

Abstract
The Epstein-Barr virus (EBV) immediate-early protein BZLF1 (Z) mediates the switch between latent and lytic EBV infection. Z not only activates early lytic viral gene transcription but also plays a direct role in lytic viral genome replication. Although a small fraction of Z is known to be sumoylated, the effects of this posttranslational modification on various different Z functions have not been well defined. In this report, we show that only the lysine at amino acid residue 12 is required for the sumoylation of Z, and that Z can be sumoylated by SUMO isoforms 1, 2, and 3. We also demonstrate that the sumo-defective Z mutants ZK12A and ZK12R have enhanced transcriptional activity. The sumoylated and nonsumoylated forms of Z were found to have a similar cellular location, both being localized primarily within the nuclear matrix. The Z sumo-defective mutants were, however, partially defective for disrupting promyelocytic leukemia (PML) bodies compared to the ability of wild-type Z. In addition, we show that lytic viral genome replication does not require the sumoylation of Z, although a Z mutant altered at both amino acids 12 and 13 is replication defective. Furthermore, we show that the sumoylation of Z is greatly increased (from less than 1 to about 11%) in lytically induced 293 cells infected with an EBV mutant virus deleted for the EBV-encoded protein kinase (EBV-PK) compared to that of 293 cells infected with wild-type EBV, and that the overexpression of EBV-PK leads to the reduced sumoylation of Z in EBV-negative cells. Our results suggest that the sumoylation of Z helps to promote viral latency, and that EBV-PK inhibits Z sumoylation during viral reactivation.
Epstein-Barr virus (EBV) is a human herpesvirus that causes infectious mononucleosis and is associated with a variety of cancers, including endemic Burkitt's lymphoma and Hodgkin's lymphoma, as well as certain epithelial cell cancers such as nasopharyngeal carcinoma (64, 78). Like all herpesviruses, EBV can infect cells in either latent or lytic forms. The EBV infection of oropharyngeal epithelial cells results in the lytic form of viral replication, while the EBV infection of B cells usually results in the establishment of viral latency (64). In healthy humans, life-long latent EBV infection persists in memory B cells, and the virus is reactivated to the lytic form during plasma cell differentiation (47). The lytic form of viral infection results in the production of infectious viral particles and allows for the virus to be transmitted from cell to cell and host to host (64).
The switch between the latent and lytic forms of EBV infection is mediated by the two EBV immediate-early proteins, BZLF1 and BRLF1 (15, 17, 46, 70, 74). Both BZLF1 (Z) and BRLF1 (R) function as transcriptional activators and are important for transactivating each other's promoters as well as activating early lytic gene expression (3, 15, 16, 18, 26, 34, 37, 45, 51, 62, 65, 74). The BZLF1 protein (Z; also called ZEBRA, Zta, and EB1) is a bZIP protein that binds to and transcriptionally activates promoters containing BZLF1-responsive elements (ZREs) (13, 25, 28, 29, 50). Z preferentially binds to and activates the methylated forms of certain cellular and viral promoters, including the BRLF1 promoter, that have CpG-containing ZREs (8, 9, 21, 39, 44), thus promoting the efficient reactivation of lytic viral gene transcription even when the viral genome is methylated. In addition to activating lytic viral gene transcription, Z also binds directly to the EBV OriLyt origin of replication and is required for viral replication (36, 67, 68). Z function in viral replication can be separated from its role in transcriptional activation, since certain transcriptionally competent mutants are unable to mediate lytic viral genome replication (22, 55). Z directly interacts with several EBV replication proteins, and these interactions likely are important for lytic viral genome replication (49, 76).
Recently, a number of different lytic herpesvirus proteins, including the EBV Z and R proteins, the human cytomegalovirus (HCMV) immediate-early proteins IE1 and IE2, and the Kaposi's sarcoma-associated herpesvirus (KSHV) K-bZIP protein, have been shown to be covalently modified by small ubiquitin-like modifier (SUMO) proteins during viral infection (2, 4, 12, 40, 42, 57). SUMO is covalently attached to many cellular and viral proteins and functions to regulate a variety of cellular processes, including transcriptional gene regulation, protein localization, cell cycle regulation, and protein-protein interactions (30). The sumoylation of transcriptional regulatory proteins often leads to their functioning as repressors (30). For example, the sumoylation of the KSHV K-bZIP protein (a homologue of BZLF1) results in the repression of viral gene expression (42). Nevertheless, in some cases, sumoylation is associated with enhanced transcriptional activity. For example, the sumoylation of the human cytomegalovirus IE1 and IE2 proteins is required for their ability to activate the transcription of lytic viral early genes (4, 40, 48, 60). The effect of sumoylation on the function of the EBV R protein remains unclear, since one group reported that sumoylation enhances R transcriptional function (12) while another group reported that it decreases R function (10).
