

Abstract
Ethambutol, together with a macrolide, is the backbone for treatment of disseminated Mycobacterium avium disease. However, at the standard dose of 15 mg/kg of body weight/day, ethambutol efficacy is limited. In addition, susceptibility breakpoints have consistently failed to predict clinical outcome. We performed dose-effect studies with extracellular M. avium as well as with bacilli within human macrophages. The maximal kill rate (Emax) for ethambutol against extracellular bacilli was 5.54 log10 CFU/ml, compared to 0.67 log10 CFU/ml for intracellular M. avium, after 7 days of exposure. Thus, extracellular assays demonstrated high efficacy. We created a hollow-fiber system model of intracellular M. avium and performed microbial pharmacokinetic-pharmacodynamic studies using pharmacokinetics similar to those of ethambutol for humans. The Emax in the systems was 0.79 log10 CFU/ml with 7 days of daily therapy, so the kill rates approximated those encountered in patients treated with ethambutol monotherapy. Ratio of peak concentration to MIC (Cmax/MIC) was linked to microbial kill rate. The Cmax/MIC ratio needed to achieve the 90% effective concentration (EC90) in serum was 1.23, with a calculated intramacrophage Cmax/MIC ratio of 13. In 10,000 patient Monte Carlo simulations, doses of 15, 50, and 75 mg/kg achieved the EC90 in 35.50%, 76.81%, and 86.12% of patients, respectively. Therefore, ethambutol doses of ≥50 mg/kg twice a week would be predicted to be better than current doses of 15 mg/kg for treatment of disseminated M. avium disease. New susceptibility breakpoints and critical concentrations of 1 to 2 mg/liter were identified for the determination of ethambutol-resistant M. avium in Middlebrook broth. Given that the modal MIC of clinical isolates is around 2 mg/liter, most isolates should be considered ethambutol resistant.
Bacteria of the Mycobacterium avium complex (MAC) are not only a cause of important opportunistic infections in patients with AIDS and cancer but also an important cause of pulmonary disease in “immunocompetent” patients (5, 17, 26, 31). In immunocompromised patients, disseminated disease is common and lethal (8, 10). Current therapy is based on a backbone of ethambutol, concurrent with either azithromycin or clarithromycin, administered for many months to several years (17, 18). This therapy fails in up to half of patients and therefore still needs to be optimized.
The first test used to examine the therapeutic potential of ethambutol, as for all other antibiotics, is MIC determination. With this test, it is hoped that discrimination can be made as to whether ethambutol should be used in the patient from whom M. avium was isolated. Unfortunately, susceptibility studies have failed to predict the effectiveness of ethambutol in patients with disseminated M. avium disease in the past (5, 34, 39). We hypothesize that part of the problem has been an arbitrarily chosen susceptibility breakpoint, which did not take into account population pharmacokinetics and microbial pharmacokinetics-pharmacodynamics (PK/PD) (2, 3). Attempts have been made to examine the efficacy of ethambutol in the beige mouse model. Unfortunately, when the model was used to predict the efficacy of ethambutol versus clofazimine versus rifampin monotherapy, clofazimine was found to have the greatest efficacy and to be much better than ethambutol (40). However, in AIDS patients with disseminated M. avium disease, ethambutol monotherapy at 15 mg/kg of body weight/day led to a decrease of 0.6 log10 CFU/ml of blood over 4 weeks (or an average of 0.15 log10 CFU/ml/week), while 200 mg a day of clofazimine had no significant effect (32). Thus, results in the beige mouse model may be difficult to translate to the clinic. The reasons are unclear. We created an in vitro PK/PD model of disseminated intracellular M. avium, which we utilized to examine the relationship between ethambutol exposure, dose schedule, and M. avium response. The results were then used for Monte Carlo simulations to determine a new susceptibility breakpoint for M. avium and an optimal ethambutol dose.
MATERIALS AND METHODS
Cells and culture conditions.
Stock cultures of M. avium (ATCC 700898) were stored in Middlebrook 7H9 broth and 10% glycerol at −80°C. The bacterial stock was thawed and incubated in a water bath in Middlebrook 7H9 broth at 37°C under shaking conditions for 4 days to achieve exponential-phase growth. Human-derived THP-1 macrophages (ATCC TIB-202) were cultured in prewarmed RPMI 1640 medium with 10% heat-inactivated fetal bovine serum (FBS) at 37°C under 5% CO2.
Materials.
Ethambutol (Sigma-Aldrich) was dissolved in sterile water to the drug concentrations required for each study. Hollow-fiber cartridges were purchased from Fibercell. RPMI medium and 10% fetal bovine serum were purchased from Sigma. FBS was heat inactivated at 56°C for 30 min and filtered prior to use.
