

Abstract
The hexapeptide WRWYCR was previously identified on the basis of its ability to inhibit bacteriophage lambda integrase-mediated recombination by trapping and preventing resolution of the Holliday junction intermediate. This peptide inhibits several unrelated DNA repair enzymes that bind to and process Holliday junctions and branched DNA substrates. WRWYCR and its d stereoisomer, wrwycr, are bactericidal against both Gram-positive and Gram-negative bacteria, causing the accumulation of DNA breaks, chromosome segregation defects, and the filamentation of cells. DNA repair is a novel target of antibiotics. In the present study, we examined the ability of the peptides to inhibit the growth of Salmonella in mammalian cells. J774A.1 macrophage-like cells and murine peritoneal macrophages were infected with Salmonella enterica serovar Typhimurium and grown in the presence or absence of peptide. We found that peptide wrwycr reduced the number of Salmonella cells recovered after 24 h growth in J774A.1 cells by 100 to 1,000 times, depending on the multiplicity of infection. The peptide also inhibited Salmonella growth in peritoneal macrophages, and although higher doses were required, these were not toxic to the host cells. The apparent lower level of potency of the peptide paralleled the lower level of replication of Salmonella and the lower level of permeation of the peptide in the peritoneal macrophages than in the J774.1 cells. Treatment with peptide wrwycr elicited the SOS response in a significant fraction of the intracellular bacteria, as would be expected if the mechanism of bacterial killing was the same in pure culture and in host cells. These results represent a proof of principle of the antimicrobial activities of compounds that target DNA repair.
The hexapeptide WRWYCR was previously identified on the basis of its ability to inhibit bacteriophage lambda integrase (Int)-mediated site-specific recombination by accumulating the Holliday junction (HJ) intermediates during the reaction (4). Holliday junctions are also central intermediates in homologous recombination-dependent DNA repair, which often but not exclusively involves RecA-dependent strand invasion. The double-strand breaks that are generated by oxidative damage, irradiation, or interstrand cross-links or nicks that have been converted to double-strand breaks during replication require repair of the break by use of the homology on the sister chromosome (14, 15, 28). During the repair process, chromosome dimers may arise. Such dimeric chromosomes cannot be properly segregated to daughter cells, unless they are converted to monomers by the Int-related bacterial site-specific recombinases XerC and XerD, which also generate Holliday junction intermediates during recombination at dif sites (3). These processes are extremely important, if not essential; for example, up to 50% of recA mutant cells are nonviable, and the remainder are hypersensitive to any DNA damage (14). Although mutations in XerC and/or XerD are not lethal, they are detrimental to cell growth, particularly under conditions that cause DNA damage (32). DNA repair has been inferred to be extremely important for the survival of Salmonella during infections, on the basis of the finding that recA and recBC mutants are avirulent (they have a more than 4-log-unit greater 50% lethal dose in mice than Rec+ strains) and that Salmonella isolates growing within macrophages incur a variety of DNA lesions, including DNA breaks (8, 9, 30). Peptide WRWYCR may thus inhibit bacterial growth, particularly under physiological conditions in which DNA repair is necessary.
Peptides WRWYCR and its d stereoisomer, wrwycr, do not require contact with proteins but, rather, are structure selective for branched DNAs. As they are dimers via a disulfide bridge, they recognize and bind to four-arm (Holliday) junctions (Kd, 12 to 14 nM) and bind with a reduced affinity and lesser stability to three-arm DNA structures that mimic replication forks (Kd, 64 to 132 nM) (25, 26; J. Boldt, R. Saha, J. Arciniega, and A. M. Segall, unpublished results). This enables the peptides to inhibit a number of structurally and mechanistically unrelated proteins that process HJs, D loops, and related DNA intermediates of DNA repair (10, 22, 25, 26).
Both WRWYCR and wrwycr inhibit bacterial growth in a dose-dependent manner, with wrwycr showing greater potency at longer incubation times, presumably due to its resistance to proteases. MIC assays showed that Gram-negative bacteria (Escherichia coli, Salmonella) were less sensitive to the peptide (64 μM) than Gram-positive bacteria (Bacillus subtilis, Lactococcus lactis, Listeria monocytogenes, Staphylococcus aureus, methicillin-resistant S. aureus) and Gram-negative bacteria with mutationally shortened lipopolysaccharide (LPS) chains (8 to 32 μM), while viability assays revealed that the peptides are bactericidal at 1× to 2× MICs (22). More recently, Debora Barnett Foster's laboratory showed that the peptide has activity against a number of clinical E. coli strains in vitro (M. Lino, Z. Naqvi, S. D. Goodman, A. M. Segall, and D. Barnett Foster, unpublished data).
Peptide wrwycr and its close relatives stabilize HJ intermediates of phage lambda site-specific recombination in E. coli and inhibit the tyrosine recombinase-dependent excision of prophages in both E. coli and Salmonella enterica LT2 (23). Treatment of E. coli with the peptide also resulted in the accumulation of filamentous cells and a 10-fold increase in anucleate cells, suggesting that the loss of bacterial viability was due to the combined effects of DNA breaks and chromosome segregation defects (22). However, while XerCD-dependent recombination in vitro is inhibited by wrwycr with a 50% inhibitory concentration of ∼50 nM, mutants with neither a single nor a double xerC and xerD mutation were resistant to the peptide. The dose of peptide treatment was positively correlated with the fraction of cells with fragmented DNA, measured either by fluorescently labeling the free 3′ OH DNA ends inside the bacteria (22) or by directly visualizing double-strand breaks by pulsed-field gel electrophoresis (C. W. Gunderson and A. M. Segall, unpublished data). The cumulative effect of the DNA damage resulting from peptide treatment induced the SOS response (22).
The idea of using antimicrobial peptides (AMPs) as antibiotics is not new. Naturally occurring AMPs are extremely abundant and diverse. They are found in most organisms, from insects to humans and plants, as well as some bacteria and fungi, and are part of the innate immunity system (5, 7, 35, 38). Many of these peptides kill by forming pores in the bacterial membrane, such as melittin (27), defensins (37), and cecropin (12). Other peptides, such as mersacidin, inhibit peptidoglycan synthesis (6). Still others have intracellular targets (5, 38). Moreover, several peptide-based drugs are being developed for clinical use.
Peptide wrwycr is the first antibiotic to date to inhibit DNA repair, which is a novel target of antimicrobial action (22). In the face of the ever growing number of antibiotic-resistant bacteria, the antibacterial properties of our peptides, which target DNA repair, a novel target of antimicrobial action, merit further investigation. In the study described here, we showed that peptide wrwycr inhibits the growth of Salmonella inside macrophage-like cells and murine peritoneal macrophages. Treatment with the peptide induces the SOS response in a significant fraction of the bacteria growing inside macrophages. These results suggest that the peptide may be worth exploring as the lead compound in a new class of antimicrobial compounds.
MATERIALS AND METHODS
Salmonella enterica serovar Typhimurium strains.
The virulent ATCC 14028S strain of Salmonella enterica serovar Typhimurium (Segall laboratory designation, strain G488) was obtained from Ferric Fang via Stanley Maloy. Strain SDT1416 was constructed by transformation of strain G488 with plasmid pFPV25.1, which encodes a constitutively expressed, bacterium-adapted high-level-fluorescence green fluorescent protein (GFP) driven by the rRNA (rrn) promoter (36). The MIC of wrwycr against SDT1416 was determined, according to the NCCLS protocol for broth microdilutions, to be 64 μM (19, 22). Strain G785 [ATCC 14028S Φ(sulA+-lacZY+)111] carries a chromosomal copy of the lacZ gene driven by the sulA promoter in an otherwise SulA-positive (SulA+) background (the construct was the generous gift of James Slauch, University of Illinois). Unless otherwise specified, all media were purchased from Fisher Scientific, Pittsburgh, PA.
Mammalian cells.
The murine macrophage-like cell line J774A.1 was grown in Dulbecco modified Eagle medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; BioWhittaker, Walkersville, MD) at 37°C with 5% CO2. The cells were grown in 75-cm2 flasks (Corning Inc., Corning, NY) with vented caps and passaged when the cell density reached ∼80%. To passage the cells, the medium was removed and the cells were washed with 10 ml of 1 mM EDTA dissolved in 1× phosphate-buffered saline (PBS). The cells were incubated with a trypsin solution (0.5% trypsin with 0.2 mM EDTA in 1× PBS) for 10 min at 37°C with 5% CO2 to release them from the tissue culture flask. Unless otherwise specified, all chemicals were purchased from Sigma-Aldrich, St. Louis, MO, or Fisher Scientific.
To harvest peritoneal murine macrophages, 8- to 12-week-old female BALB/c mice were injected in the peritoneal cavity with 1 ml of 4% thioglycolate to induce macrophage proliferation and were killed 4 days later. The thioglycolate solution was prepared by dissolving thioglycolate powder in boiling water, and the mixture was then autoclaved. Peritoneal macrophages were harvested by washing the peritoneal space with 10 ml of DMEM (without FBS) and collecting the effluent. The cells were centrifuged for 5 min at 4°C (Sorvall RT6000 centrifuge) at 700 rpm and were then resuspended in DMEM-10% FBS with penicillin and streptomycin (Invitrogen). The cells were counted with a hemocytometer and plated at the appropriate densities. All relevant federal guidelines and institutional policies concerning the use and handling of animals were strictly followed.
Peptide uptake. (i) Localization by microscopy.
