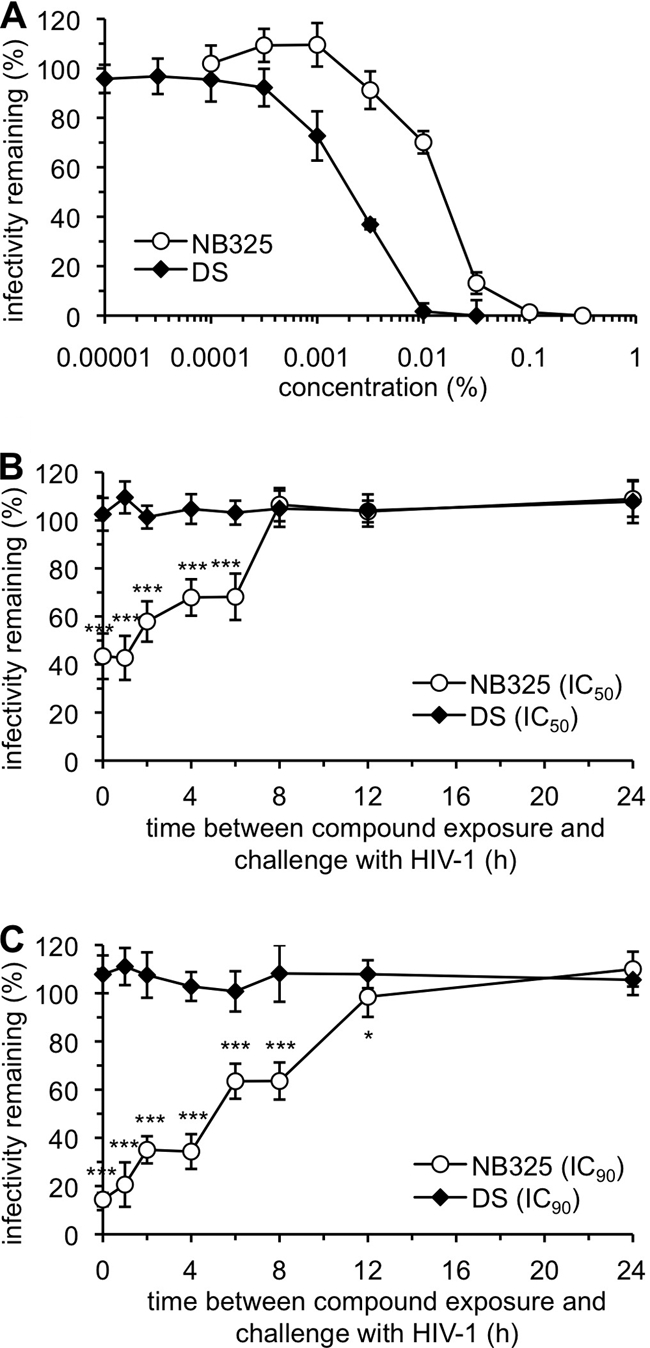
NB325 pre-exposure results in persistent protection from infection by HIV-1 strain IIIB. (A) Stimulated CD4+ T lymphocytes were infected with HIV-1 strain IIIB in the absence or presence of NB325 or dextran sulfate (DS) for 1 h at 37°C. These results were used to establish the NB325 IC50 (0.018%) and IC90 (0.05%) for subsequent experiments. The IC50 and IC90 for DS were established similarly. Stimulated CD4+ T lymphocytes were incubated with the IC50 (B) or IC90 (C) of NB325 or DS for 1 h at 37°C. Cells were then challenged with HIV-1 IIIB immediately after compound washout (0 h) or at up to 24 h after exposure and washout. The remaining infectivity is expressed as the percentage relative to the results for infected, mock-exposed cells. Each graph is representative of data from two independent experiments in which each data point was examined in triplicate. Statistical significance was calculated in comparison to results for mock-exposed cells (unless otherwise indicated) using the two-tailed, unpaired Student's t test (*, P ≤ 0.05, and ***, P ≤ 0.001).