Determination of MIC and mutation frequency.
MIC studies were performed as described by the Clinical and Laboratory Standards Institute (11). The ethambutol concentrations examined were 0, 0.125, 0.25, 0.5, 1.0, 2, 4, 8, 16, and 32 mg/liter. An Etest (AB Biodisk) was utilized as a confirmatory test. Mutation frequency studies were performed by adjusting the turbidity of exponential-phase-growth bacteria to a McFarland standard of 0.5 (1 × 107 CFU/ml) and then plating the bacteria onto Middlebrook 7H10 agar supplemented with ethambutol at 3 times the MIC.
Infection of THP-1 macrophages with M. avium.
THP-1 cells were cultured and prewarmed in RPMI medium-10% FBS to achieve a density of 1.5 ×106 macrophages per ml. These cells were then infected with M. avium at 1.5 ×105 CFU/ml overnight by coincubation at 37°C under 5% CO2, giving a multiplicity of infection of 10. Infected macrophages were washed twice with warm RPMI medium-10% FBS by centrifugation at 100 × g for 5 min and then examined for viability using trypan blue and counted using a hemocytometer. A bacillary burden of 4 log10 to 5 log10 CFU/ml was obtained with this process of intracellular infection.
Exposure-effect study of extracellular MAC in 24-well plates.
To study the activity of ethambutol against extracellular M. avium, we grew bacteria in log-phase growth for 4 days to a density of 1.5 ×103 CFU/ml. A bacterial suspension in Middlebrook 7H9 broth was coincubated with an ethambutol concentration of either 16, 8, 4, 2, 1.0, 0.5, 0.25, 0.125, or 0 mg/liter in triplicate. After 7 days of incubation at 37°C under 5% CO2, the cultures were serially diluted, plated on Middlebrook 7H10 agar, and incubated at 37°C under 5% CO2 and colonies counted 14 days later.
Exposure-effect study with intracellular M. avium in 24-well plates.
Infected macrophages were prepared as described above and then coincubated with ethambutol concentrations in 24-well plates for 7 days, as described above. On day 7, 1 ml of cell suspension was centrifuged at 100 × g for 5 min, and the supernatant was discarded and then reconstituted with 1 ml of 0.5% Triton X-100 in phosphate-buffered saline. The reconstituted suspension was vortex agitated to obtain macrophage lysate. Serial dilutions of macrophage lysate suspension were performed, plated at 0.2 ml on Middlebrook 7H10 agar, and incubated for 14 days as described above.
Pharmacokinetic-pharmacodynamic model of disseminated M. avium.
Hollow-fiber systems have been used for microbial PK/PD studies of various bacterial groups (6, 7, 23, 25, 42). The full model has been described in detail for extracellular bacteria in the past (21, 24). The system is used to mimic the human concentration-time profiles of antibiotics in the central compartment, which circulates RPMI medium-10% FBS, but is cell free. Twenty milliliters of THP-1 macrophages infected with M. avium was inoculated into the external compartment, where they were continuously bathed in medium from the central compartment, which freely crosses the semipermeable hollow fibers. The cells are too big to pass across the semipermeable membrane and thus stay confined to the peripheral compartment, where they are harvested for cell counts and quantitative cultures. This system was able to mimic the bacillary burden encountered in severely immunocompromised patients with disseminated M. avium disease (9, 14, 16, 43).
For dose-effect studies, each hollow-fiber system was treated once daily with ethambutol under the control of a computerized syringe pump. Concentration-time profiles achieved by human doses equivalent to 0, 6, 12.5, 20, 25, 50, 100, and 200 mg/kg/day were mimicked. Computer-controlled peristaltic pumps pumped in fresh RPMI medium-10% FBS and pumped used medium out of the systems to achieve drug dilution rates that resulted in the ethambutol's multiphasic kinetics: a time to maximum concentration of 2 h, a half-life of 3 h during the first 11 h, and a half-life of 12 h thereafter (35, 36, 45). Concentrations achieved in each system were examined by sampling the central compartment at 0, 2, 5, 8, 11, 16, and 24 h after the 7th infusion. The peripheral compartment was sampled on days 2 and 7. Macrophage viability was assessed, after which macrophages were ruptured, and bacterial density was determined as described above. The ethambutol-resistant subpopulation was determined by incubating the cultures on Middlebrook agar that had been supplemented with ethambutol at 3 times the MIC.