J774A.1 cells or peritoneal macrophages were seeded in four-well (LabTek-II; Fisher Scientific, Pittsburgh, PA) chamber slides at 200,000 cells per well and the slides were incubated at 37°C in 5% CO2 overnight to allow adhesion. The medium was removed and replaced with fresh DMEM containing N-terminal rhodamine (Rh)-labeled peptides (Sigma-Genosys, St. Louis, MO), and the slides were incubated for the indicated length of time. The medium was removed after incubation, and the cells were washed three times with 1× PBS and then fixed with 4% paraformaldehyde for 30 min. After removal of the paraformaldehyde and washing of the cells once more with 1× PBS, the chambers were detached from the slide. The slide was inverted onto a coverslip with the SlowFade reagent (Invitrogen Molecular Probes, Carlsbad, CA), added to minimize photobleaching. Series of images were collected with a TCS SP2 confocal microscope (Leica Microsystems Inc., Bannockburn, IL).
(ii) Analysis by HPLC and MALDI-TOF.
J774A.1 cells and murine peritoneal macrophages were seeded in six-well tissue culture plates at 300,000 cells per well, and the plates were incubated for 24 h at 37°C in 5% CO2 to allow adhesion. The medium was replaced with fresh DMEM with peptide, and the plates were incubated for an additional 24 h. The cells were washed three times with ice-cold 1× PBS. Cell extracts were made by lysing the cells with 100 μl lysis buffer (0.71% Nonidet P-40, 71 mM Tris, pH 7.5, 0.71 mM EDTA, 212 mM NaCl) per well and physical scraping. Large cell debris was removed from the extract by spinning the samples for 1 min in a microcentrifuge at 16.2 relative centrifugal force. The samples were fractionated by reverse-phase high-performance liquid chromatography (HPLC) and analyzed on a high-performance liquid chromatograph (Gold BioEssential 126/168 system; Beckman Coulter) to determine the presence and abundance of the peptide. Samples were manually injected via a 300-μl loop onto a Jupiter 4-μm Proteo 90-Å column (Phenomenex, Torrance, CA) and then eluted with 0.1% trifluoroacetic acid in 100% acetonitrile at a flow rate of 1 ml per minute. The acetonitrile gradient was 0 to 30% from 5 to 13.5 min, 30 to 45% from 13.5 to 28.5 min, and up to 100% from 28.5 to 60 min. Fractions were collected from 6 to 25 min at 0.2-min intervals in a UV-transparent 96-well microtiter plate and were subsequently analyzed on a SpectraMax plate reader (Molecular Devices, Sunnyvale, CA) at 280 nm (see Fig. S1A in the supplemental material). Samples with the highest absorbances in the 17- to 19-min range (the expected elution time for the pure peptide) were suspected to contain the peptide wrwycr, on the basis of comparisons with the chromatographic profile of pure wrwycr. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry (performed at the Scripps Center for Mass Spectrometry, La Jolla, CA) of the HPLC fractions presumed to contain the peptide, on the basis of the separation pattern of the pure peptide, confirmed the identities of the wrwycr-dependent peaks, which comprised two prominent peaks that corresponded to the expected molecular masses of the peptide monomer (968 Da) and the peptide dimer (1,933 Da) (see Fig. S1B in the supplemental material). The monomer and dimer peaks obtained by the HPLC method used prior to mass spectrometry were too close to be separated into different HPLC fractions. The peptide concentrations were determined by first obtaining the total amount of peptide in each sample by calculating the area under the peptide peaks by using the integration function of the 32 Karat software (Beckman Coulter) accompanying the HPLC system and then comparing them to the values on a standard curve. Once the total quantity of peptide in each sample was determined, the intracellular peptide concentration was calculated by dividing the amount by the number of cells per sample (300,000) and the typical cell volume of 1 pl per J774A.1 cell (13). The volume of peritoneal macrophages was assumed to be similar.
Salmonella and wrwycr colocalization.
J774A.1 cells were seeded in four-well (LabTek-II; Fisher Scientific) chamber slides at 200,000 cells per well, and the slides were incubated at 37°C in 5% CO2 overnight to allow adhesion. The cells were infected with Salmonella strain SDT1416, as described above, at a multiplicity of infection (MOI) of 5. After 1 h of incubation with 100 μg/ml gentamicin to kill the extracellular bacteria, the cells were incubated with 50 μM rhodamine-labeled wrwycr in DMEM with 15 μg/ml gentamicin for 3 h at 37°C in 5% CO2. The peptide-containing medium was removed, and the cells were washed three times with 1× PBS and then fixed with 4% paraformaldehyde for 10 min. After removal of the paraformaldehyde and washing of the cells again with 1× PBS, the chambers were detached from the slide. The slide was inverted onto a coverslip, SlowFade reagent (Invitrogen Molecular Probes) was added to minimize photobleaching, and image series were collected with a TCS SP2 confocal microscope (Leica Microsystems Inc.).
Salmonella invasion assay.
The Salmonella invasion assay was adapted from a gentamicin protection assay (17). J774A.1 or peritoneal macrophage cells were seeded in 24-well flat-bottom tissue culture plates at 200,000 cells per well, and the plates were then incubated overnight at 37°C with 5% CO2. J774A.1 cells were released from the growing surface by either physical scraping or treatment with 1% trypsin (Invitrogen)-2 mM EDTA in 1× PBS. An overnight culture of strain SDT1416 (100 μl) was added to each well to achieve an MOI of between 1 and 2. After invasion was allowed to occur for 1 h, the Salmonella-containing medium was removed. The cells were washed three times with 1× PBS and were then given fresh DMEM containing 15 μg/ml gentamicin. The cells were incubated in the gentamicin solution for 1 h to kill the extracellular Salmonella cells. The medium was then removed and replaced with fresh DMEM containing 15 μg/ml gentamicin (to prevent residual extracellular bacterial growth) with or without peptide. At this time (time 0 h), samples were collected and plated (see below), while the rest of the samples were incubated for an additional 24 h at 37°C in 5% CO2. The medium was then removed and the cells were washed with 1× PBS. The cells were incubated with 500 μl of 1% Triton X-100 in PBS at room temperature for 5 min to lyse the macrophages. An additional 500 μl of 1× PBS was then added to each well; and the samples were collected, diluted, plated on LB agar plates, incubated overnight at 37°C, and counted.
Assessing Salmonella DNA damage inside macrophages.
J774A.1 cells and peritoneal macrophages were infected with Salmonella strain G785 and treated with peptide, and the bacteria were released from the eukaryotic cells, as described above for the invasion assay. The recovered bacteria were resuspended in 100 μl 33 μM 5-dodecanoylaminofluorescein di-β-d-galactopyranoside (C12FDG; Invitrogen) in LB medium, and the suspension was incubated at 37°C for 2 h. The lipophilic 12-carbon moiety of C12FDG helps the otherwise non-membrane-permeant FDG (fluorescein di-β-d-galactopyranoside) to penetrate the cell membrane. Each sample was then diluted with 500 μl of 1× PBS and analyzed on a FACSAria cell sorter (Becton Dickinson, San Jose, CA). In order to determine the appropriate incubation time with the C12FDG substrate, 1 μg/ml (final concentration) of the DNA cross-linking agent mitomycin C (MMC), a potent inducer of the SOS response, was added to exponentially growing Salmonella cultures for 3 h to induce DNA damage. The bacteria were then incubated with the C12FDG substrate for various times and analyzed by flow cytometry. In order to improve its ability to permeate the cells, the C12FDG substrate was mixed with 0.2 mM EDTA; the EDTA by itself did not induce the SOS response (data not shown). More than 60% of strain ATCC 14028S Φ(sulAp-lacZY+)111 cells were positive for β-galactosidase activity following the treatment with 0.5 μg/ml MMC (data not shown). By comparison, less than 1% of the cells that either were not treated with MMC or did not carry the sulAp-lacZ+ construct (G488) were positive for β-galactosidase activity. In bacteria grown in pure culture with wrwycr treatments, the fraction of Lac+ cells peaked after 2 h of incubation with the C12FDG substrate and decreased to nearly 0 by 3 h, possibly due to self-quenching of the intracellular fluorescein and/or to overwhelming DNA damage and the inhibition of transcription (data not shown). Note that none of the wrwycr concentrations resulted in the labeling of 100% of the bacteria with fluorescein, and fewer labeled bacteria were observed at 150 μM wrwycr than at 100 μM wrwycr, perhaps due to the inhibition of transcription due to peptide-dependent damage of the template (20).
Cell viability. (i) MTT reduction assay.
J774A.1 cells or peritoneal macrophages were seeded in 96-well plates at 40,000 cells per well, and the plates were incubated overnight at 37°C in 5% CO2. The medium was removed and replaced with fresh DMEM containing the appropriate treatments. The cells were treated for 24 h. Following treatment, 20 μl of a 4-mg/ml 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) solution in 1× PBS was added to each well. The cells were incubated with the MTT solution at 37°C in 5% CO2 for up to 3 h to allow the reduction of the MTT. One hundred microliters of lysis buffer (20% SDS in 1:1 water-dimethyl formamide, 2% glacial acetic acid, 2% 1 M hydrochloric acid) was added to each well, and the plates were incubated at 37°C overnight to aid with dispersion of the dye. On the following day, the optical density of the reaction mixtures was read at 575 nm with a Molecular Devices SpectraMax Plus plate reader (Sunnyvale, CA).
(ii) Analysis by flow cytometry with Live/Dead assay kit.