While we previously showed that the Z protein can be sumoylated on lysine residue 12 (2), the effect(s) of Z sumoylation on its transcriptional and replication functions remains unclear. Although a previous paper reported that the overexpression of SUMO-1 decreases the ability of Z to activate a subset of early lytic viral promoters in reporter gene assays (including the early lytic BMRF1 promoter), the same study found that SUMO-1 enhances the ability of Z to induce BMRF1 expression from the endogenous viral genome (1). Furthermore, it was not clear in this study whether the effect of SUMO-1 overexpression was mediated though enhanced Z sumoylation per se or the enhanced sumoylation of other viral or cellular proteins.
The effect of Z sumoylation on its replication function also is unknown. A Z mutant altered at both residues 12 and 13 previously was reported to be transcriptionally competent but defective for supporting the replication of an EBV oriLyt-containing plasmid (66), suggesting the possibility that Z sumoylation actually is required for its replication function. Nevertheless, the phenotype of a Z K12 mutant has not yet been reported in the context of the intact viral genome. Thus, the transcriptional and replication effects of Z sumoylation remain poorly defined.
Z also has been shown to be posttranslationally modified by phosphorylation in vivo at amino acid residues threonine 14, serine 167, serine 173, and serine 186, along with minor phosphorylation at amino acid residues 6, 7, and 8 (23). In addition, Z has been reported to interact with, and become phosphorylated at, amino acid residue 209 by the EBV BGLF4-encoded protein kinase (EBV-PK) (6), suggesting that EBV-PK plays a role in regulating Z function. Interestingly, the KSHV-encoded protein kinase (a homologue of EBV-PK) not only binds to and phosphorylates the KSHV K-bZIP protein (a homologue of Z) but also reduces K-bZIP sumoylation (43). However, whether EBV-PK likewise affects Z sumoylation has not been studied.
In this study, we have examined the role(s) of Z sumoylation with regard to its transcriptional and replication functions and determined if EBV-PK modulates Z sumoylation. We show that the sumoylation of lysine 12 inhibits Z transcriptional activity and is not required for Z-mediated viral replication in the context of the intact viral genome. We also show that Z can be sumoylated by SUMO-2 and SUMO-3 as well as SUMO-1, and that the subnuclear localization of Z (which is largely associated with the nuclear matrix) is not significantly altered by sumoylation. Finally, we demonstrate that the presence of EBV-PK decreases the extent of Z sumoylation via a mechanism that does not require Z serine residue 209, the site reported to be phosphorylated by EBV-PK. These results suggest that Z sumoylation promotes the establishment of viral latency, and that EBV-PK helps to reverse Z sumoylation during viral reactivation.
MATERIALS AND METHODS
Cell lines.
293 and HeLa cells were maintained in Dulbecco's modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin. EBV-infected 293 cells (a gift from H.-J. Delecluse) have been described previously (20, 26) and were maintained in DMEM supplemented with hygromycin (100 μg/ml), 10% FBS, and penicillin-streptomycin. Raji cells (ATCC) were maintained in RPMI 1640 supplemented with 10% FBS and penicillin-streptomycin. Hone-Akata cells (an EBV-positive nasopharyngeal carcinoma line; a gift from Lawrence Young) were maintained in RPMI 1640 supplemented with G418 (400 μg/ml), 10% FBS, and penicillin-streptomycin. All cells were grown at 37°C in a humidified 5% CO2 incubator.
Plasmids.