Next, we determined the best ethambutol dosing schedule. On the basis of the dose-effect study, the weekly cumulative doses that achieved 10%, 80%, and 90% of the maximal kill rate (Emax) (10% effective concentration [EC10], EC80, and EC90) were administered using one of two dose schedules, either 7 equal doses daily for the week or the whole weekly dose every 7 days. In addition, the cumulative weekly dose that achieved the EC80 was also divided into two equal doses and administered every 3.5 days. These concentrations were chosen so as to satisfy both the optimal information and the doses used in the clinic. The treatment was for 14 days. Pharmacokinetic sampling was performed at 13 different time points over the last 48 h of the study. On days 0, 7, and 14, bacterial cultures were performed in each system for total bacterial counts and the ethambutol-resistant population, as described above.
Measurement of ethambutol concentrations.
Samples were diluted 1:100 with deionized water, and a 20-μl sample volume was injected directly without further processing. A liquid chromatography-tandem mass spectrometry (LC/MS/MS) method was used to analyze samples on a Shimadzu high-performance liquid chromatography (HPLC) system with an ODS-3 Inertsil Varian column (50 by 2.1 mm [particle size, 5 μm]) at 40°C. The isocratic mobile phase (0.2 ml/h) consisted of 50% 0.1% formic acid in deionized water and 50% 0.1% formic acid in methanol (vol/vol). Detection was accomplished using an API 3000 mass spectrometer that was programmed in multiple-reaction-monitoring mode, with monitoring of the transition of the mass/charge ratio from 205.10 m/z for the precursor ion to 116.10 m/z for the product ion for ethambutol. The accuracy was evaluated at 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500, 600, 700, 800, 900, and 1,000 ng/ml. A reproducibility test (n = 5) for each concentration was performed on 3 different days. The results for the method were linear from 0.1 to 1,000 ng/ml, with a correlation coefficient of 0.999 and accuracy rates within ±5% between and within days. The lower limit of quantitation was 0.1 ng/ml.
Pharmacokinetic-pharmacodynamic modeling.
Pharmacokinetic analysis was performed using ADAPT II software (13). A one-compartment open model with first-order input and elimination was chosen. Estimation employed weighted least squares. Values for microbial PK/PD exposure parameters, such as ratio of peak concentration to MIC (Cmax/MIC), ratio of 0- to 24-h area under the concentration-time curve to MIC (AUC0-24/MIC), and percentage of time that the concentration persisted above the MIC (TMIC), were then calculated. The relationship between bacterial burden (Y) and exposure was examined using the inhibitory sigmoid Emax model in GraphPad Prism 5 software, with 1/Y2 weighting. The relationship between exposure and microbial kill rate was utilized to calculate the exposure associated with 90% of the maximal kill rate (EC90), which was considered the optimal kill rate.
Identification of optimal dose and susceptibility breakpoint.
The exposure-effect studies described above pertain to M. avium isolates with the same MIC as our isolate and for patients with pharmacokinetic parameter values identical to those in our hollow-fiber systems. In the clinical situation, there is a distribution of MICs among clinical isolates, as documented by broth susceptibility tests with 103 clinical isolates examined by Heifets et al. (28). In addition, pharmacokinetic variability in patients is always present, due to variability of xenobiotic metabolism and many other factors. Therefore, when an ethambutol dose is administered to patients, there will be a distribution of exposure parameters. The optimal clinical dose is that dose which would achieve the EC90 in the majority of patients. We utilized population pharmacokinetic data published by Peloquin et al. (36, 45) (ka [absorption constant], 0.84 ± 0.51/h; volume, 9.14 ± 10.47 liters/kg; and systemic clearance, 2.17 ± 1.29 liters/h/kg) as prior data in the PRIOR subroutine of the ADAPT 5 program. With this, we performed 10,000 patient Monte Carlo simulations to determine how likely ethambutol doses of 15 mg/kg, 25 mg/kg, 50 mg/kg, or 75 mg/kg would achieve the EC90 exposures in the serum of ≥90% of patients with disseminated M. avium disease, given the pharmacokinetic and MIC variability. In addition to determining the best therapeutic dose, we were also interested in establishing an ethambutol susceptibility breakpoint for M. avium. The susceptibility breakpoint was defined as the lowest MIC that allowed the maximum tolerated dose to achieve the EC90 in >90% of patients (2, 4, 44). The critical concentration of the drug, as defined by Clinical and Laboratory Standards Institute guidelines (11), was then set at 1 tube dilution higher than this MIC.
RESULTS
The ethambutol MIC was 8 mg/liter. The mutation frequency to 3 times the MIC was 7.25 × 10−7 ± 2.94 × 10−7. The relationships between ethambutol concentration and microbial kill for both extracellular and intracellular M. avium are shown in Fig. 1. Figure 1a demonstrates the extensive killing of M. avium by ethambutol which was an Emax of 5.54 (95% confidence interval [CI], 4.76 to 6.32) log10 CFU/ml in only 7 days, with an EC50 of 5.38 (95% CI, 3.68 to 7.08) mg/liter. However, while the EC50 did not change from 7.93 (95% CI, 5.19 to 10.66) mg/liter, the Emax declined to only 0.67 (95% CI, 0.54 to 0.81) log10 CFU/ml for intracellular M. avium. Thus, studies of ethambutol effect on extracellular bacilli overestimated the efficacy.