J774A.1 cells or peritoneal macrophages were seeded in six-well tissue culture plates at 300,000 cells per well, and the plates were incubated at 37°C in 5% CO2 to allow adhesion. The medium was replaced with fresh DMEM with or without peptide, and the plates were incubated for an additional 24 h. The cells were released from the plate by trypsinization and were then treated with the reagents in the Live/Dead viability/cytotoxicity kit for mammalian cells (Invitrogen Molecular Probes), according to the manufacturer's instructions, and analyzed on a FACSAria cell sorter (Becton Dickinson).
Statistical analysis.
Statistical analysis was performed by using the SAS system (version 9.1; SAS, Cary, NC). The Salmonella invasion results for both J774A.1 and peritoneal macrophages were log transformed to improve the normal distribution of the data. Unpaired t tests were used to assess the significance of the differences in bacterial growth between peptide treatments. Paired t tests were used to assess the significance of differences in the MTT assay results for both J774A.1 cells and peritoneal macrophages. Due to the small sample sizes, the Wilcoxon exact t test (nonparametric test) was used to assess the significance of differences in Salmonella DNA damage and the Live/Dead assay for cell viability. For all statistical analyses, a P value of <0.05 was considered significant. P values are displayed only when they indicated significant differences.
RESULTS
Entry of wrwycr into macrophage and macrophage-like cells.
In order to determine whether the peptide may be effective against intracellularly growing pathogens, the ability of Liss-Rh-labeled wrwycr to enter the host cells was initially examined by confocal laser microscopy. Although these modified peptides are ∼20 times less potent at inhibiting bacteriophage lambda recombination (J. Boldt and A. Segall, unpublished results), they were useful as tracers for localization studies. J774A.1 cells and peritoneal macrophages were incubated with rhodamine-labeled wrwycr for various lengths of time and were then analyzed by microscopy for the assessment of peptide uptake. The labeled peptide first became visible in J774A.1 cells at approximately 5 min. The fluorescence increased with increasing time of incubation, reaching the maximum intensity by 4 h (Fig. 1B to D). In contrast, Rh-labeled wrwycr entered the peritoneal macrophages more slowly and was visible only once it was inside these macrophages when a 10-fold higher peptide concentration was used (Fig. 1E and F). To address the possibility that rhodamine facilitated the uptake of the peptide, we tested and showed that the uptake of phalloidin, which is a non-membrane-permeant mushroom toxin that binds to filamentous actin and which is similar in size to our peptide, is not stimulated by rhodamine (see Fig. S2A and B in the supplemental material). However, peripheral cellular staining was evident when tetramethyl rhodamine isocyanate (TRITC)-phalloidin was coincubated with 100 μM wrwycr (see Fig. S2D in the supplemental material), suggesting that high peptide concentrations may affect membrane permeability in some way. Free rhodamine, even at very high concentrations (150 μM), did not enter eukaryotic cells (S. Patra and A. M. Segall, unpublished results).
FIG. 1.
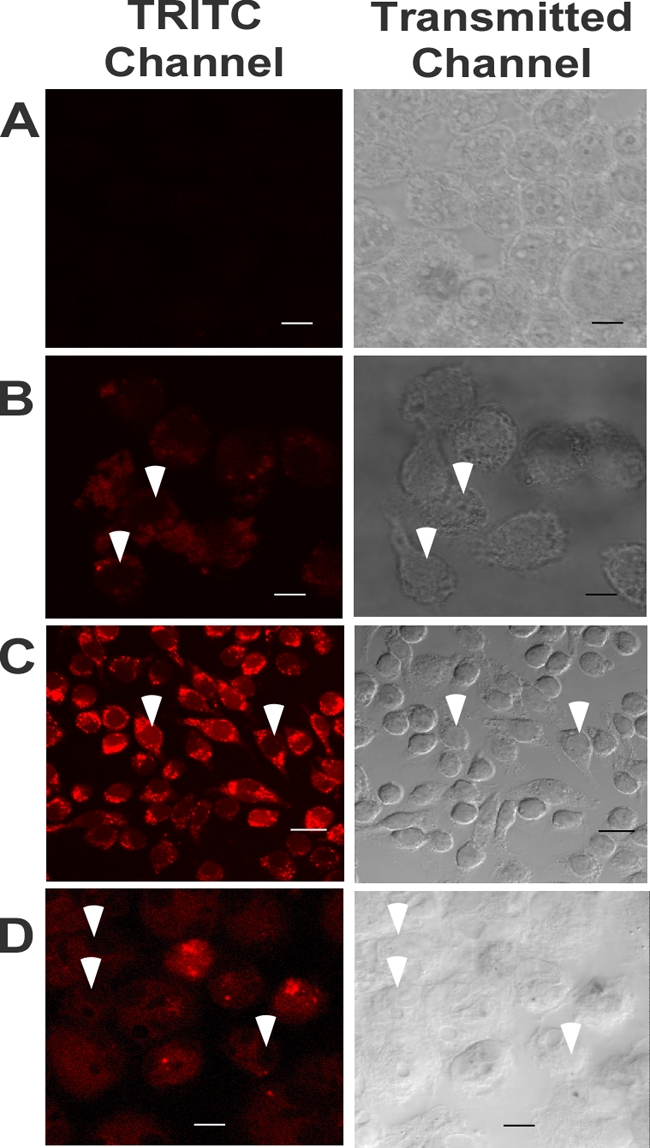
Uptake of rhodamine-labeled peptide. J774A.1 cells were incubated at 37°C either with no peptide (A), with 0.5 μM rhodamine-labeled wrwycr for 5 min (B), or with 1 μM rhodamine-labeled wrwycr for 4 h (C). The cells and labeled peptide were visualized by confocal microscopy. Peritoneal macrophages were incubated for 5 h with 10 μM (D) labeled peptide. The images were obtained by confocal laser microscopy. The image shown in each panel represents an optical slice near the center of the cell. White arrowheads indicate the nuclei.
The effects of temperature and inhibitors of endocytosis on peptide uptake were tested to investigate the basic mechanism of uptake. Lowering of the incubation temperature from 37°C to 4°C reduced the uptake of rhodamine-labeled peptides to undetectable levels (Fig. 2A and B), suggesting that peptide entry is an active process. To assess the role of endocytosis in peptide uptake, the phosphoinositide 3-kinase (PI3K) inhibitor wortmannin was used to prevent early endosomal vesicle fusion (33). Cotreatment with 100 nM wortmannin reduced but did not eliminate the intracellular levels of peptide, implicating a role of endocytosis in peptide uptake (Fig. 2C and D).
FIG. 2.
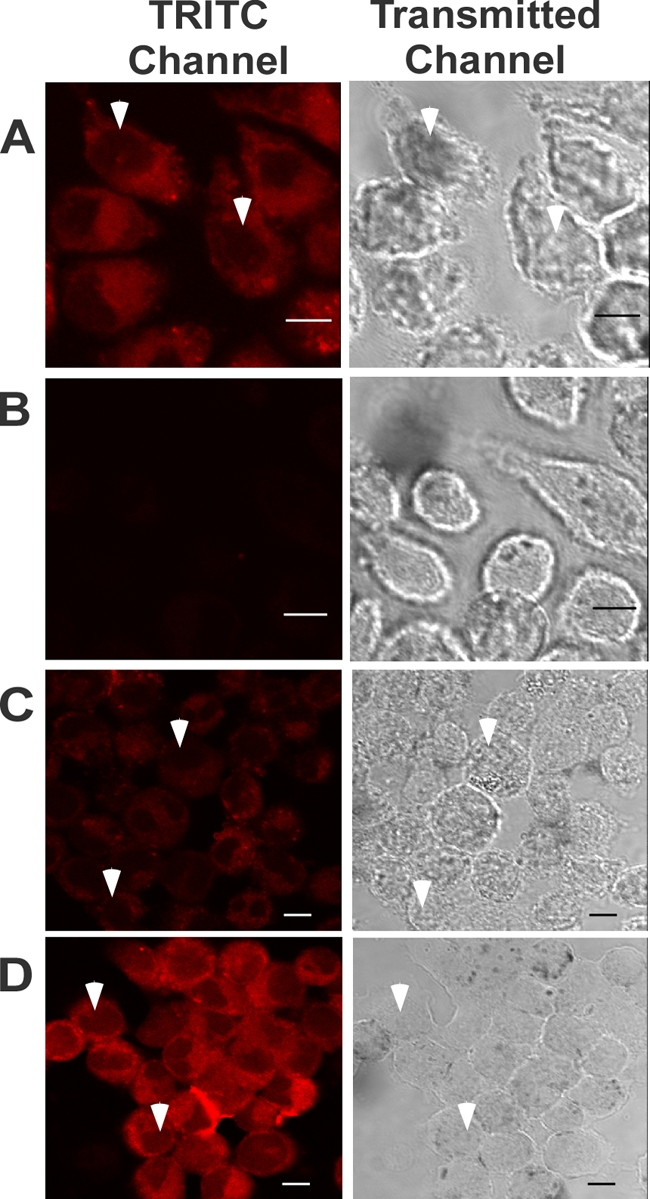
Effect of temperature or wortmannin on peptide uptake. J774A.1 cells were incubated with 10 μM rhodamine-labeled wrwycr at either 37°C (A) or 4°C (B) for 4 h and were then visualized by confocal microscopy. J774A.1 cells were treated with 10 μM rhodamine-labeled wrwycr for 5 h either in the presence (C) or in the absence (D) of 100 nM wortmannin and were then visualized by confocal laser microscopy. The image shown in each panel represents an optical slice near the center of the cells. White arrowheads indicate the nuclei.