Pre-exposure to the IC50 of NB325 resulted in a 50% reduction in HIV-1 infection when virus was added immediately or 30 min after compound washout (Fig. 2B). Beyond 30 min after washout, the level of PVI was time dependent; by 6 h after exposure, the antiviral activity of NB325 was diminished (32% inhibition) relative to its activity when incubated simultaneously with cells and virus. No PVI was observed at time points following 6 h after exposure to NB325 at this concentration. Dextran sulfate (DS), which interacts with the viral envelope as its mechanism of action (16), provided no protection from subsequent infection following removal of the compound (Fig. 2B and C). Application of NB325 at its IC90 provided persistent inhibition of infection up to 8 h after exposure when challenged with HIV-1 (Fig. 2C). These results, which are consistent with the proposed mechanism of action for NB325 (20), demonstrated that the magnitude of PVI provided by NB325 was both time and dose dependent.
Pre-exposure to NB325 is not associated with increased or delayed cytotoxicity.
To demonstrate that compound toxicity was not affecting the results of the preceding compound washout assays, CD4+ T lymphocytes were assayed for cytotoxicity following NB325 exposure and washout. NB325 cytotoxicity following a 1-h exposure (Fig. 3A) was evident only at 0.1% and above (above the NB325 IC90). In addition, there was no delayed cytotoxicity associated with NB325 exposure, since the concentration-dependent levels of cytotoxicity measured immediately after exposure were identical to those measured at 24 h after exposure (Fig. 3A). Cells were also exposed to the IC50 and IC90 of NB325 for 1 h and assayed for cytotoxicity at time points after washout identical to those used in the previous experiment. No cell necrosis or apoptosis was observed as a consequence of NB325 exposure at any of the time points up to 24 h after exposure to NB325 (Fig. 3B). These results indicate that compound cytotoxicity did not play a role in the persistent protection from infection afforded by NB325.
FIG. 3.
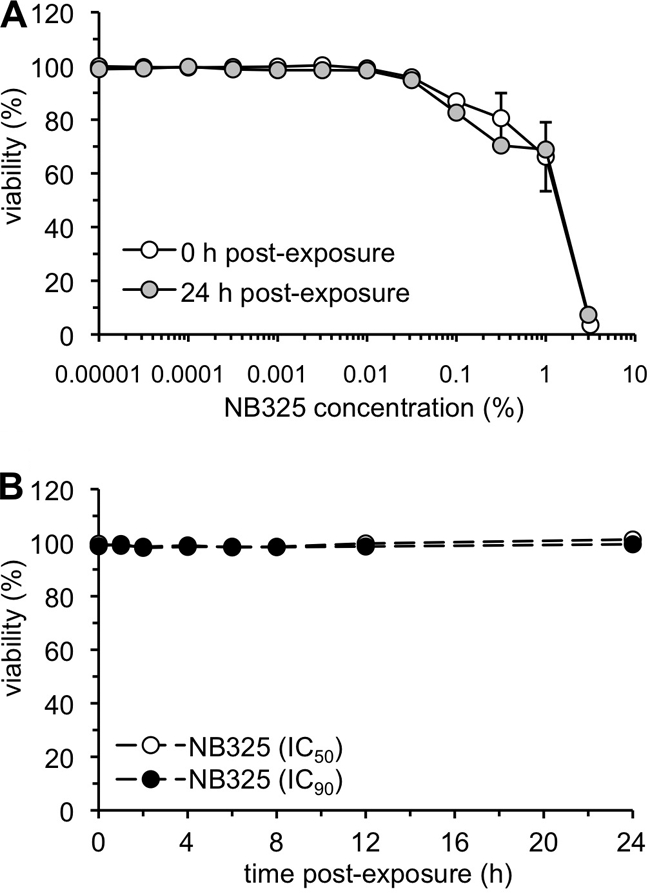
IC50 and IC90 of NB325 have no effect on cell viability. Stimulated CD4+ T lymphocytes were incubated with compound for 1 h at 37°C and then washed thoroughly. (A) Cytotoxicity at the indicated concentrations was assessed (as described in Materials and Methods) immediately following incubation or at 24 h after exposure. Error bars show standard deviations. (B) NB325 cytotoxicity at the calculated IC50 (0.018%) and IC90 (0.05%) was assessed at the indicated times after exposure. Viability (%) is expressed with respect to that of mock-exposed cells. Each graph is representative of data from two independent experiments in which each data point was examined in triplicate.