FIG. 1.

Relationship between ethambutol exposure and effect on extracellular (a) and intracellular (b) Mycobacterium avium.
The ethambutol concentration-time profiles achieved in the microbial PK/PD model of disseminated M. avium are shown in Fig. 2, which demonstrates ethambutol's multiphasic profile. The median values for the pharmacokinetic parameter estimates were as follows: ke (elimination rate constant), 0.153 h−1; volume, 342.8 liters; serum clearance, 95.1 liters·h−1; ka, 8.1 h−1; kcp, 6.9 h−1; and, kpc, 178.9 h−1 (r2 = 0.99) (kcp and kpc are the intercompartmental transfer constants from central to peripheral compartment and vice versa). The concentrations achieved had no effect on the viability of macrophages. In terms of microbial pharmacodynamics, there was no microbial kill by any ethambutol dose with 2 days of daily dosing; the microbial effect commenced later. The relationship between daily dose and day 7 M. avium burden was
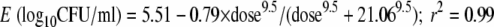 |
(1) |
where “dose” is the human equivalent daily dose (mg/kg). An examination of bacterial burden for all dose groups revealed that the effect of ethambutol was merely to constrain the rate of increase of bacterial burden since the bacterial burdens on day 7 were actually higher than those at the start of therapy (4.1 log10 CFU/ml at time zero). Thus, ethambutol was only bacteriostatic.
FIG. 2.

Ethambutol concentration-time profile in an in vitro PK/PD model of disseminated Mycobacterium avium disease.
The pharmacokinetic profile achieved in dose scheduling studies is shown in Fig. 3. The inhibitory sigmoid Emax relationship between day 7 bacterial burden and TMIC demonstrated an r2 of 0.18, an r2 of 0.56 for AUC/MIC ratio, and an r2 of 0.79 for Cmax/MIC ratio, with an EC50 (Cmax/MIC ratio) of 0.28. Furthermore, a reexamination of equation 1 from the dose-effect study using the Cmax/MIC ratio values achieved in that study identified an EC50 (Cmax/MIC ratio) of 0.32. The EC90 was calculated as a Cmax/MIC ratio of 1.23 in serum. In terms of resistance, by day 14 all treated arms had the same bacterial density as the nontreated control due to emergence of resistance, with no discernible pattern.
FIG. 3.

Ethambutol pharmacokinetics with dose scheduling study design.
In a 10,000-subject Monte Carlo simulation, a log-normal distribution best recapitulated the original pharmacokinetic parameter estimates as follows: ka, 0.84 ± 0.51/h; volume, 9.14 ± 10.47 liters/kg; and systemic clearance, 2.17 ± 1.29 liters/h. The median peak concentration for 19 mg/kg was 2.54 mg/liter, similar to the 2.11-mg/liter value observed in the original study (45). The probabilities of achieving the EC90 with different doses are shown in Fig. 4. The target attainment probability for 15 mg/kg was 35.50%, that for 25 mg/kg was 54.40%, that for 50 mg/kg was 76.81%, and that for 75 mg/kg was 86.12%. If it is conceded that the EC90 cannot be safely achieved in an acceptable number of patients with disseminated M. avium disease, and we settle for a lower target, equal to the EC50, then that target was achieved by 15 mg/kg in 80.33%, 25 mg/kg in 90.31%, and ≥50 mg/kg in ≥97.06% of patients. In fact, 15 mg/kg achieved a target attainment of >90% only when the exposure target was reduced to that associated with EC25 or 0.19 log10 CFU/ml per week, a kill rate similar to that observed in the clinic when patients were treated with 15 mg/kg/day. In terms of susceptibility breakpoints, the maximum MICs that allowed the doses to achieve the EC90 in >90% of patients were 0.475 mg/liter for 15 to 25 mg/kg and 0.95 mg/liter for 50 mg/kg. This means the critical concentrations should be ∼1.0 mg/liter with 15 to 25-mg/kg dosing and about 2 mg/liter when a 50-mg/kg dose is utilized.
FIG. 4.

Target attainment by different ethambutol doses in 10,000 simulated patients.
DISCUSSION
We have created a model that has utility in studying the relationship between antimicrobial exposure and intracellular M. avium response. Contrary to the high efficacy identified when extracellular M. avium was exposed to static concentrations of ethambutol in liquid culture, microbial kill indices in the in vitro PK/PD system approximated the modest effect achieved in blood cultures of patients treated with ethambutol monotherapy. Therefore, our model holds promise in the examination of efficacy of both new and old drugs that have the potential to treat M. avium infections.