To quantitatively assess peptide uptake and to circumvent any potential effects of rhodamine on peptide uptake, an HPLC-based method was used to directly measure the amount of unlabeled peptide inside cells. The HPLC assay detects peptide wrwycr on the basis of the characteristic absorbance at 280 nm by the aromatic amino acids tryptophan and, to a lesser extent, tyrosine. Peptide wrwycr standards typically elute off the column at about 17.5 min for the monomer and ∼17.75 min for the dimer (see the red trace in Fig. S1 in the supplemental material). Consistent with earlier observations, addition of the reducing agent dithiothreitol (DTT) abolishes the 17.75-min peak and increases the 17.5-min peak as the peptide dimers are converted to monomers (see the blue trace in Fig. S1 in the supplemental material). To determine the uptake of unlabeled wrwycr, the cells were incubated with unlabeled peptide at 37°C for 24 h. After removal of the peptide-containing medium (which was also analyzed) and washing of the cells with PBS, cell lysates were prepared and analyzed by HPLC to determine the amount of peptide internalized. The amount of peptide present in each sample was determined by integrating the area under the peaks at 17.5 and 17.75 min and comparing those to the areas on a standard curve. Intracellular peptide concentrations were calculated by dividing the total peptide in each sample by the number of cells per sample and the cell volume. The average volume of a J774A.1 cell is 1 pl (13); the typical volume of murine peritoneal macrophages was assumed to be the same. Consistent with the microscopy data, larger amounts of total peptide were detected in the J774A.1 cells than in peritoneal macrophages (Table 1). Less expected, however, was the finding that the intracellular peptide concentrations were several hundred times higher than those in the extracellular medium, supporting the notion that the peptide is being concentrated within the cells. This could be due to either active transport into cells or passive transport followed by trapping of the peptide inside the cells and binding to cellular constituents, preventing its equilibration with the medium. The protocol for making cell lysates excluded membranes, and thus, the peptide detected is intracellular rather than peptide only peripherally or integrally associated with the membrane.
TABLE 1.
HPLC analysis of wrwycr concentration after 24 h of incubation at 37°C
| Source (peptide concn units)b | Cell type | Result for the following total input wrwycr concna: |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 μM |
50 μM |
100 μM |
150 μM |
||||||
| [Pep] | D/M ratio | [Pep] | D/M ratio | [Pep] | D/M ratio | [Pep] | D/M ratio | ||
| Intracellular (mM) | J774A.1 | 0 | NAc | 7.2 | 1.7 | 20.8 | 2.9 | 30.1 | 12.5 |
| Intracellular (mM) | Peritoneal Mφ | 0 | NA | 2.5 | 0.9 | 7.4 | 0.7 | 24.8 | 11.4 |
| Medium (μM) | J774A.1 | 0 | NA | 11.6 | 3.8 | 24.7 | 4.1 | 55.4 | 2.5 |
| Medium (μM) | Peritoneal Mφ | 0 | NA | 8.8 | 6.4 | 21.0 | 6.7 | 53.4 | 13.8 |
The total peptide concentration ([Pep]) is expressed by using the molecular mass of the peptide monomer rather than that of the peptide dimer. Values are averages of three determinations. D, peptide dimer; M, peptide monomer.
The peptide source was either cell lysates (intracellular) or medium recovered from treated cells. Note that the intracellular concentration of peptide was in the mM range, while the concentration recovered from the medium in which the cells were treated was in the μM range.
NA, not applicable.
The peptide dimer-to-monomer ratios were also determined, since the dimer species is 50 times more active in vitro than the monomer (4). With 50 μM wrwycr treatment, the peptide dimers outnumbered the peptide monomers almost 2 to 1 in J774A.1 cells (Table 1). As the peptide treatment concentrations increased, the ratio of intracellular peptide dimer to monomer also increased. In contrast, the relative abundance of peptide dimers to monomers was similar in the 50 and 100 μM peptide-treated peritoneal macrophage lysates. However, with 150 μM wrwycr treatment, intracellular peptide was predominantly in the dimeric form in both J774A.1 cells and peritoneal macrophages (Table 1). The peptide stock solution added to the medium contained at least 67% dimers (a ratio of at least 2:1 dimer to monomer) and as much as 85% dimers; the peptide quantified in the medium in which J774A.1 cells were treated remained in the dimeric form, while the medium in which peritoneal macrophages were treated was even further enriched in dimers, for unknown reasons (Table 1). The ramification of the peptide monomer and dimer distributions in the two cell types will be discussed below.
A caveat regarding the intracellular peptide concentration is that it was calculated on the basis of the number of cells at the beginning of the experiment, which poses a potential problem for the analysis of J774A.1 cells. Since cell numbers were not determined at the end of the experiment, the replication or death of the cells was not taken into account. Under conditions that are more conducive to cell replication (i.e., 24 h of incubation at 37°C and a low peptide concentration), the calculated intracellular peptide concentration is likely to be an overestimation. At high peptide concentrations, at which cells are more likely to die than replicate, the actual intracellular concentrations may have been underestimated. These caveats do not apply to the peritoneal macrophages, since they do not replicate in culture and are only marginally sensitive to the peptides (discussed below).
In order to address whether the peptide is taken up actively or passively, peptide treatment and quantitation were also undertaken at 4°C, a temperature at which active transport mechanisms should operate poorly, if at all. In J774A.1 cells incubated with 50 or 100 μM peptide, there was virtually no difference in the amount of peptide internalized by cells after 5 h at 4°C compared to the amount internalized by cells incubated at 37°C. With 150 μM peptide treatment, there was about twice as much peptide present in the lysate of cells incubated at 37°C as in the lysate of cells incubated at 4°C (Table 2). This is in stark contrast to the microscopy results, which showed that lowering of the incubation temperature to 4°C abolished all intracellular staining by the rhodamine-labeled peptides. The same trend was seen even after 24 h: cells incubated at 4°C had at least as much intracellular peptide as those incubated at 37°C and also had a higher proportion of dimers than monomers (Table 2).
TABLE 2.
Effect of temperature on wrwycr uptake by J774A.1 cells
| Temp (°C) | Result for the indicated input total wrwycr concn at the following time of incubationa: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 h |
24 h |
|||||||||||
| 50 μM |
100 μM |
150 μM |
50 μM |
100 μM |
150 μM |
|||||||
| [Pep]b | D/M ratio | [Pep] | D/M ratio | [Pep] | D/M ratio | [Pep] | D/M ratio | [Pep] | D/M ratio | [Pep] | D/M ratio | |
| 37 | 0.9 | 2.1 | 8.0 | 2.6 | 28.9 | 11.0 | 1.4c | 4.3 | 16.9 | 3.1 | 30.7 | 12.5 |
| 4 | 1.2 | 2.3 | 7.7 | 2.7 | 15.8 | 8.8 | 10.1 | 7.4 | 32.9 | 11.5 | 47.7 | 15.5 |
The total peptide concentration is expressed by using the molecular mass of the peptide monomer rather than that of the peptide dimer. D, peptide dimer; M, peptide monomer.
[Pep], measured intracellular peptide concentration (mM).
This concentration of peptide is unusually lower than the average for the same treatments described in Table 1.
Salmonella and wrwycr colocalized in J774A.1 cells.
To determine whether the peptide penetrated the same subcellular compartments as Salmonella, we infected J774A.1 cells with a derivative of S. enterica serovar Typhimurium strain ATCC 14028S that constitutively expresses GFP (strain SDT1416) and treated these infected cells with Rh-labeled wrwycr. Confocal microscopy revealed that there is an overlap between GFP-expressing S. enterica serovar Typhimurium and Rh-labeled wrwycr inside J774A.1 cells (Fig. 3), indicating that the peptide should be able to reach the intracellular bacteria and exert its antibacterial effects. However, not all of the Salmonella colocalized with the labeled peptide. Three categories of localization patterns between Salmonella and the labeled peptide were found. Some of the bacterial cells appeared yellow throughout the range of focal planes spanning the entire thickness of the cell, suggesting that they had taken up the labeled peptide. Other bacterial cells appeared either yellow or green, depending on the focal plane, and we believe that these cells were surrounded by but did not take up the peptide. When the focal plane was near the center of the bacterial cells, they appeared green. When the focal plane was near the edge of the Salmonella cells, the green fluorescence from the bacteria overlapped with the red fluorescence from the surrounding peptide, causing the bacteria to appear yellow. Finally, some of the Salmonella cells appeared green in all focal planes and presumably were not in the same compartment(s) as the peptide. Approximately half of the intracellular Salmonella cells had taken up the labeled peptide, while another third colocalized with but had not taken up the peptide.
FIG. 3.

Colocalization of peptide and Salmonella. J774A.1 cells were infected with GFP-expressing Salmonella (strain SDT1416) at an MOI of 5 and were then incubated with 50 μM rhodamine-labeled wrwycr (red). Shown are two sets of confocal Z-series images of optical slices near the center of the cells for two separate fields of view (A to D and E to H). Salmonella bacteria that did not colocalize with peptide appear green (black arrowheads) in all optical slices, while those bacteria that presumably took up the peptide appear yellow (white arrowheads) in all optical slices. Salmonella bacteria that colocalized with but that did not take up the peptide appear green in some optical slices and yellow in others (arrow).
wrwycr inhibits intracellular Salmonella growth in J774A.1 cells and peritoneal macrophages.