Epitope-specific disruption of CXCR4 detection caused by NB325 pre-exposure is apparent up to 24 h after exposure.
Previous studies demonstrated that the mechanism underlying the immediate activity of NB325 involves domain-specific interactions with CXCR4 (20). First noted in experiments using P4-R5 MAGI cells (a HeLa-based HIV-1 indicator cell line) and PM-1 cells (a T lymphocyte cell line that naturally expresses both CXCR4 and CCR5) (N. Thakkar and F. C. Krebs, unpublished observations), the epitope-specific mechanism of HIV-1 inhibition was subsequently documented in experiments involving primary human CD4+ T lymphocytes (20). To determine whether the same mechanism was associated with PVI, the impact of NB325 exposure on CXCR4 epitope recognition on memory CD4+ T lymphocytes was examined.
Following a 1-h exposure, the frequency of memory CD4+ T lymphocytes with CXCR4 detected by clone 12G5 (specific to a conformation-dependent epitope involving ECL1 and ECL2) was decreased in a concentration-dependent manner (Fig. 4A and B), as previously observed following a 2-h exposure (20). Exposure to NB325 at its IC50 resulted in a time-dependent decrease that returned to nearly mock-exposed levels by 8 h after exposure. Exposure to NB325 at its IC90 also resulted in a time-dependent decrease in detection. However, the decreased epitope recognition by clone 12G5 did not return to mock-exposed levels through 24 h after exposure, but instead reached a plateau (∼80%) by 8 to 12 h after exposure.
FIG. 4.

Epitope-specific disruption of CXCR4 detection caused by pre-exposure to NB325 is apparent up to 24 h after exposure. Stimulated CD4+ T lymphocytes were incubated in the absence or presence of NB325 for 1 h at 37°C. Cells were incubated immediately or up to 24 h after NB325 exposure and washout with fluorochrome-conjugated antibodies specific for CD4 (clone RPA-T4), CD45RO (clone UCHL1), and CXCR4 (clones 1D9, 12G5, and 173). The graphs and selected scatter plots (from assays at 0 h and 8 h after exposure to the IC50 and IC90 of NB325) display the frequencies and numbers (respectively) of CD45RO+ CD4+ T lymphocytes expressing CXCR4 as detected by clones 12G5 (A, B), 173 (C, D), or 1D9 (E), as well as the frequency of CD4 detection (F). Each graph is representative of data from two independent assays in which each concentration was examined in duplicate. Scatter plots depict results from a single, representative experiment. The number in each scatter plot quadrant indicates the percentage of cells (with respect to the total number of sampled cells) found in that quadrant.
A time-dependent reduction in the recognition of the ECL2 epitope (by antibody 173) was also observed (Fig. 4C and D). The greater reduction in ECL2 epitope recognition compared to that observed with antibody 12G5 was also consistent with previous observations after a 2-h NB325 exposure (20). Exposure to the IC50 of NB325 resulted in decreased recognition of CXCR4 through 24 h after exposure, with a plateau in recognition at times later than approximately 4 h after exposure. Exposure to NB325 at its IC90 also resulted in a time-dependent decrease in epitope recognition which persisted through 24 h after exposure and also appeared to plateau at approximately 6 h after exposure.
In contrast, no changes were detected in the frequency of memory CD4+ T lymphocytes expressing CXCR4 as determined through the use of the 1D9 monoclonal antibody, which recognizes the CXCR4 N terminus (Fig. 4E). These results were identical to observations made in previous experiments after a 2-h exposure to NB325 (20). CXCR4 N terminus epitope recognition remained unchanged through 24 h after exposure to NB325.