We utilized the model to examine the ethambutol microbial PK/PD relationships. Unlike with Mycobacterium tuberculosis (20-25, 29, 30, 38, 41), no antimicrobial PK/PD studies have been performed with M. avium in the past. The ratio of peak concentration to MIC (Cmax/MIC) best explained the microbial effect of ethambutol on intracellular M. avium. In recent studies of Mycobacterium tuberculosis, the AUC/MIC ratio (a measure of total drug exposure) best explained the microbial kill rate of ethambutol, although in one experiment, both Cmax/MIC ratio and AUC/MIC ratio could have explained the effect (41). In reactivation tuberculosis, M. tuberculosis is predominantly an extracellular pathogen, as opposed to the intracellular pathogen M. avium (15). We speculate that the concentration-time profile of ethambutol within the infected macrophage may explain the microbial response pattern that we observed. Indeed, if one considers that the extracellular Cmax/MIC ratio associated with 90% of the optimal kill rate is only 1.23, barely above the MIC, the importance of the intracellular or intramacrophage concentration becomes clearer. This is because ethambutol is concentrated 11- to 21-fold in macrophages (12, 27), so we calculate that the corresponding optimal intracellular Cmax/MIC ratio would be at least 13. Such a Cmax/MIC exposure ratio is more in the range with that associated with optimal efficacy for a number of other antibiotics (3, 33). Whatever the explanation, our dose scheduling study suggests that at least 50 mg/kg twice a week would be better than 15 mg/kg daily and that perhaps even 75 mg/kg once or twice a week would be more effective. The finding that high peak concentrations associated with once-a-week therapy are superior to low peak concentrations associated with daily therapy suggests that ethambutol may have a considerable postantibiotic effect against intracellular M. avium.
A concern with higher ethambutol doses is that involving ocular toxicity. Recently, Griffith et al. examined 229 patients from six prospective clinical trials who had received either 25 mg/kg three times a week or 15 mg/kg daily as part of their treatment regimen (19). Dosing schedule was the only significant predictor of toxicity; 6% of patients on daily therapy were diagnosed with ocular toxicity, versus 0% with the intermittent therapy. This strongly suggests either a time above the threshold or AUC-driven toxicity. Conversely, this means that peak ethambutol concentrations are not the most important drivers of toxicity. This harmonizes with prior studies using 50 mg/kg administered twice a week, which were also associated with little to no ocular toxicity (1, 37). Thus, it may be that intermittent dosing would drive both efficacy and safety. Nevertheless, the safety of a dose of 75 mg/kg is as of yet unknown. Thus, while likely to lead to higher efficacy, this higher dose still needs to be proven to be safe and cannot yet be recommended for routine use. In addition, the lower doses of 50 mg/kg may work satisfactorily in practice since ethambutol is actually administered in combination with a macrolide.
M. avium susceptibility breakpoints were developed from those for M. tuberculosis (28). However, these breakpoints have failed to predict clinical success (5, 34, 39). Our study suggests that the breakpoints should be set lower, so that the critical concentrations would be 1.0 mg/liter with current dosing and 2 mg/liter for ≥50 mg/kg. It may be that the lack of correlation between ethambutol susceptibility and clinical outcome in patients treated with 15 mg/kg in the past was observed because most isolates were already ethambutol resistant. With this dose, the probability of target attainment in all patients infected with isolates with MICs above the 1.0-mg/liter breakpoint would be only 27.8%. Thus, a small proportion of patients infected by isolates with MICs of 2 to 4 mg/liter (“resistant”) would still respond to therapy since a high enough Cmax/MIC ratio is achieved in some patients on the basis of pharmacokinetic variability. However, this proportion of patients is clearly too low, and thus, these isolates should be deemed resistant. The predictive power of our proposed breakpoint will need to be examined for a large number of patients treated with the higher dose of ethambutol.
In summary, we created an in vitro model of disseminated M. avium. The model was utilized to study the microbial PK/PD properties of ethambutol. These studies revealed that the effect of ethambutol against M. avium was Cmax/MIC ratio linked. On the basis of these studies, a dose of ≥50 mg/kg twice a week for disseminated M. avium is proposed. In addition, a 1-mg/liter critical concentration for resistance testing is proposed.
Footnotes
Published ahead of print on 15 March 2010.