In order to test the susceptibility of intracellularly growing Salmonella to peptide, J774A.1 cells were infected with S. enterica serovar Typhimurium at an MOI of 1 for 1 h, washed, and treated with gentamicin to kill the bacteria that did not invade the cells. The cells were then grown in the presence or absence of peptide (and with continued gentamicin treatment to prevent the residual growth of extracellular bacteria) at concentrations near or up to 2.5-fold higher than the MIC values determined previously (22). Initial tests confirmed that the d-isoform peptide had a greater effect on bacterial growth than the l-isoform peptide (data not shown), presumably due to the resistance of the former to degradation. These preliminary experiments also showed the antibacterial effects after 24-h peptide treatments to be more pronounced than those after 12-h treatments (a greater than 100-fold reduction in the numbers of CFU/ml after 24 h of treatment with 100 μM peptide compared to a less than a 50-fold reduction after 12 h of treatment; data not shown). We observed a 68.5% reduction in bacterial growth with 50 μM peptide treatment and an 85.1% reduction with 100 μM peptide treatment (Fig. 4A). Since peptide wrwycr was dissolved in dimethyl sulfoxide (DMSO), the effects of the solvent were also tested. No statistically significant effects were observed, although there was a trend toward a dose-dependent enhancement of bacterial growth. The peptide inhibited intracellular bacterial growth in a concentration-dependent manner. Although peptide treatments significantly reduced the level of Salmonella growth relative to that in the untreated samples, they did not reduce the bacterial numbers below those in the initial inoculum, suggesting that the peptide is bacteriostatic rather than bactericidal inside host cells or that the intracellular bacteria are exposed to an insufficient amount of (active) peptide due to differential localization. Due to cytotoxic effects (discussed below), peptide concentrations higher than 100 μM were not tested with J774A.1 cells.
FIG. 4.

Effects of wrwycr (also known as d8) on intracellular Salmonella growth. J774A.1 cells (A) and peritoneal macrophages (B) were infected with S. enterica serovar Typhimurium ATCC 14028S at an MOI of 1 and were then incubated with wrwycr at the indicated concentration for 24 h. Gentamicin (15 μg/ml) was included in the peptide treatments to prevent extracellular bacterial growth, which ensured that the effects of the peptide observed were due to the inhibition of intracellular Salmonella growth. The effects of DMSO and the peptide were normalized to the effects on the DMEM-treated controls. The inoculum at time zero is indicated by the horizontal dashed line. Shown are the average levels of bacterial growth inhibition and standard errors. *, statistical significance with P < 0.05; **, statistical significance with P < 0.005.
J774A.1 macrophage-like cells resemble inactive macrophages and are very permissive for Salmonella growth. Since wrwycr inhibited Salmonella growth inside these cells, we tested whether wrwycr can similarly inhibit bacterial growth inside thioglycolate-elicited murine peritoneal macrophages. The peptide exhibited dose-dependent inhibition of bacterial growth in the peritoneal macrophages cells as well (Fig. 4B), although it was less potent than it was in the J774A.1 cells: bacterial growth was reduced by only 34% and 45% with 50 μM and 100 μM peptide treatments, respectively. Equivalent concentrations of DMSO did not significantly affect intracellular bacterial growth. Treatment with 150 μM wrwycr reduced bacterial growth 72.3% compared to that detected in the DMEM-treated peritoneal macrophages and by nearly 83% compared to that achieved with the equivalent DMSO concentration (1.5%). In this case, the DMSO treatment significantly stimulated bacterial growth compared to the level of growth in DMEM alone. The peptide was bacteriostatic in the peritoneal macrophages as well as in the J774A.1 cells, as none of the peptide treatments reduced the bacterial numbers below those in the initial inoculum (indicated by the dotted line in Fig. 4).
Salmonella bacteria growing inside peptide-treated macrophages induce the SOS response.
Testing of whether the mechanism of bacterial growth inhibition by the peptide inside macrophages was the same as the mechanism of bacterial growth inhibition in pure culture was made more difficult by the fact that we have been unable to isolate peptide-resistant mutants of either Salmonella or E. coli and cannot use such mutants to test whether they are also resistant to peptide inside macrophage cells. Instead, we used a chromosomal SOS-inducible β-galactosidase reporter to determine whether the bacteria inside the peptide-treated cells were experiencing DNA damage [using the SOS reporter construct present in strain ATCC 14028S Φ(sulAp+-lacZY+)111, designated strain G785). LacZ activity was monitored by determination of the level of production of fluorescein from the nonfluorescent substrate C12FDG. Fluorescein is trapped inside cells, presumably through the interactions of the long carbon chain with the lipid bilayer.
To test whether intracellular bacteria accumulated DNA damage and induced the SOS response after the host cells were treated with peptide, we performed the invasion assay at an MOI of 10 using strain G785. The bacteria collected from the murine cells were incubated with C12FDG and analyzed by flow cytometry for induction of the SOS response. The effect of the peptide on bacterial growth was tested in parallel by taking a small aliquot from each sample for colony count determination. Times of 4 and 24 h postinfection were used to track the changes in both bacterial numbers and the induction of the SOS response. At 4 h postinfection, the numbers of Salmonella bacteria in cells grown in the absence of treatments had increased by 8-fold, with only about 1% of them having induced the SOS response (Fig. 5A and Table 3). Neither Salmonella growth nor SOS induction was significantly affected by any of the DMSO concentrations used (0.5%) or by treatment with 50 μM wrwycr. Treatment of the macrophages with both 100 μM and 150 μM wrwycr resulted in a significant increase in the numbers of SOS-positive Salmonella bacteria and a decrease in bacterial growth compared to the numbers of bacteria and level of growth achieved with the DMSO treatment and, in the case of the higher peptide concentration, compared to the numbers of bacteria and level of growth achieved with DMEM-treated cells as well.
FIG. 5.

Induction of the SOS response upon wrwycr (also known as d8) treatment in intracellular bacteria. J774A.1 cells were infected with G785 (carrying an SOS reporter gene fusion) at an MOI of 10 and incubated with the indicated treatments for 4 or 24 h. Gentamicin (15 μg/ml) was included to prevent extracellular bacterial growth. The colony counts of intracellular Salmonella from J774A.1 cells were determined at 4 h (A) and 24 h (B) postinfection. The inoculum at time zero is indicated by the horizontal dashed line. Each data point shown represents the colony counts from a single well, and the means are indicated by the horizontal bars. The P values shown are the results of Wilcoxon exact one-tailed t tests. Statistical analysis was performed on all pairwise combinations, but only significant (or nearly significant) differences are shown. *, statistical significance with P < 0.05; **, statistical significance with P < 0.005; hpi, hours postinfection.
TABLE 3.
Fraction of Salmonella bacteria inside J774A.1 cells in which the SOS response was induced at 4 h and 24 h postinfection
| Treatment | 4 h postinfection |
24 h postinfection |
||
|---|---|---|---|---|
| % bacteriaa | % SOS+ cellsb | % bacteria | % SOS+ cells | |
| DMEM | 66.7 ± 4.4 | 1.4 ± 1.3 | 74.3 ± 1.7 | < 0.01 |
| 0.6% DMSO | 80.2 ± 3.5 | 0.3 ± 0.2 | 57.8 ± 08 | < 0.01 |
| 50 μM wrwycr | 57.1 ± 5.8 | 0.3 ± 0.1 | 45.2 ± 10.2 | 0.4 ± 0.2 |
| 1.2% DMSO | 76.1 ± 4.0 | 0.1 ± 0 | 57.1 ± 13.3 | 0.03 ± 0.1 |
| 100 μM wrwycr | 19.0 ± 1.7 | 1.3 ± 0.6 | 2.8 ± 0.5 | 16.5 ± 4.8 |
| 1.8% DMSO | 82.5 ± 3.7 | 0.1 ± 0.1 | 54.2 ± 9.8 | < 0.01 |
| 150 μM wrwycr | 3.0 ± 0.9 | 8.5 ± 3.1 | 1.9 ± 0.2 | 28.4 ± 5.2 |
% bacteria, the fraction of Salmonella bacteria, determined by reactivity with LPS antibody, among the number of particles released by lysis of J774A.1 cells that fit the forward-scatter versus side-scatter parameters characteristic of bacterial cells. The other particles may be mitochondria and/or other nonbacterial particles.
% SOS+ cells, the fraction of fluorescein-positive Salmonella bacteria (LPS-positive particles). All the differences between the percentage of SOS response-positive cells in 1.2% DMSO versus the percentage obtained after treatment with 100 μM peptide and in 1.8% DMSO versus the percentage obtained after treatment with 150 μM peptide at either 4 h or 24 h are significant (P values of 0.0022 in all cases, in Wilcoxon exact one-tailed t tests).
By 24 h postinfection, the numbers of intracellular Salmonella bacteria in J774A.1 cells grown in the absence of peptide treatments had increased by over 240-fold, a more than 30-fold increase since the 4-h time point (Fig. 5B). On average, only 0.6% of these bacteria were positive for an SOS response (Table 3). Treatments with 100 μM and 150 μM peptide inhibited intracellular Salmonella growth by at least 99% (Fig. 5B) and resulted in significantly higher fractions of SOS-positive Salmonella bacteria (Table 3). However, despite the >99% decrease in growth, the peptide treatments did not reduce the bacterial levels much below those in the initial inoculum, which is indicated by the dotted line in Fig. 5.
wrwycr toxicity toward J774A.1 cells and peritoneal macrophages.