The effects of NB325 on CXCR4 recognition across the population of memory CD4+ T lymphocytes were confirmed on a per-cell basis by determining the mean fluorescence intensity (MFI) for each antibody at the time points indicated in Fig. 4 following a 1-h exposure to the IC50 or IC90 of NB325. The analyses verified no significant change in the MFI of CXCR4 as detected by clone 1D9 through 24 h after exposure (data not shown). In contrast, NB325 exposure caused time-and concentration-dependent decreases in the MFI of CXCR4 (data not shown), as detected by monoclonal antibody clones 12G5 and 173, similar to those illustrated in Fig. 4.
To demonstrate that changes in cell surface protein recognition following NB325 exposure were limited to CXCR4, both the frequency of cells with detectable CD4 and the MFI of CD4 detection were determined at time points up to 24 h after exposure. No significant changes in the detection of CD4, as indicated by cell frequency (Fig. 4F) or MFI (data not shown), were noted up to 24 h after exposure. Similar results (data not shown) were obtained using the CD4-specific monoclonal antibody OKT4, which binds to an epitope distinct from that recognized by the antibody RPA-T4 (19). These results support the conclusion that PVI involves the viral coreceptor CXCR4 but not the CD4 receptor.
Pre-exposure to NB325 inhibits CXCL12-induced chemotaxis.
Previous studies (20) presented inhibition of CXCL12-mediated chemotaxis as further evidence that NB325 was interacting with CXCR4. By extension, chemotaxis inhibition experiments were similarly designed to provide additional evidence that NB325 PVI involved a durable and functional interaction between NB325 and CXCR4. In experiments in which cells were pre-exposed to the IC50 or IC90 of NB325, the inhibition of CXCL12 chemotaxis by a 1-h pre-exposure and washout was comparable to the inhibition achieved by a 2-h concurrent exposure to both NB325 and CXCL12 (Fig. 5A). In extended experiments, NB325 effectively inhibited CD4+ T lymphocyte chemotaxis induced by CXCL12 up to 6 h after exposure and washout (Fig. 5B). NB325 introduced at the IC90 was particularly effective, achieving complete inhibition of chemotaxis up to 12 h after exposure and approximately 80% inhibition compared to that of mock-exposed cells at 24 h after exposure. At its IC50, NB325 was somewhat less effective up to 6 h after exposure (relative to the results with its IC90) and increasingly less effective between 8 and 24 h after exposure. In these experiments, NB325 alone did not induce any measurable chemotaxis (data not shown).
FIG. 5.
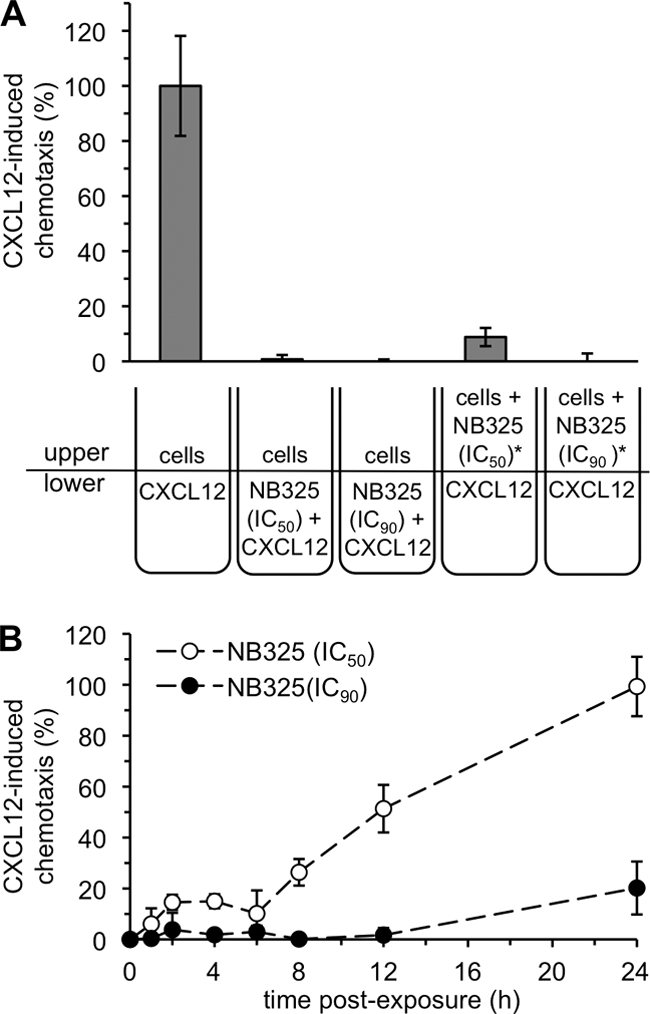
Pre-exposure to NB325 inhibits CXCL12-induced chemotaxis through CXCR4. (A) Chemotaxis of stimulated CD4+ T lymphocytes was induced for 2 h in the presence of CXCL12 alone or CXCL12 and NB325. In a variation of the experiment (*), cells were pre-exposed to NB325 for 1 h, washed, and then immediately exposed to CXCL12 in a transmigration chamber. In this experiment, 100% chemotaxis equated to migration of 470,000 cells. (B) Stimulated CD4+ T lymphocytes were incubated with NB325 or mock exposed for 1 h. Following exposure, chemotaxis of pretreated or mock-exposed cells was induced by CXCL12 in a transmigration chamber in the absence of NB325. Induction by CXCL12 was immediate or at the indicated time points after washout. The extent of chemotaxis (%) was calculated with respect to the number of cells that migrated in response to CXCL12 alone (213,000 cells). Results displayed are representative of two independent assays, in which each condition was examined in triplicate. Error bars show standard deviations.