Plasmid DNA was purified on maxi-prep columns according to the manufacturer's protocol (Qiagen). pSG5 and pCDNA3.1 were obtained from Stratagene and Invitrogen, respectively. pSG5-Z, containing the genomic Z sequence transcribed from a simian virus 40 promoter, was a kind gift from Diane Hayward (66). Site-directed mutagenesis was performed according to the manufacturer's protocol (Stratagene) using pSG5-Z as the template DNA to generate the following constructs: pSG5-ZK12R (ZK12R forward, 5′-TCTGAAGATGTACGATTTACACCTGAC-3′; reverse, 5′-GTCAGGTGTAAATCGTACATCTTCAGA-3′), pSG5-ZK12A (ZK12A forward, 5′-TCTGAAGATGTAGCATTTACACCTGAC-3′; reverse, 5′-GTCAGGTGTAAATGCTACATCTTCAGA-3′), pSG5-ZT14A (ZT14A forward, 5′-GTAAAATTTGCACCTGACCCA-3′; reverse, 5′-TGGGTCAGGTGCAAATTTTAC-3′), pSG5-ZK161R (K161R forward, 5′-CTGGCTGTTGTGGTCTCCGTGTGCGTCG-3′; reverse, 5′-CGACGCACACGGAGACCACAACAGCCAG-3′), pSG5-ZK178R (K178R forward, 5′-CACCCGATTCTTGTATCGCCTTATTTCTAGTTCAGAATC-3′; reverse, 5′-GATTCTGAACTAGAAATAAGGCGATACAAGAATCGGGTG-3′), pSG5-ZK181R (K181R forward, 5′-GGAAGCCACCCGATTCCTGTATCGCTTTATTTC-3′; reverse, 5′-GAAATAAAGCGATACAGGAATCGGGTGGCTTCC-3′), pSG5-ZK188R (K188R forward, 5′-CTTGGCCCGGCATCTTCTGGAAGCCAC-3′; reverse, 5′-GTGGCTTCCAGAAGATGCCGGGCCAAG-3′), pSG5-ZK192R (K192R forward, 5′-CAGCAGTTGCTTAAACCTGGCCCGGCATTTTCTG-3′; reverse, 5′-CAGAAAATGCCGGGCCAGGTTTAAGCAACTGCTG-3′), pSG5-ZK194R (K194R forward, 5′-CTGCAGCAGTTGCCTAAACTTGGCCCGGC-3′; reverse, 5′-GCCGGGCCAAGTTTAGGCAACTGCTGCAG-3′), pSG5-ZK207R (K207R forward, 5′-GTCATTTTCAGATGATCTGGCAGCAGCCACCTG-3′; reverse, 5′-CAGGTGGCTGCTGCCAGATCATCTGAAAATGAC-3′), pSG5-ZS209A (ZS209A forward, 5′-CAGCCTGTCATTTTCAGCTGATTT-3′; reverse, 5′-GCTGCTGCCAAATCAGCTGAAAAT-3′), and pSG5-ZK219R (K219R forward, 5′-GGGCACATCTGCCTCAACAGGAGGCG-3′; reverse, 5′-CGCCTCCTGTTGAGGCAGATGTGCCC-3′). The pcDL-SRα296-Zm12/13 vector expresses a BZLF1 mutant altered at amino acids 12 and 13 (converting KF to AA) as previously described (66) and was a gift from Diane Hayward. pGal4-Z-WT(1-167), pGal4-ZK12A(1-167), and pGal4-ZK12R(1-167) were generated by amplifying amino acids 1 to 167 from pSG5-Z, pSG5-ZK12A, and pSG5-ZK12R, respectively, with oligonucleotides Z1-167 BamHI-5′ (5′-GCGGATCCGCATGATGGACCCAAACTCG-3′) and Z1-167 SacI-3′ (5′-GCGAGCTCCAGCGATTCTGGCTGTTG-3′), digesting the PCR fragments with BamHI and SacI, and ligating them into pSG424 (a kind gift from M. Green) containing the Gal4 DNA binding domain digested with BamHI and SacI. pGal4-E1B-CAT (a kind gift from M. Green) contains five copies of the Gal4 binding motif upstream of the E1B minimal TATA promoter and CAT reporter gene. pcDNA-BGLF4-FLAG has been described previously and was a gift from M. Marschall (31, 53). pHA-SUMO-2 and pHA-SUMO-3 were a gift from Shigeki Miyamoto. pSENP1 and pmyc-SUMO-1 were provided by Grace Gill. The pGS284 shuttle vector (56) and the EBV wild-type bacmid p2089 (20) were described previously and were gifts from W. Hammerschmidt. pRK-BALF4 codes for the EBV glycoprotein 110 and was a gift from H.-J. Delecluse (59). pCINeo-PMLIV expresses the human promyelocytic leukemia (PML) protein (7) and was a gift from Keith Leppard.
EBV-PK viral mutant.