To determine whether the peptide effects are directly on the bacteria or are indirect due to their toxicity to the host cells, we assessed their effects on eukaryotic cells using the MTT metabolic assay, in which the yellow compound MTT is reduced to a purple formazan derivative by lactate and succinate dehydrogenases (29). Compared to treatment with DMEM, treatment with 50 μM wrwycr did not reduce the metabolic activity of J774A.1 cells (Fig. 6A). Surprisingly, 0.5% DMSO treatment resulted in a borderline-significant increase in the MTT signal of nearly 29%. While 100 μM wrwycr treatment resulted in a 17.3% decrease in the metabolic activity of J774A.1 cells compared to that achieved with DMEM treatment, the same effect was observed with treatment 1% DMSO, the amount of peptide solvent present in 100 μM wrwycr. The peritoneal macrophages were much less sensitive to the peptide. Up to 100 μM wrwycr and 1.5% DMSO had no significant effect on the metabolic activity of the peritoneal macrophages (Fig. 6B). Treatment with 150 μM wrwycr, however, resulted in a significant increase (18%) in the metabolic activity of the peritoneal macrophages (Fig. 6B).
FIG. 6.

Effects of wrwycr (also known as d8) on host cell viability. MTT reduction assay results for J774A.1 cells (A) and peritoneal macrophages (B) are shown. Cells were grown in 96-well plates and treated with wrwycr and DMSO at the indicated concentrations for 24 h. Following treatment, the cells were allowed up to 3 h to convert MTT to formazan, as measured by determination of the optical density at 575 nm. Shown are the average effects of the treatments on MTT reduction with standard errors normalized to the results for the untreated (DMEM-treated) control. The means are indicated by the short horizontal bars. The P values shown are the results of Wilcoxon one-tailed tests. Statistical analysis was performed on all pairwise combinations, but only significant (or nearly significant) differences are shown. *, statistical significance with P < 0.05; **, statistical significance with P < 0.005. (C) Sample dot plots from flow cytometric analysis of effects of wrwycr on J774A.1 cell viability by the Live/Dead assay. (Top left quadrant) Cells not stained with the Live/Dead stain do not autofluoresce; (top right quadrant) live cells (untreated) appear green on the basis of their ability to generate calcein from calcein AM and to exclude the ethidium homodimer; (bottom left quadrant) dead cells (killed with methanol for 5 min for 37°C) were unable to process calcein AM and stain red with the ethidium homodimer; (bottom right quadrant) J774A.1 cells treated with wrwycr for 24 h stain green or red or double stain both red and green. The cells that were positive for both calcein and the ethidium homodimer were in the earlier stages of death; they were not able to recover when they were replated in peptide-free medium. The results for the complete treatment sets are shown in Table 4.
While the MTT reduction assay showed that wrwycr at 100 μM has detrimental effects on the overall metabolic activity of J774A.1 cells, the assay cannot distinguish between the possibility that 17% of the cells died and were therefore metabolically inactive while the rest were normal or there was an average of a 17% decrease in the metabolic activity of all cells. Therefore, we tested the viability of individual peritoneal macrophages and J774A.1 cells using a fluorescence-based viability/cytotoxicity assay and analyzed individual cells by flow cytometry. This assay discriminates between live and dead cells on the basis of both the integrity of the membrane (penetration of the membrane-impermeant ethidium homodimer) and the enzymatic activity of esterases that cleave the nonfluorescent membrane-permeant calcein acetoxymethyl ester (calcein AM) to produce the green fluorescent molecule calcein. Dead cells are permeable to the ethidium homodimer and lack active esterases (e.g., Fig. 6C). Cells that were positive for both calcein and the ethidium homodimer were in the early stages of death, on the basis of the observation that they did not recover when they were sorted and replated in peptide-free medium (data not shown). The Live/Dead assay reported that 35% of the J774A.1 cells died as a result of 100 μM wrwycr treatment, an effect greater than that which we saw by the MTT reduction assay (Table 4). Peritoneal macrophages were less sensitive to the peptide, just as the MTT reduction assay indicated, with only ∼10% cell death achieved with 150 μM wrwycr (Table 4).
TABLE 4.
Effect of wrwycr on viability of J774A.1 cells and peritoneal macrophages determined by the Live/Dead assay
| Treatment | % cells |
|||
|---|---|---|---|---|
| J774A.1 |
Peritoneal macrophages |
|||
| Livea | Deadb | Live | Dead | |
| DMEM | 95.6 ± 1.9 | 1.7 ± 1.1 | 94.1 ± 1.9 | 4.3 ± 1.0 |
| 0.5% DMSO | NTc | NT | 94.8 ± 0.5 | 3.2 ± 0.2 |
| 50 μM wrwycr | 93.0 ± 0.4 | 4.3 ± 0.7 | 94.2 ± 0.9 | 3.2 ± 1.0 |
| 1.0% DMSO | NT | NT | 93.9 ± 0.6 | 3.5 ± 0.9 |
| 100 μM wrwycr | 58.9 ± 11.1 | 35.3 ± 10.3 | 94.4 ± 0.7 | 3.1 ± 0.3 |
| 1.5% DMSO | NT | NT | 94.9 ± 1.1 | 3.3 ± 1.0 |
| 150 μM wrwycr | NT | NT | 87.9 ± 7.0 | 9.2 ± 7.9 |
Live, cells that processed calcein AM to calcein in the Live/Dead assay by the use of cellular esterases.
Dead, the fraction of cells that are permeable to propidium iodide, indicating membrane disruption.
NT, not tested.
DISCUSSION
In order to investigate its potential as an antibiotic, we examined the ability of peptide wrwycr to enter murine macrophage cells and to affect the growth of the bacterial pathogen Salmonella Typhimurium, in addition to examining the peptide's cytotoxicity toward the host cells.
Peptide wrwycr readily entered J774A.1 murine macrophage-like cells at low micromolar concentrations within minutes. In contrast, entry into peritoneal macrophages was much less efficient: a concentration of rhodamine-labeled peptide up to 10-fold higher was needed to see intracellular staining. The uptake of the labeled peptide by J774A.1 cells was greatly reduced, but not eliminated, by the endocytosis inhibitor wortmannin. It is possible that the peptides also entered cells by increasing the permeability of the plasma membrane by using mechanisms similar to those of other cationic antimicrobial peptides, such as melittin (5, 7, 27, 35, 37); however, wrwycr has no predicted alpha helix-forming propensity, like melittin and other pore-forming antimicrobial peptides. The amino acid composition of peptide wrwycr chemically resembles the cell-penetrating class of peptides (CPPs) that are derived from a number of eukaryotic proteins, including HIV-1 Tat and the Drosophila Antennapedia homeodomain-derived penetratin peptide Antp (39). These peptides are noted for their amphipathic nature and their positive charge at physiological pH (they are composed of from 17 to 100% positively charged amino acids [39]). CPPs use both endocytotic mechanisms and temperature- and endocytosis-independent membrane translocation and require endosomal acidification for efficient entry into the cytoplasm (16, 20, 24, 34, 35). In the case of peptide wrwycr, HPLC analysis revealed intracellular concentrations several hundred times higher than the input concentrations, even at 4°C; this temperature-independent uptake is similar to that of CPPs. In contrast, the rhodamine-labeled peptide did not enter cells at 4°C, and its entry may depend on endocytosis rather than direct translocation for its entry. Trypan blue dye exclusion showed that cell membrane integrity was not compromised during the extended incubation at 4°C and thus does not account for peptide uptake at this temperature (data not shown). It is also possible that the peptide is effluxed to a greater extent at 37°C than at 4°C. More detailed analyses of the mechanisms of peptide transport, efflux, and subcellular localization, including how the peptide colocalizes with Salmonella, are future goals.
Fifty, 100, and 150 nmol of peptide are present in 1 ml of the 50, 100, and 150 μM peptide treatments, respectively. Typically, the amount of total peptide recovered (in the lysate and spent medium) at the end of the 24 h of incubation at 37°C ranged from 7 to 65 nmol, accounting for approximately 14 to 43% of the total peptide added. The rest of the peptide was most likely bound to membranes or was sequestered in various vesicles and lost in the cell debris pellet. Because the peptides used are composed of d-conformation amino acids, degradation of the peptide backbone should not occur, but it is possible that some degradation of the side chains may account for some of the peptide loss.
Peptide effect on Salmonella growth inside murine macrophages.
Peptide wrwycr exhibited dose-dependent inhibition of Salmonella growth in both J774A.1 cells and peritoneal macrophages. The inhibitory effect in J774A.1 cells was dependent on the MOI of the bacterial infection: the reduction in bacterial colony counts observed with 100 μM wrwycr, for example, was almost 10-fold greater when the Salmonella MOI used was 10 (Fig. 5B) than when the MOI was 1 (Fig. 4A). Thus, the peptide concentration per bacterial cell is not limiting inside the macrophages. We do not know why the MOI should affect the efficiency of the peptide. One possibility is that at greater MOIs, the physiological state of the intracellular Salmonella bacteria makes them more susceptible to killing by our peptide, while another is that the Salmonella bacteria stimulate the host cell response in a manner that increases peptide-dependent killing. Certainly, higher Salmonella multiplicities of infection have been shown to cause more cytotoxicity in mammalian macrophages and macrophage-like cells (31). This observation predicts that we would observe greater peptide-dependent killing in peritoneal macrophages as well (in which infection was done only at an MOI of 1), and this will be tested in the future.