NB325 binds persistently to CD4+ T lymphocytes.
Cellular retention of TMC120 and UC781 has been shown to be one mechanism by which these compounds provide persistent antiviral activity. To determine if NB325 retention by the cell is also the cause of its persistent antiviral activity, flow cytometry-based experiments were performed with fluorescently labeled NB325. Assays of cytotoxicity and antiviral activity demonstrated that the labeled compound was indistinguishable from the unlabeled compound (data not shown). Incorporation of the fluorescent label also had no impact on CXCR4 epitope recognition (data not shown), indicating that the labeled compound was mechanistically identical to the parent compound within the limits of the experimental conditions examined.
In these experiments, the frequency of memory CD4+ T lymphocytes that retained labeled NB325 decreased in a time-and concentration-dependent manner (Fig. 6A and B). By 8 h after exposure to the IC50 of labeled NB325, the frequency of memory CD4+ T lymphocytes that retained labeled NB325 decreased to approximately 10% and remained stable through 24 h after exposure. While the frequency of NB325-positive cells dropped considerably during the first 4 h after exposure to the IC50 of labeled NB325, the decline in the amount of NB325 retained following exposure to the IC90 was much less over the 24-h period (55% NB325-positive cells at 24 h). As expected, the MFI of NB325-positive cells immediately following exposure to the IC90 was much greater than that following exposure to the IC50 (Fig. 6C). Concentration-dependent differences in MFI were only apparent out to 8 h after exposure to labeled NB325. In combination, these results indicate that NB325 is retained on the surface of memory CD4+ T lymphocytes up to 24 h after exposure; however, the amount of NB325 retained on each cell is greatly reduced by approximately 8 h after exposure.
FIG. 6.

NB325 binds persistently to CD4+ T lymphocytes in association with CXCR4. Stimulated CD4+ T lymphocytes were incubated with fluorochrome-conjugated NB325 for 1 h at 37°C. Following exposure, cells were washed, incubated with fluorochrome-conjugated monoclonal antibodies specific for CD4, CD45RO, and CXCR4, and collected for flow cytometry. (A and B) The graph (A) and selected scatter plots (from assays at 0 h and 8 h after exposure to the IC50 and IC90 of NB325) (B) display the frequencies and numbers (respectively) of memory CD4+ T lymphocytes retaining NB325. (C) Graph depicts the MFI of NB325 on the surface of memory CD4+ T lymphocytes. The graphed results are representative of three independent assays in which each condition was examined in triplicate. Scatter plots depict results from a single representative experiment. The number in each scatter plot quadrant indicates the percentage of cells (with respect to the total number of sampled cells) found in that quadrant.