A stop codon was introduced into the EBV-PK open reading frame (BGLF4) of the wild-type EBV bacmid p2089 in Escherichia coli by allelic exchange as previously described (69, 73). Wild-type EBV sequence 122904 to 124359 flanking the PK start site was PCR amplified (BGLF4-122904, 5′GCGGATCCCTTTAGCCGCACATCCAGCATCTT-3′; BGLF4-124359, 5′-GCTCTAGATACCCACTGCGGTTTATACACCAT-3′), digested with BamHI and XbaI, and cloned into pSP65 digested with BamHI and XbaI to make pSP65-PKreg. Site-directed mutagenesis was performed on pSP65-PKreg to mutate the ATG translation start codon to a TGA translation stop codon (BGLF4STOP-A-S, 5′-CTCGAGCCATTTGAGGAACTGAGATGTGAATATGGCTGCGGAG-3′; BGLF4STOP-A-AS, 5′-CTCCGCAGCCATATTCACATCTCAGTTCCTCAAATGGCTCGAG-3′) according to the manufacturer's protocol (Stratagene). A second translation stop codon mutation also was introduced, changing the fifth amino acid residue from a methionine to a stop codon, using site-directed mutagenesis (BGLF4STOP-B-S, 5′-GAGGAACTGAGATGTGAATTGAGCTGCGGAGTTGAGCCCGAC-3′; BGLF4STOP-B-AS, 5′-GTCGGGCTCAACTCCGCAGCTCAATTCACATCTCAGTTCCTC-3′). The mutated PK open reading frame was cut out of pSP65 using BamHI and XbaI restriction enzymes and ligated into the shuttle vector pGS284 digested with BglII and NheI to yield pGS284-PKStop. pGS284-PKStop in S17λpir E. coli was conjugated with the wild-type EBV bacmid p2089 in GS500 E. coli. Cointegrates were selected in LB containing carbenicillin and chloramphenicol. Cultures then were recovered in LB containing chloramphenicol only and plated on LB agar plates containing 5% sucrose and chloramphenicol. Colonies were screened by PCR using oligonucleotides to detect mutated DNA sequence (BGLF4STOP-DET, 5′-GAGCCATTTGAGGAACTGA-3′; BGLF4-122904, 5′-GCGGATCCCTTTAGCCGCACATCCAGCATCTT-3′). Colonies containing the correct mutations were further screened by the DNA sequencing of the PK region and restriction enzyme analysis comparing the PKStop bacmid with wild-type bacmid DNA by independent digestions with BamHI-, HindIII-, SalI-, and EcoRI-independent digests.
Generation of EBV latent cell lines.
Wild-type and PKStop bacmid DNAs were transfected in parallel into 293 cells using Lipofectamine 2000 reagent (Invitrogen) to generate wild-type EBV (EBV-WT) and EBV-PKmut cell lines. Latently infected stable cell clones were isolated individually, selected in hygromycin (100 μg/ml), and screened for the ability to produce infectious virus following transfection with BZLF1 and PK expression vectors.
DNA transfection.
DNA was transfected into 293 cells using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's protocol. HeLa cells were transfected with DNA using Fugene 6 reagent (Roche) according to the manufacturer's protocol.
Immunoblotting.
Immunoblotting was performed as previously described (2, 8). Cell lysates were harvested in SUMO buffer containing protease inhibitor cocktail (Roche) and quantified by the SUMO protein assay (Bio-Rad). Equivalent amounts of protein were separated in sodium dodecyl sulfate, 10% polyacrylamide gels and transferred to membranes. Membranes were blocked in phosphate-buffered saline (PBS) containing 5% milk and 0.1% Tween 20 solution and incubated with primary antibody. Dilutions of primary antibodies were as follows: 1:250 anti-EAD (BMRF1; Vector), 1:250 anti-BRLF1 (Argene), 1:250 anti-ZEBRA (BZ-1; sc-53904; Santa Cruz), 1:500 anti-c-myc (Santa Cruz), 1:500 anti-hemagglutinin (HA) (Santa Cruz), 1:5,000 anti-β-actin (Sigma), 1:2,000 anti-tubulin (Sigma), 1:200 anti-lamin B (Santa Cruz), 1:2,000 anti-FLAG (Sigma), and 1:1,500 anti-histone H3 (35), a gift from Jeff Ross. Following primary antibody incubation, membranes were washed in PBS containing 0.1% Tween 20 (PBST) and incubated in the appropriate horseradish peroxidase secondary antibody (Thermo Scientific) at a 1:10,000 dilution. Membranes then were washed with PBST and visualized by ECL treatment (Pierce) and exposure to film. The quantitation of immunoblots was performed using Adobe Photoshop. The mean and pixel counts for each band were multiplied and background signal subtracted from the measurements to yield the absolute intensity. The absolute intensity then was used to calculate the percentage of sumoylated Z for Fig. 9.
FIG. 9.