Because the intracellular peptide concentration is at least 20 times the in vitro bactericidal concentration, it is possible that much of the intracellular peptide is not available to the Salmonella bacteria. Alternatively (and not mutually exclusively), Salmonella bacteria growing intracellularly may be more resistant to the peptide than those grown in pure culture, perhaps due to membrane modifications that interfere with the peptide's uptake by bacterial cells. In order to reach the Salmonella cells, either the peptide must enter Salmonella-containing vesicles from the cytoplasm or peptide-containing endosomes must fuse with Salmonella-containing vesicles, perhaps limiting the access of the peptide to the bacteria. Confocal microscopy with the rhodamine-labeled peptide indicated that ∼50% of the bacteria had taken up the peptide inside J774A.1 cells and another ∼30% were in close proximity to it, leaving open the possibility that some intracellular bacteria either were not exposed to the peptide or were exposed to levels too low to elicit the bactericidal effects seen in free-growing bacterial cells.
The potency of peptide wrwycr against Salmonella growing inside murine peritoneal macrophages was somewhat less than that of Salmonella growing in J774A.1 cells. The peptide entered peritoneal macrophages less efficiently than it did J774A.1 cells: twice as much peptide was required to achieve the same intracellular concentration in peritoneal macrophages that was present in J774A.1 cells when the peptide input concentration was 50 μM, and only at the highest concentration (150 μM) did the peptide dimer concentrations exceed the peptide monomer concentrations. However, when the bacterial growth inhibition effects were normalized to the intracellular peptide concentration, peptide wrwycr was indeed more potent against Salmonella growth in peritoneal macrophages than in J774A.1 cells. We expected the greater oxidative response elicited by Salmonella in peritoneal macrophages (11) to cause more bacterial DNA damage, leading to an increased concentration of DNA repair intermediates and, thus, more targets for the peptide (22). In pure culture, hydrogen peroxide and the peptide synergistically kill E. coli in culture (Y. Xu and A. Segall, unpublished results).
Inferring DNA damage from the bacterial SOS response.
To test whether the peptide's mechanism of Salmonella growth inhibition inside tissue culture cells is the same as that in pure culture, we used a Salmonella strain carrying the lacZ gene driven by the sulA promoter, a reporter for the SOS response. When LacZ activity specifically among LPS-positive particles was monitored by flow cytometry, a dose-dependent induction of the SOS response was detected in the bacteria isolated from peptide-treated J774A.1 cells but not in bacteria isolated from DMEM- or DMSO-treated J774A.1 cells. This is consistent with the proposed mechanism of peptide killing in pure culture, namely, interference with DNA repair and the accumulation of unrepaired DNA damage (22). In pure culture, only about a 4-fold induction of the SOS response in the whole population was detected by use of the reporter genes; flow cytometry for detection of the induction of the SOS response on an individual-cell basis also showed that the SOS response was induced in only a fraction of the population (Table 3 and data not shown; Y. Xu, I. Naili, and A. Segall, unpublished results). It is possible that the peptide has multiple targets in bacteria and that only some cells die of unrepaired DNA damage and induce the SOS response. Alternatively, it is possible that at high peptide concentrations, all the bacteria in pure culture or grown intracellularly die of unrepaired DNA damage but that a high proportion of these sustain so much damage that transcription of the SOS reporter is inhibited. A third possibility is that some of the DNA damage caused by the peptide does not induce the SOS response (alkylation damage, for example, does not induce the SOS response).
The fact that bacteria grown in DMEM- or DMSO-treated J774A.1 cells did not induce the SOS response under parallel conditions is consistent with the low oxidative burst of these cells in the absence of added gamma interferon (γ-IFN), but a microarray-based study by Hinton and colleagues of gene expression in Salmonella enterica growing inside macrophages identified a number of moderately induced SOS genes (3- to 5-fold greater expression relative to that of the control at 4 h or 12 h), including lexA, dinF, uvrB, recN, and dinB (18). However, most members of the SOS regulon were not similarly induced, for example, the uvrA and uvrC genes (0.95 to 1.24-fold induction at 4 h or 12 h). Notably, the sulA gene, assayed in our study, was induced only 1.6-fold relative to that for the control at 4 h and ∼2-fold at 12 h (12 h was the latest time point evaluated in the study of Hinton and colleagues [18]); this subtle level of induction may not be easily detectable in our assay. The greatest level of SOS gene induction in the study of Hinton and colleagues was observed at 8 h (and was as much as 30% greater than that at 4 or 12 h), followed by decreased gene expression at 12 h; by 24 h, the time point at which we saw the greatest sulA induction, the Salmonella bacteria inside untreated macrophages may have repaired the DNA damage that caused the increase in the level of expression of the SOS response gene, and the induction of sulA in the peptide-treated bacteria was most reasonably due to persistent damage that was not or that could not be repaired. Another technical difference between the microarray study and our own was the very large MOI used by Hinton and colleagues (100 Salmonella per macrophage infected), which may also have contributed to the differences between our observed lack of induction in untreated bacterial cells compared to the results of the study of Hinton and colleagues (18); as discussed above, the greater bacterial load may cause a much greater response on the part of the host cells. Finally, the greater oxidative burst of the peritoneal macrophages would be expected to induce the SOS response even in bacteria not treated with peptide, thereby hiding the peptide effect. Preliminary experiments that used IFN-γ to stimulate J774A.1 cells and a Salmonella MOI of 1 showed that the stimulated macrophage-like cells killed the bacteria to similar levels with or without the peptide. However, those experiments will be repeated in the future with a larger Salmonella MOI, at which we saw greater killing by the peptide.
The toxicity of the peptide for the murine cells was assessed by the population-based MTT reduction assay, which measures metabolic activity, and a flow cytometry-based viability assay. Together, those assays showed that the J774A.1 cells (a transformed cell line) were more sensitive to the peptide than the primary peritoneal macrophages. Even with treatment with 150 μM peptide, over 90% of the peritoneal macrophages remained viable. In contrast to the MTT data, 50 μM peptide treatment did not cause increased J774A.1 cell death, as determined by the Live/Dead assay. This suggests that low concentrations of wrwycr may reduce cell metabolic activity rather than cause death. Even with 100 μM peptide treatment, there was virtually no increase in the proportion of dead cells; however, at this peptide concentration, up to 40% of the J774A.1 cells, in some cases, had become positive for staining with both the live and the dead stains (a representative dot plot is shown in Fig. 6C). The inability of these cells, after they were sorted, to even adhere to the plates, much less replicate, indicated that they were nonviable cells that retained residual enzymatic activities. On the basis of these results, these cells were categorized as “dead” cells in Table 4. In aggregate, these results indicate that peritoneal macrophages are much more resistant to peptide treatment than J774A.1 cells. This result is consistent with the results of other studies in which we have found that primary or slowly dividing cells, for example, mouse cardiac myocytes or low-passage-number cells of the IMR-90 cell line, are quite resistant to the peptide, while relatively fast-dividing cell lines, such as HeLa and U2OS cells, are much more susceptible (L. Su, S. Patra, and A. Segall, unpublished data; S. Patra, R. N. Authement, and A. Segall, unpublished data).
Although the rhodamine-labeled peptides localized primarily in the cytoplasm, nuclear staining was apparent at 4 h and later time points, albeit at reduced levels compared to the level of cytoplasmic staining. Nuclear entry and interference with the repair of endogenous DNA damage may be one of the major mechanisms of peptide wrwycr toxicity in mammalian cells (Su et al., unpublished; Patra et al., unpublished). Slower and reduced levels of entry into the nucleus may spare the host cells from the peptide's predominant toxic effects, supported by the greater toxicity of peptides to which nine N-terminal arginines (R9), a CPP tag which promotes the permeation of cargo proteins in mammalian cells (21), were added. These R9-tagged peptides entered cells and concentrated in the nucleus extremely efficiently and were much more toxic than the unmodified peptides (L. Su, D. Fujimoto, and A. Segall, unpublished data). Reducing the nuclear permeability without decreasing the binding activities toward branched DNA repair intermediates should decrease the toxicities of these antibiotics and enhance their usefulness.
The activity of peptide wrwycr at killing intra- and extracellular bacteria is a promising proof of principle for this inhibitor that binds to intermediates of DNA repair, a novel target for antibiotics. The peptide binds to branched DNA molecules, with the peptide having the highest affinities for Holliday junctions and somewhat lower levels of affinity for complete or partial replication forks (those that have only the leading or the lagging strand) (25, 26). Such structures are expected to arise when replication forks regress or when they collapse when they encounter double-strand breaks or nicks; covalent protein-DNA intermediates, such as those that occur during abortive topoisomerase reactions; and interstrand cross-links, among others (14, 15, 28). Branched DNA structures arise in several independent ways, making it difficult to prevent their occurrence without rendering the cell extremely debilitated. Even if they arise, bacterial mutants defective in DNA repair would very likely be hypersensitive to killing by the immune system. Moreover, the peptide does not appear to depend on any single uptake system for transport, since neither deletions of the major peptide uptake system in B. subtilis nor deletion of any single nonessential E. coli gene present in the Keio knockout collection (2) confers resistance to the peptide (22; S. Orchard and A. Segall, unpublished results). No spontaneous mutants have been isolated in a hyperpermeable Salmonella strain with short LPS chains (1) (the maximum sensitivity of the assay was 1 in 1010 [J. Patelzick and A. Segall, unpublished results]). In aggregate, these properties suggest that the peptide may be a very effective antibiotic from the standpoint of the low-frequency evolution of resistance-conferring mutations. A possible disadvantage of the peptide's targets is that DNA repair intermediates in bacteria may be similar enough to those in the mammalian host to cause toxic side effects. However, as discussed above, inefficient nuclear transport and the chromatin structure itself may protect eukaryotic host cells. These features may be improved upon when future generations of these antimicrobial compounds are designed in order to increase the differential toxicities between bacteria and mammalian cells.