DISCUSSION
In the studies presented here, we demonstrated that the biguanide-based compound NB325 provides persistent protection from HIV-1 infection in a time-and concentration-dependent manner up to 12 h after exposure. The results of previous experiments demonstrated that the mechanism underlying the immediate antiviral activity of NB325 involved a domain-specific interaction with CXCR4 (20). The present studies demonstrated that the interaction between NB325 and CXCR4 persists in a time- and concentration-dependent manner through 24 h after exposure. Furthermore, these studies demonstrated that NB325 is maintained on the surface of memory CD4+ T lymphocytes up to 24 h after exposure, suggesting that the persistent retention of NB325 is, in large part, responsible for its PVI and its persistent inhibition of CXCL12-induced chemotaxis.
In comparing the duration of the persistent antiviral activity and the persistence of reductions in epitope recognition, it was apparent that measurable reductions in CXCR4 epitope recognition were observed in the absence of antiviral activity. For example, persistent antiviral activity was not evident beyond 12 h after exposure to NB325 at its IC90 (Fig. 2C), despite clear reductions in CXCR4 detection up to 24 h after exposure to the same concentration of NB325 (Fig. 4A through D). These observations suggest that a minimum threshold of interactions between NB325 and CXCR4 is required for a measurable reduction in HIV-1 infection. These results, which may be a function of the stoichiometry of interactions between NB325 and CXCR4, will need to be explored in further investigations focused on mechanisms of action.
Compounds shown (or presumed) to confer PVI differ in the mechanisms that provide this effect. PSC-RANTES, which is a CCR5-specific HIV-1 entry inhibitor, presumably provides persistent antiviral activity through prolonged intracellular sequestration of CCR5 (11). PVI attributed to the NNRTIs TMC120 and UC781 (2, 15, 17, 21, 22) is likely a consequence of compound penetration of the host cell membrane (15) and intracellular inhibition of reverse transcription (10, 15). Alternatively, these compounds may penetrate the envelopes of cell-free virions by the same mechanism and inhibit the reverse transcriptase molecules packaged in the virion even before the virus has attached to and entered the cell (2, 17).
The probable mechanism responsible for NB325 PVI is the retention of NB325 on the cell surface and durable interactions with CXCR4. This mechanism is consistent with persistent reductions in CXCR4 epitope recognition (Fig. 4A through D) and retention of labeled NB325 on the cell surface (Fig. 6). Prolonged inhibition of infection does not appear to involve CXCR4 internalization, since detection of the CXCR4 N terminus was unchanged following NB325 exposure and washout (Fig. 4E). The current results, however, cannot be used to preclude the effects of nonspecific interactions between NB325 and the cell membrane. Studies of the BG-based molecule polyhexamethylene biguanide (PHMB), which differs from NB325 in the linkers separating the biguanide units (14), demonstrated nonspecific interactions with anionic phospholipids contained in the cellular membrane (3-6, 12). Interactions such as these between NB325 and an HIV-1-susceptible cell might promote the retention of NB325 on the cell surface and increase the local concentration of NB325 available for interactions with CXCR4.
Regardless of the mechanism, persistent retention of the compound as a consequence of either an extended duration of exposure or repetitive application might be anticipated to increase the otherwise low cytotoxicity of NB325. However, experimental results to date do not support this hypothesis and indicate only limited toxicity under these circumstances. As shown by the results described above, there was no delayed cytotoxicity associated with chronic NB325 retention (Fig. 3B), nor was concentration-dependent cytotoxicity affected by long-term (up to 24 h) retention of the compound (Fig. 3A). Similarly, in vitro experiments involving extended, continuous exposure (2, 4, 8, or 24 h) of HeLa or P4-R5 MAGI cells to NB325 demonstrated relatively small increases in concentration-dependent cytotoxicity even after the longest exposure to NB325 (data not shown). Furthermore, continuous exposure to PEHMB (NB325) for 10 min or 2, 4, 8, or 24 h resulted in minimal epithelial disruption and inflammation of the cervical mucosa in experiments involving a mouse model of cervicovaginal toxicity (7).