REFERENCES
- 1.Albert, R. K., J. A. Sbarbaro, L. D. Hudson, and M. Iseman. 1976. High-dose ethambutol: its role in intermittent chemotherapy. A six-year study. Am. Rev. Respir. Dis. 114:699-704. [DOI] [PubMed] [Google Scholar]
- 2.Ambrose, P. G. 2006. Monte Carlo simulation in the evaluation of susceptibility breakpoints: predicting the future: insights from the society of infectious diseases pharmacists. Pharmacotherapy 26:129-134. [DOI] [PubMed] [Google Scholar]
- 3.Ambrose, P. G., S. M. Bhavnani, C. M. Rubino, A. Louie, T. Gumbo, A. Forrest, and G. L. Drusano. 2007. Pharmacokinetics-pharmacodynamics of antimicrobial therapy: it's not just for mice anymore. Clin. Infect. Dis. 44:79-86. [DOI] [PubMed] [Google Scholar]
- 4.Ambrose, P. G., A. K. Meagher, J. A. Passarell, S. A. Van Wart, B. B. Cirincione, S. M. Bhavnani, and E. Ellis-Grosse. 2009. Application of patient population-derived pharmacokinetic-pharmacodynamic relationships to tigecycline breakpoint determination for staphylococci and streptococci. Diagn. Microbiol. Infect. Dis. 63:155-159. [DOI] [PubMed] [Google Scholar]
- 5.Anonymous. 2001. First randomised trial of treatments for pulmonary disease caused by M avium intracellulare, M malmoense, and M xenopi in HIV negative patients: rifampicin, ethambutol and isoniazid versus rifampicin and ethambutol. Thorax 56:167-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaser, J., B. B. Stone, and S. H. Zinner. 1985. Efficacy of intermittent versus continuous administration of netilmicin in a two-compartment in vitro model. Antimicrob. Agents Chemother. 27:343-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaser, J., B. B. Stone, and S. H. Zinner. 1985. Two compartment kinetic model with multiple artificial capillary units. J. Antimicrob. Chemother. 15(Suppl. A):131-137. [DOI] [PubMed] [Google Scholar]
- 8.Chaisson, R. E., J. E. Gallant, J. C. Keruly, and R. D. Moore. 1998. Impact of opportunistic disease on survival in patients with HIV infection. AIDS 12:29-33. [DOI] [PubMed] [Google Scholar]
- 9.Chaisson, R. E., P. Keiser, M. Pierce, W. J. Fessel, J. Ruskin, C. Lahart, C. A. Benson, K. Meek, N. Siepman, and J. C. Craft. 1997. Clarithromycin and ethambutol with or without clofazimine for the treatment of bacteremic Mycobacterium avium complex disease in patients with HIV infection. AIDS 11:311-317. [DOI] [PubMed] [Google Scholar]
- 10.Chaisson, R. E., R. D. Moore, D. D. Richman, J. Keruly, and T. Creagh. 1992. Incidence and natural history of Mycobacterium avium-complex infections in patients with advanced human immunodeficiency virus disease treated with zidovudine. The Zidovudine Epidemiology Study Group. Am. Rev. Respir. Dis. 146:285-289. [DOI] [PubMed] [Google Scholar]
- 11.Clinical and Laboratory Standards Institute. 2003. Susceptibility testing of mycobacteria, nocardiae, and other aerobic actinomycetes. Approved standard. Clinical and Laboratory Standards Institute, Wayne, PA. [PubMed]
- 12.Conte, J. E., Jr., J. A. Golden, J. Kipps, E. T. Lin, and E. Zurlinden. 2001. Effects of AIDS and gender on steady-state plasma and intrapulmonary ethambutol concentrations. Antimicrob. Agents Chemother. 45:2891-2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Argenio, D. Z., and A. Schumitzky. 1997. ADAPT II. A program for simulation, identification, and optimal experimental design. User manual. Biomedical Simulations Resource. University of Southern California, Los Angeles, CA.