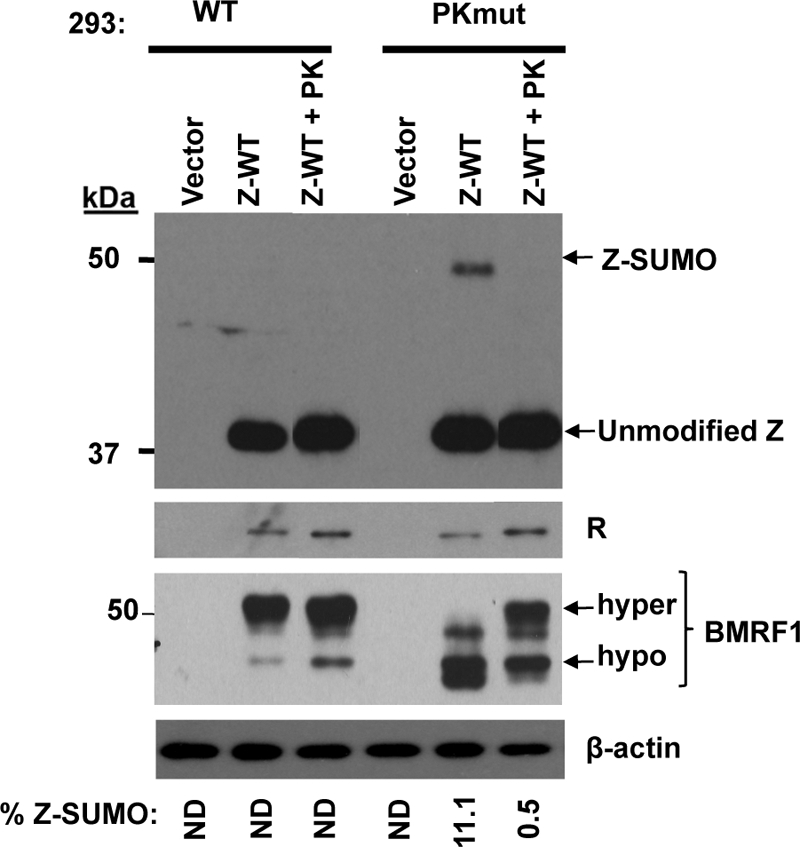
Z is more sumoylated in 293 cells infected with a PK-deleted EBV mutant (PKmut). 293 cells stably infected with wild-type or PKmut (293-PKmut) virus were transfected with a wild-type Z expression plasmid with or without an EBV-PK expression plasmid. Immunoblot analysis was performed to determine the amounts of sumo-modified Z, BMRF1, and R. The hypo- and hyperphosphorylated forms of BMRF1 are indicated. As expected, the PKmut cells did not express the hyperphosphorylated form of BMRF1 unless transfected with an EBV-PK expression plasmid (14, 32). β-Actin was used as a loading control. The percentage of Z protein sumo-modified in each condition is indicated; ND indicates that sumo-modified Z was not detectable.
CAT assays.
Chloramphenicol acetyltransferase (CAT) assays were performed as previously described (33). HeLa cells were transfected with the indicated expression vectors and harvested in reporter lysis buffer (Promega) 48 h following transfection. Lysates were incubated with acetyl-coenzyme A and [14C]chloramphenicol, and acetyltransferase activity was determined following thin-layer chromatography. Activity was quantified on a Storm 840 phosphorimager (Molecular Dynamics).
Virus titration assay.
Virus titration assays were performed as previously described (41). Supernatant from 293 EBV-infected cells was harvested 72 h posttransfection with BZLF1 and BALF4 expression vectors and filtered through a 0.8-μm-pore-size filter. Raji cells (2 × 105 cells/infection) were infected with various amounts of virus and incubated at 37°C. Phorbol-12-myristate-13-acetate (TPA; 20 ng/ml) and sodium butyrate (3 mM final concentration) were added 24 h after infection. Green fluorescent protein-positive Raji cells were counted 48 h postinfection to determine viral titers.
Subcellular fractionation assay.
Cellular fractionation was performed as previously described (61, 63). HeLa cells were transfected with the appropriate expression vectors and harvested 48 h posttransfection. Cells were washed with PBS, scraped off the dish, and lysed in CSKT buffer [10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), pH 6.8; 100 mM NaCl; 300 mM sucrose; 3 mM MgCl2; 1 mM EGTA; 1 mM dithiothreitol; 0.5% Triton X-100; and protease inhibitor cocktail; from Roche Molecular Diagnostics]. After a 5-min incubation, the lysed cells were centrifuged for 3 min at 7,500 rpm, and the supernatant was saved as the soluble fraction. The pellet was washed with CSK buffer (CSKT without Triton X-100) and resuspended in CSK buffer containing DNase I. The sample then was incubated for 2 h at 37°C, and CSK buffer containing (NH4)2SO4 was added to a final concentration of 0.25 M. Following a 5-min incubation at 4°C, the sample was centrifuged at 7,500 rpm for 3 min, and the supernatant was saved as the chromatin fraction. The remaining pellet then was suspended in CSK buffer containing 2 M NaCl and centrifuged for 3 min at 7,500 rpm. The final pellet was resuspended in buffer containing 8 M urea and 10 mM Tris-HCl, pH 8.0, and saved as the nuclear matrix fraction.
Immunofluorescence assay.
Immunofluorescence assays were performed as previously described (27). HeLa cells were grown on glass coverslips and fixed in 100% ice-cold methanol. Cells were incubated with BZLF1-specific mouse monoclonal antibody (BZ.1; Santa Cruz), HA-rabbit polyclonal antibody (Santa Cruz), or PML rabbit polyclonal antibody (Santa Cruz) at a 1:25 dilution and then incubated with a 1:50 dilution of Texas Red-conjugated goat anti-mouse antibody and fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch, West Grove, PA). Coverslips were mounted with Vectashield Hard Set and visualized using a Nikon A1R confocal microscope.