The results of previous and ongoing studies indicate that NB325 (PEHMB) functions outside HIV-1-susceptible cells as a viral entry inhibitor. Without the requirement for cell penetration, lower concentrations of this compound (or its derivatives) may be required in vivo to achieve an acceptable level of efficacy by either immediate or persistent antiviral activity. In addition, since BG-based compounds like NB325 are highly soluble, the challenges associated with compound formulation and delivery may not be as great as those associated with more lipophilic compounds. In highlighting the persistent antiviral activity attributable to NB325, the present study describes another positive aspect of NB325 as an HIV-1 inhibitor and provides further justification for the continued development of BG-based compounds and their derivatives as novel agents effective against HIV-1.
Acknowledgments
These studies were supported through a grant from the National Institute of Allergy and Infectious Diseases, National Institutes of Health (1 U19 AI076965, Mohamed Labib, principal investigator, and Brian Wigdahl, co-principal investigator). The research was also supported by research development funds provided by the Department of Microbiology and Immunology and the Institute for Molecular Medicine and Infectious Disease, Drexel University College of Medicine.
Labeled NB325 was graciously produced by Hosahudya Gopi (Drexel University College of Medicine).
Footnotes
Published ahead of print on 15 March 2010.
REFERENCES
- 1.Balzarini, J., W. G. Brouwer, D. C. Dao, E. M. Osika, and E. De Clercq. 1996. Identification of novel thiocarboxanilide derivatives that suppress a variety of drug-resistant mutant human immunodeficiency virus type 1 strains at a potency similar to that for wild-type virus. Antimicrob. Agents Chemother. 40:1454-1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borkow, G., J. Barnard, T. M. Nguyen, A. Belmonte, M. A. Wainberg, and M. A. Parniak. 1997. Chemical barriers to human immunodeficiency virus type 1 (HIV-1) infection: retrovirucidal activity of UC781, a thiocarboxanilide nonnucleoside inhibitor of HIV-1 reverse transcriptase. J. Virol. 71:3023-3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broxton, P., P. M. Woodcock, and P. Gilbert. 1984. Binding of some polyhexamethylene biguanides to the cell envelope of Escherichia coli ATCC 8739. Microbios 41:15-22. [PubMed] [Google Scholar]
- 4.Broxton, P., P. M. Woodcock, and P. Gilbert. 1984. Injury and recovery of Escherichia coli ATCC 8739 from treatment with some polyhexamethylene biguanides. Microbios 40:187-193. [PubMed] [Google Scholar]
- 5.Broxton, P., P. M. Woodcock, and P. Gilbert. 1983. A study of the antibacterial activity of some polyhexamethylene biguanides towards Escherichia coli ATCC 8739. J. Appl. Bacteriol. 54:345-353. [DOI] [PubMed] [Google Scholar]
- 6.Broxton, P., P. M. Woodcock, F. Heatley, and P. Gilbert. 1984. Interaction of some polyhexamethylene biguanides and membrane phospholipids in Escherichia coli. J. Appl. Bacteriol. 57:115-124. [DOI] [PubMed] [Google Scholar]
- 7.Catalone, B. J., T. M. Kish-Catalone, L. R. Budgeon, E. B. Neely, M. Ferguson, F. C. Krebs, M. K. Howett, M. Labib, R. Rando, and B. Wigdahl. 2004. Mouse model of cervicovaginal toxicity and inflammation for preclinical evaluation of topical vaginal microbicides. Antimicrob. Agents Chemother. 48:1837-1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fletcher, P., S. Harman, H. Azijn, N. Armanasco, P. Manlow, D. Perumal, M. P. de Bethune, J. Nuttall, J. Romano, and R. Shattock. 2009. Inhibition of human immunodeficiency virus type 1 infection by the candidate microbicide dapivirine, a nonnucleoside reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 53:487-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fletcher, P., Y. Kiselyeva, G. Wallace, J. Romano, G. Griffin, L. Margolis, and R. Shattock. 2005. The nonnucleoside reverse transcriptase inhibitor UC-781 inhibits human immunodeficiency virus type 1 infection of human cervical tissue and dissemination by migratory cells. J. Virol. 