Supplementary Material
Acknowledgments
We are grateful to Tong Xu for her assistance with the harvesting of murine peritoneal macrophages; to Geoffrey Cassell for his critical reading of the manuscript; and to Stanley Maloy, Joshua Fierer, and Nancy Buchmeier for critical discussions and advice. We thank James Slauch, University of Illinois, Urbana-Champaign, for the gift of strain G785. Lisa Morrison and Steve Barlow, the director of the SDSU EM and Fluorescence Imaging Core, were very helpful with confocal microscopy training. Ilham Naili helped with the initial examination of the induction of the intracellular SOS response in the Salmonella bacteria.
This work was supported by National Institutes of Health grants R01 A1058253 and R21 AI061351 to A.M.S. from the National Institute for Allergy and Infectious Diseases.
None of us has financial interests related to the work described here.
Footnotes
Published ahead of print on 22 February 2010.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Ames, B. N., F. D. Lee, and W. E. Durston. 1972. An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc. Natl. Acad. Sci. U. S. A. 70:782-786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baba, T., T. Ara, M. Hasegawa, Y. Okumura, M. Baba, K. A. Datsenko, M. Tomita, B. L. Wanner, and H. Mori. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barre, F.-X., and D. Sherratt. 2002. Xer site-specific recombination: promoting chromosome segregation, p. 149-161. In N. L. Craig, R. Craigie, M. Gellert, and A. M. Lambowitz (ed.), Mobile DNA II. ASM Press, Washington, DC.
- 4.Boldt, J. L., C. Pinilla, and A. M. Segall. 2004. Reversible inhibitors of lambda integrase-mediated recombination efficiently trap Holliday junction intermediates and form the basis of a novel assay for junction resolution. J. Biol. Chem. 279:3472-3483. [DOI] [PubMed] [Google Scholar]
- 5.Brogden, K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238-250. [DOI] [PubMed] [Google Scholar]
- 6.Brotz, H., G. Bierbaum, K. Leopold, P. E. Reynolds, and H. G. Sahl. 1998. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 42:154-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown, K. L., and R. E. Hancock. 2006. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 18:24-30. [DOI] [PubMed] [Google Scholar]
- 8.Buchmeier, N. A., C. J. Lipps, M. Y. So, and F. Heffron. 1993. Recombination-deficient mutants of Salmonella typhimurium are avirulent and sensitive to the oxidative burst of macrophages. Mol. Microbiol. 7:933-936. [DOI] [PubMed] [Google Scholar]
- 9.Buchmeier, N. A., S. J. Libby, Y. Xu, P. C. Loewen, J. Switala, D. G. Guiney, and F. C. Fang. 1995. DNA repair is more important than catalase for Salmonella virulence in mice. J. Clin. Invest. 95:1047-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bugreev, D. V., O. M. Mazina, and A. V. Mazin. 2006. Rad54 protein promotes branch migration of Holliday junctions. Nature 442:590-593. [DOI] [PubMed] [Google Scholar]
- 11.Chadfield, M., and J. Olsen. 2001. Determination of the oxidative burst chemiluminescent response of avian and murine-derived macrophages versus corresponding cell lines in relation to stimulation with Salmonella serotypes. Vet. Immunol. Immunopathol. 80:289-308. [DOI] [PubMed] [Google Scholar]
- 12.Christensen, B., J. Fink, R. B. Merrifield, and D. Mauzerall. 1988. Channel-forming properties of cecropins and related model compounds incorporated into planar lipid membranes. Proc. Natl. Acad. Sci. U. S. A. 85:5072-5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Closs, E. I., J. Scheld, M. Sharafi, and U. Forstermann. 2000. Substrate supply for nitric-oxide synthase in macrophages and endothelial cells: role of cationic amino acid transporters. Mol. Pharmacol. 57:68-74. [PubMed] [Google Scholar]
- 14.Cox, M. M. 2001. Recombinational DNA repair of damaged replication forks in Escherichia coli. Annu. Rev. Genet. 35:35-82. [DOI] [PubMed] [Google Scholar]
- 15.Cox, M. M., M. F. Goodman, K. N. Kreuzer, D. J. Sherratt, S. J. Sandler, and K. J. Marians. 2001. The importance of repairing stalled replication forks. Nature 404:37-41. [DOI] [PubMed] [Google Scholar]
- 16.Duchardt, F., M. Fotin-Mleczek, H. Schwarz, R. Fischer, and R. Brock. 2007. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 8:848-866. [DOI] [PubMed] [Google Scholar]
- 17.Elsinghorst, E. A. 1994. Measurement of invasion by gentamicin resistance. Methods Enzymol. 236:405-420. [DOI] [PubMed] [Google Scholar]
- 18.Eriksson, S., S. Lucchini, A. Thompson, M. Rhen, and J. C. D. Hinton. 2003. Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol. Microbiol. 47:103-118. [DOI] [PubMed] [Google Scholar]
- 19.Ferraro, M. J. 2000. National Committee for Clinical Laboratory Standards methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard H7-A5, p. 36. National Committee for Clinical Laboratory Standards, Wayne, PA.
- 20.Fischer, R., K. Kohler, M. Fotin-Mleczek, and R. Brock. 2004. A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J. Biol. Chem. 279:12625-12635. [DOI] [PubMed] [Google Scholar]
- 21.Futaki, S., T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda, and Y. Sugiura. 2001. Arginine-rich peptide: an abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 276:5836-5840. [DOI] [PubMed] [Google Scholar]
- 22.Gunderson, C. W., and A. M. Segall. 2006. DNA repair, a novel antibacterial target: Holliday junction-trapping peptides induce DNA damage and chromosome segregation defects. Mol. Microbiol. 59:1129-1148. [DOI] [PubMed] [Google Scholar]
- 23.Gunderson, C. W., J. L. Boldt, R. N. Authement, and A. M. Segall. 2009. Peptide wrwycr inhibits excision of several prophages and traps Holliday junctions inside bacteria. J. Bacteriol. 191:2169-2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiao, C. Y., D. Delaroche, F. Burlina, I. D. Alves, G. Chassaing, and S. Sagan. 2009. Translocation and endocytosis for cell-penetrating peptide internalization. J. Biol. Chem. 284:33957-33965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kepple, K. V., J. L. Boldt, and A. M. Segall. 2005. Holliday junction-binding peptides inhibit distinct junction-processing enzymes. Proc. Natl. Acad. Sci. U. S. A. 102:6867-6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kepple, K. V., N. Patel, P. Salamon, and A. M. Segall. 2008. Interactions between branched DNAs and peptide inhibitors of DNA repair. Nucleic Acids Res. 36:5319-5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuzaki, K., S. Yoneyama, and K. Miyajima. 1997. Pore formation and translocation of melittin. Biophys. J. 73:831-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michel, B., G. Grompone, M. J. Flores, and V. Bidnenko. 2004. Multiple pathways process stalled replication forks. Proc. Natl. Acad. Sci. U. S. A. 101:12783-12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosmann, T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65:55-63. [DOI] [PubMed] [Google Scholar]
- 30.Schlosser-Silverman, E., M. Elgrably-Weiss, I. Rosenshine, R. Kohen, and S. Altuvia. 2000. Characterization of Escherichia coli DNA lesions generated within J774 macrophages. J. Bacteriol. 182:5225-5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwan, W. R., X. Z. Huang, L. Hu, and D. J. Kopecko. 2000. Differential bacterial survival, replication, and apoptosis-inducing ability of Salmonella serovars within human and murine macrophages. Infect. Immun. 68:1005-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sciochetti, S. A., and P. J. Piggot. 2000. A tale of two genomes: resolution of dimeric chromosomes in Escherichia coli and Bacillus subtilis. Res. Microbiol. 151:503-511. [DOI] [PubMed] [Google Scholar]
- 33.Simonsen, A., R. Lippe, S. Christoforidis, J.-M. Gaullier, A. Brech, J. Callaghan, B.-H. Toh, C. Murphy, M. Zerial, and H. Stenmark. 1998. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394:494-498. [DOI] [PubMed] [Google Scholar]
- 34.Ter-Avertisyan, G., G. Tünnemann, D. Nowak, M. Nitschke, A. Herrmann, M. Drab, and M. C. Cardoso. 2009. Cell entry of arginine-rich peptides is independent of endocytosis. J. Biol. Chem. 284:3370-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toke, O. 2005. Antimicrobial peptides: new candidates in the fight against bacterial infections. Biopolymers 80:717-735. [DOI] [PubMed] [Google Scholar]
- 36.Valdivia, R. H., and S. Falkow. 1996. Bacterial genetics by flow cytometry: rapid isolation of Salmonella typhimurium acid-inducible promoters by differential fluorescence induction. Mol. Microbiol. 22:367-378. [DOI] [PubMed] [Google Scholar]
- 37.Wimley, W. C., M. E. Selsted, and S. H. White. 1994. Interactions between human defensins and lipid bilayers: evidence for formation of multimeric pores. Protein Sci. 3:1362-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389-395. [DOI] [PubMed] [Google Scholar]
- 39.Zorko, M., and U. Langel. 2005. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 57:529-545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.