RESULTS
Z sumoylation occurs primarily on lysine 12.
We previously reported that the EBV Z protein is posttranslationally modified by SUMO-1, and that a Z mutant altering amino acid residues 12 and 13 is not sumoylated (2). However, since Z sumoylation results in the formation of multiple larger forms of Z as detected by immunoblot analysis (2), it is possible that one or more of these larger forms are due to Z sumoylation occurring at more than one lysine residue. To examine the potential contribution of other Z lysine residues, we generated a series of Z mutants in which every lysine in Z was individually mutated to an arginine (R). In the case of ZK12, both ZK12R and ZK12A mutants were constructed. As shown in Fig. 1A, only the Z lysine 12 mutants, ZK12R and ZK12A, were not posttranslationally modified. Furthermore, none of the larger forms of sumoylated Z were significantly reduced by the mutation of lysine residues other than K12. These data indicate that only Z lysine residue 12 substantially contributes to Z sumoylation.
FIG. 1.

Z is sumoylated at lysine 12. (A) HeLa cells were transfected with an empty vector control, wild-type Z, and wild-type Z and expression plasmid SENP1, ZK12R, ZK12A, ZK161R, ZK178R, ZK181R, ZK188R, ZK192R, ZK194R, ZK207R, or ZK219R as indicated. Cell lysates were prepared in SUMO buffer 48 h posttransfection, and immunoblot analysis was performed using an antibody against Z. The sumo-modified forms of Z are indicated by brackets. (B) HeLa cells were transfected with an empty vector control, wild-type Z, ZK12A, or ZT14A expression plasmid. Sumoylated forms of Z were assessed as described for panel A. β-Actin was used as a loading control.
To further confirm that the higher-molecular-weight isoforms of Z represent various different sumoylated forms, the Z protein was transfected into HeLa cells in the presence or absence of a sentrin protease 1 (SENP1) expression vector (Fig. 1A). In the presence of cotransfected SENP1 (which removes SUMO moieties from proteins) (72), the higher-migrating forms of Z were no longer detected. These results suggest that both of the larger modified forms of Z are conjugated to SUMO and that SENP1 can cleave the SUMO off Z.
Threonine 14 is not required for Z sumoylation.
Sumoylation has been reported to occur at several consensus motifs, including a phosphorylation-dependent SUMO motif (PDSM) (5). Although Z does not contain the consensus PDSM sequence (ΨKxExxSP), the threonine located at residue 14 has been shown to be phosphorylated in vivo (23) and could regulate the sumoylation of ZK12. To test this hypothesis, HeLa cells were transfected with expression plasmids encoding either wild-type Z (Z-WT) or the mutant ZT14A. As shown in Fig. 1B, the ZT14A mutant was sumoylated similarly to wild-type Z. These data indicate that phosphorylation at ZT14 is not required for the sumoylation of Z.
Z is sumoylated by SUMO-1, SUMO-2, and SUMO-3.
Sumoylation can be mediated by either SUMO-1 or the closely related isoforms (SUMO-2/3); some proteins are modified only by certain SUMO isoforms (38). Although our previous studies demonstrated that Z can be sumoylated by SUMO-1, it was unknown whether Z also can be modified by SUMO-2/3. To examine this, HeLa cells were transfected with Z, ZK12A, and ZK12R expression plasmids in the presence or absence of myc-tagged SUMO-1 (Fig. 2A), HA-tagged SUMO-2 (Fig. 2B), and HA-tagged SUMO-3 (Fig. 2C). As expected, the conjugation of the myc-tagged SUMO-1 to wild-type Z, but not ZK12A or ZK12R, was detected. SUMO-1 conjugation to the cellular RanGAP1 protein, which migrates at approximately 75 kDa (52, 54), also was detected in SUMO-1-transfected cells. The HA-tagged forms of SUMO-2 and SUMO-3 also complexed with wild-type Z but not the ZK12A and ZK12R mutants. Taken together, these data indicate that Z can be modified by any of the SUMO types at lysine 12.
FIG. 2.