79:11179-11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenhead, P., P. Hayes, P. S. Watts, K. G. Laing, G. E. Griffin, and R. J. Shattock. 2000. Parameters of human immunodeficiency virus infection of human cervical tissue and inhibition by vaginal virucides. J. Virol. 74:5577-5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartley, O., H. Gaertner, J. Wilken, D. Thompson, R. Fish, A. Ramos, C. Pastore, B. Dufour, F. Cerini, A. Melotti, N. Heveker, L. Picard, M. Alizon, D. Mosier, S. Kent, and R. Offord. 2004. Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc. Natl. Acad. Sci. U. S. A. 101:16460-16465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda, T., A. Ledwith, C. H. Bamford, and R. A. Hann. 1984. Interaction of a polymeric biguanide biocide with phospholipid membranes. Biochim. Biophys. Acta 769:57-66. [DOI] [PubMed] [Google Scholar]
- 13.Jones, K. H., and D. A. Kniss. 1987. Propidium iodide as a nuclear counterstain for immunofluorescence studies on cells in culture. J. Histochem. Cytochem. 35:123-125. [DOI] [PubMed] [Google Scholar]
- 14.Krebs, F. C., S. R. Miller, M. L. Ferguson, M. Labib, R. F. Rando, and B. Wigdahl. 2005. Polybiguanides, particularly polyethylene hexamethylene biguanide, have activity against human immunodeficiency virus type 1. Biomed. Pharmacother. 59:438-445. [DOI] [PubMed] [Google Scholar]
- 15.Motakis, D., and M. A. Parniak. 2002. A tight-binding mode of inhibition is essential for anti-human immunodeficiency virus type 1 virucidal activity of nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 46:1851-1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moulard, M., H. Lortat-Jacob, I. Mondor, G. Roca, R. Wyatt, J. Sodroski, L. Zhao, W. Olson, P. D. Kwong, and Q. J. Sattentau. 2000. Selective interactions of polyanions with basic surfaces on human immunodeficiency virus type 1 gp120. J. Virol. 74:1948-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Njai, H. F., P. J. Lewi, C. G. Janssen, S. Garcia, K. Fransen, L. Kestens, G. Vanham, and P. A. Janssen. 2005. Pre-incubation of cell-free HIV-1 group M isolates with non-nucleoside reverse transcriptase inhibitors blocks subsequent viral replication in co-cultures of dendritic cells and T cells. Antivir. Ther. 10:255-262. [PubMed] [Google Scholar]
- 18.O'Malley, L. P., K. Z. Hassan, H. Brittan, N. Johnson, and A. N. Collins. 2006. Characterization of the biocide polyhexamethylene biguanide by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. J. Appl. Polym. Sci. 102:4928-4936. [Google Scholar]
- 19.Reinherz, E. L., P. C. Kung, G. Goldstein, and S. F. Schlossman. 1979. Separation of functional subsets of human T cells by a monoclonal antibody. Proc. Natl. Acad. Sci. U. S. A. 76:4061-4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thakkar, N., V. Pirrone, S. Passic, W. Zhu, V. Kholodovych, W. Welsh, R. F. Rando, M. E. Labib, B. Wigdahl, and F. C. Krebs. 2009. Specific interactions between the viral coreceptor CXCR4 and the biguanide-based compound NB325 mediate inhibition of human immunodeficiency virus type 1 infection. Antimicrob. Agents Chemother. 53:631-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Herrewege, Y., J. Michiels, J. Van Roey, K. Fransen, L. Kestens, J. Balzarini, P. Lewi, G. Vanham, and P. Janssen. 2004. In vitro evaluation of nonnucleoside reverse transcriptase inhibitors UC-781 and TMC120-R147681 as human immunodeficiency virus microbicides. Antimicrob. Agents Chemother. 48:337-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Herrewege, Y., G. Vanham, J. Michiels, K. Fransen, L. Kestens, K. Andries, P. Janssen, and P. Lewi. 2004. A series of diaryltriazines and diarylpyrimidines are highly potent nonnucleoside reverse transcriptase inhibitors with possible applications as microbicides. Antimicrob. Agents Chemother. 48:3684-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vermes, I., C. Haanen, H. Steffens-Nakken, and C. Reutelingsperger. 1995. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184:39-51. [DOI] [PubMed] [Google Scholar]