- 14.Dube, M. P., F. R. Sattler, F. J. Torriani, D. See, D. V. Havlir, C. A. Kemper, M. G. Dezfuli, S. A. Bozzette, A. E. Bartok, J. M. Leedom, J. G. Tilles, and J. A. McCutchan. 1997. A randomized evaluation of ethambutol for prevention of relapse and drug resistance during treatment of Mycobacterium avium complex bacteremia with clarithromycin-based combination therapy. California Collaborative Treatment Group. J. Infect. Dis. 176:1225-1232. [DOI] [PubMed] [Google Scholar]
- 15.Eum, S. Y., J. H. Kong, M. S. Hong, Y. J. Lee, J. H. Kim, S. H. Hwang, S. N. Cho, L. E. Via, and C. E. Barry III. 2010. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137:122-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gordin, F. M., P. M. Sullam, S. D. Shafran, D. L. Cohn, B. Wynne, L. Paxton, K. Perry, and C. R. Horsburgh, Jr. 1999. A randomized, placebo-controlled study of rifabutin added to a regimen of clarithromycin and ethambutol for treatment of disseminated infection with Mycobacterium avium complex. Clin. Infect. Dis. 28:1080-1085. [DOI] [PubMed] [Google Scholar]
- 17.Griffith, D. E. 2007. Therapy of nontuberculous mycobacterial disease. Curr. Opin. Infect. Dis. 20:198-203. [DOI] [PubMed] [Google Scholar]
- 18.Griffith, D. E., T. Aksamit, B. A. Brown-Elliott, A. Catanzaro, C. Daley, F. Gordin, S. M. Holland, R. Horsburgh, G. Huitt, M. F. Iademarco, M. Iseman, K. Olivier, S. Ruoss, C. F. von Reyn, R. J. Wallace, Jr., and K. Winthrop. 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 175:367-416. [DOI] [PubMed] [Google Scholar]
- 19.Griffith, D. E., B. A. Brown-Elliott, S. Shepherd, J. McLarty, L. Griffith, and R. J. Wallace, Jr. 2005. Ethambutol ocular toxicity in treatment regimens for Mycobacterium avium complex lung disease. Am. J. Respir. Crit. Care Med. 172:250-253. [DOI] [PubMed] [Google Scholar]
- 20.Gumbo, T. 2008. Integrating pharmacokinetics, pharmacodynamics and pharmacogenomics to predict outcomes in antibacterial therapy. Curr. Opin. Drug Discov. Devel. 11:32-42. [PubMed] [Google Scholar]
- 21.Gumbo, T., A. Louie, M. R. Deziel, and G. L. Drusano. 2005. Pharmacodynamic evidence that ciprofloxacin failure against tuberculosis is not due to poor microbial kill but to rapid emergence of resistance. Antimicrob. Agents Chemother. 49:3178-3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gumbo, T., A. Louie, M. R. Deziel, W. Liu, L. M. Parsons, M. Salfinger, and G. L. Drusano. 2007. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob. Agents Chemother. 51:3781-3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gumbo, T., A. Louie, M. R. Deziel, L. M. Parsons, M. Salfinger, and G. L. Drusano. 2004. Selection of a moxifloxacin dose that suppresses drug resistance in Mycobacterium tuberculosis, by use of an in vitro pharmacodynamic infection model and mathematical modeling. J. Infect. Dis. 190:1642-1651. [DOI] [PubMed] [Google Scholar]
- 24.Gumbo, T., A. Louie, W. Liu, P. G. Ambrose, S. M. Bhavnani, D. Brown, and G. L. Drusano. 2007. Isoniazid's bactericidal activity ceases because of the emergence of resistance, not depletion of Mycobacterium tuberculosis in the log phase of growth. J. Infect. Dis. 195:194-201. [DOI] [PubMed] [Google Scholar]
- 25.Gumbo, T., C. S. Siyambalapitiyage Dona, C. Meek, and R. Leff. 2009. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: A paradigm for faster assessment of new antituberculosis drugs. Antimicrob. Agents Chemother. 53:3197-3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han, X. Y., J. J. Tarrand, R. Infante, K. L. Jacobson, and M. Truong. 2005. Clinical significance and epidemiologic analyses of Mycobacterium avium and Mycobacterium intracellulare among patients without AIDS. J. Clin. Microbiol. 43:4407-4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hand, W. L., R. M. Boozer, and N. L. King-Thompson. 1985. Antibiotic uptake by alveolar macrophages of smokers. Antimicrob. Agents Chemother. 27:42-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heifets, L. B., M. D. Iseman, and P. J. Lindholm-Levy. 1986. Ethambutol MICs and MBCs for Mycobacterium avium complex and Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 30:927-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jayaram, R., S. Gaonkar, P. Kaur, B. L. Suresh, B. N. Mahesh, R. Jayashree, V. Nandi, S. Bharat, R. K. Shandil, E. Kantharaj, and V. Balasubramanian. 2003. Pharmacokinetics-pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob. Agents Chemother. 47:2118-2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jayaram, R., R. K. Shandil, S. Gaonkar, P. Kaur, B. L. Suresh, B. N. Mahesh, R. Jayashree, V. Nandi, S. Bharath, E. Kantharaj, and V. Balasubramanian. 2004. Isoniazid pharmacokinetics-pharmacodynamics in an aerosol infection model of tuberculosis. Antimicrob. Agents Chemother. 48:2951-2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasperbauer, S. H., and C. L. Daley. 2008. Diagnosis and treatment of infections due to Mycobacterium avium complex. Semin. Respir. Crit. Care Med. 29:569-576. [DOI] [PubMed] [Google Scholar]
- 32.Kemper, C. A., D. Havlir, D. Haghighat, M. Dube, A. E. Bartok, J. P. Sison, Y. Yao, B. Yangco, J. M. Leedom, J. G. Tilles, et al. 1994. The individual microbiologic effect of three antimycobacterial agents, clofazimine, ethambutol, and rifampin, on Mycobacterium avium complex bacteremia in patients with AIDS. J. Infect. Dis. 170:157-164. [DOI] [PubMed] [Google Scholar]
- 33.Kim, M.-K., and D. P. Nicolau. 2007. Aminoglycosides, p. 147-175. In C. H. Nightangle, P. G. Ambrose, G. L. Drusano, and T. Murakawa (ed.), Antimicrobial pharmacodynamics in theory and practice. Informa Healthcare USA, New York, NY.