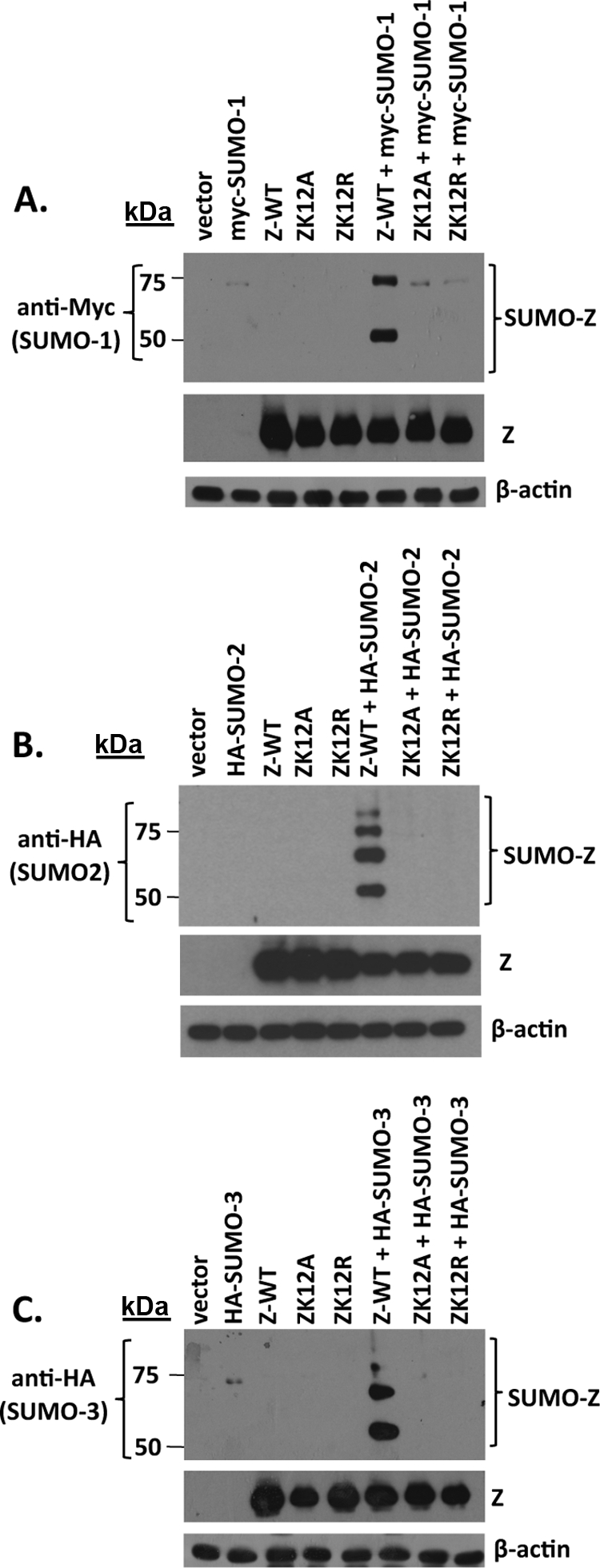
Z is sumoylated by SUMO-1, SUMO-2, and SUMO-3. HeLa cells were cotransfected with expression plasmids encoding wild-type Z, ZK12A, or ZK12R in the presence or absence of myc-SUMO-1 (A), HA-SUMO-2 (B), or HA-SUMO-3 (C), and cell lysates were prepared 48 h posttransfection for immunoblot analysis. Myc or HA antibodies were used to detect myc-SUMO-1, HA-SUMO-2, or HA-SUMO-3 modifications. Z antibody was used to examine Z expression. β-Actin was used as a loading control.
Sumoylation does not alter Z localization.
Sumoylation has been demonstrated to alter the localization of some proteins. Previous studies showed that the Zm12/13 mutant localizes to the nucleus (2), but the subnuclear localization of a sumo-defective Z mutant has not been closely examined. To determine if sumoylation affects the subnuclear localization of Z, the locations of wild-type Z and the ZK12 mutants were examined using biochemical fractionation. HeLa cells were transfected with expression plasmids encoding wild-type Z, ZK12A, and ZK12R. Cell lysates subsequently were fractionated to obtain soluble (both cytoplasmic and nuclear), chromatin-associated, and nuclear matrix fractions. As shown in Fig. 3, both wild-type Z and the Z sumoylation mutants ZK12A and ZK12R were located predominantly in the nuclear matrix fraction. These results suggest that Z sumoylation does not significantly alter Z localization.
FIG. 3.
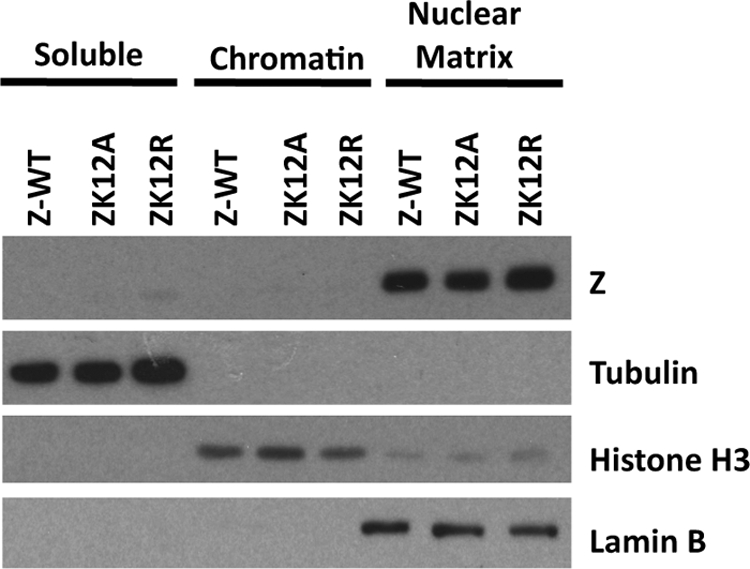
Sumoylation does not alter Z subcellular localization. HeLa cells were transfected with expression plasmids encoding wild-type Z, ZK12A, and ZK12R. Forty-eight hours posttransfection, cells were fractionated as described in Materials and Methods to yield soluble, chromatin, and nuclear matrix fractions. Immunoblot analysis was performed to detect Z fractionation using antibody against Z. Antibodies against tubulin, histone H3, and lamin B were used as controls for proper fractionation to detect the soluble, chromatin, and nuclear matrix fractions, respectively.