- 34.Kobashi, Y., K. Yoshida, N. Miyashita, Y. Niki, and M. Oka. 2006. Relationship between clinical efficacy of treatment of pulmonary Mycobacterium avium complex disease and drug-sensitivity testing of Mycobacterium avium complex isolates. J. Infect. Chemother. 12:195-202. [DOI] [PubMed] [Google Scholar]
- 35.Lee, C. S., D. C. Brater, J. G. Gambertoglio, and L. Z. Benet. 1980. Disposition kinetics of ethambutol in man. J. Pharmacokinet. Biopharm. 8:335-346. [DOI] [PubMed] [Google Scholar]
- 36.Peloquin, C. A., A. E. Bulpitt, G. S. Jaresko, R. W. Jelliffe, J. M. Childs, and D. E. Nix. 1999. Pharmacokinetics of ethambutol under fasting conditions, with food, and with antacids. Antimicrob. Agents Chemother. 43:568-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sbarbaro, J. A., and L. D. Hudson. 1974. High dose ethambutol; an oral alternate for intermittent chemotherapy. Am. Rev. Respir. Dis. 110:91-94. [DOI] [PubMed] [Google Scholar]
- 38.Shandil, R. K., R. Jayaram, P. Kaur, S. Gaonkar, B. L. Suresh, B. N. Mahesh, R. Jayashree, V. Nandi, S. Bharath, and V. Balasubramanian. 2007. Moxifloxacin, ofloxacin, sparfloxacin, and ciprofloxacin against Mycobacterium tuberculosis: evaluation of in vitro and pharmacodynamic indices that best predict in vivo efficacy. Antimicrob. Agents Chemother. 51:576-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sison, J. P., Y. Yao, C. A. Kemper, J. R. Hamilton, E. Brummer, D. A. Stevens, and S. C. Deresinski. 1996. Treatment of Mycobacterium avium complex infection: do the results of in vitro susceptibility tests predict therapeutic outcome in humans? J. Infect. Dis. 173:677-683. [DOI] [PubMed] [Google Scholar]
- 40.Sison, J. P., Y. Yao, C. A. Kemper, J. R. Hamilton, E. Brummer, D. A. Stevens, and S. C. Deresinski. 1996. Treatment of Mycobacterium avium complex infection: does the beige mouse model predict therapeutic outcome in humans? J. Infect. Dis. 173:750-753. [DOI] [PubMed] [Google Scholar]
- 41.Srivastava, S., S. Musuka, C. Sherman, C. Meek, R. Leff, and T. Gumbo. 2010. Efflux-pump-derived multiple drug resistance to ethambutol monotherapy in Mycobacterium tuberculosis and the pharmacokinetics and pharmacodynamics of ethambutol. J. Infect. Dis. 201:1225-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tam, V. H., A. Louie, M. R. Deziel, W. Liu, R. Leary, and G. L. Drusano. 2005. Bacterial-population responses to drug-selective pressure: examination of garenoxacin's effect on Pseudomonas aeruginosa. J. Infect. Dis. 192:420-428. [DOI] [PubMed] [Google Scholar]
- 43.Torriani, F. J., C. A. Behling, J. A. McCutchan, R. H. Haubrich, and D. V. Havlir. 1996. Disseminated Mycobacterium avium complex: correlation between blood and tissue burden. J. Infect. Dis. 173:942-949. [DOI] [PubMed] [Google Scholar]
- 44.Turnidge, J., and D. L. Paterson. 2007. Setting and revising antibacterial susceptibility breakpoints. Clin. Microbiol. Rev. 20:391-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu, M., W. J. Burman, J. R. Starke, J. J. Stambaugh, P. Steiner, A. E. Bulpitt, D. Ashkin, B. Auclair, S. E. Berning, R. W. Jelliffe, G. S. Jaresko, and C. A. Peloquin. 2004. Pharmacokinetics of ethambutol in children and adults with tuberculosis. Int. J. Tuber. Lung Dis. 8:1360-1367. [PubMed] [Google Scholar]