To further examine the potential effects of Z sumoylation on its subnuclear localization, we cotransfected HeLa cells with expression plasmids encoding wild-type Z or the ZK12A mutant in the presence or absence of tagged SUMO-1, SUMO-2, or SUMO-3 expression vector and performed immunofluorescence assays 48 h posttransfection. In each case, the wild-type Z and ZK12A proteins were dispersed throughout the nucleus (excluding nucleoli) in the presence or absence of cotransfected SUMO proteins (Fig. 4A and data not shown). Interestingly, wild-type Z decreased the localization of all three SUMO isoforms within punctate dots in the nucleus, but this effect was partially attenuated with the ZK12A mutant.
FIG. 4.

Z sumo-defective mutants are partially inhibited for PML body disruption. (A) Immunofluorescence analysis was performed on HeLa cells transfected with expression plasmids encoding wild-type Z, ZK12A, and HA-SUMO-2. Slides were costained with primary antibodies against Z (shown in red) and HA tags (shown in green). Images were visualized by confocal microscopy. (B) HeLa cells were transfected with expression vectors encoding wild-type Z, ZK12A, or Zm12/13 (500 ng) in the presence or absence of a cotransfected PML expression vector (15 ng). Slides were costained with primary antibodies against PML (shown in green) and Z (shown in red) and visualized by confocal microscopy. Arrows indicate Z-expressing cells.
Sumo-defective Z mutants are partially defective in dispersing PML bodies.
Since SUMO proteins are known to be localized within PML bodies and Z can disperse PML bodies (2), we also analyzed the ability of wild-type and mutant Z proteins to disperse PML bodies. The sumoylation of the PML protein is required for the formation of PML bodies (58, 77), and wild-type Z (but not the sumo-defective Zm12/13 mutant) was shown previously to compete with cotransfected PML for sumoylation (2). HeLa cells were cotransfected with wild-type Z, ZK12A, or Zm12/13 expression plasmid with or without a small amount of a PML expression plasmid, and immunofluorescence assays were performed 48 h following transfection (Fig. 4B). Although the ability of the wild-type and mutant Z proteins to disperse endogenous PML bodies was similar, the ZK12A and Zm12/13 mutants were impaired in the ability to disperse PML bodies when the PML protein was cotransfected with the Z vectors. Similar results were obtained using ZK12R (data not shown). These results suggest that when the amount of Z protein is limiting (relative to the level of PML), the Z sumoylation mutants are partially defective for dispersing PML bodies.
Sumoylation decreases Z transcriptional function.
Although the sumoylation of transcription factors often inhibits their ability to activate transcription, sumoylation has the opposite effect in some cases. To determine if Z sumoylation affects Z transcriptional function, reporter gene assays were performed using expression vectors in which the amino acid residues 1 to 167 of the Z, ZK12A, or ZK12R transcriptional domain was fused to the Gal4 DNA binding domain. HeLa cells were cotransfected with the Gal4 fusion constructs and an expression plasmid containing the Gal4 DNA binding site and a minimal E1B promoter driving the expression of the CAT reporter gene (Fig. 5). The ZK12A and ZK12R transcriptional activation domains functioned in this assay as much stronger transcriptional activators than did the wild-type Z transcriptional activation domain (Fig. 5A). Immunoblot analysis demonstrated that the wild-type and mutant Z proteins accumulated to similar levels in the cells (Fig. 5B). Since the Z-gal4 fusion proteins contain only the transcriptional activation domain of Z (and not the Z DNA binding domain), these data suggest that sumoylation decreases Z function as a transcriptional activator.
FIG. 5.

Sumoylation decreases Z transcriptional activity. (A) HeLa cells were transfected with Gal reporter constructs expressing gal fused to Z-WT(1-167), ZK12A(1-167), or ZK12R(1-167) and an expression construct encoding a CAT reporter gene downstream of the Gal4 DNA binding domain and E1b promoter. CAT assays were performed on the lysates 48 h after transfection and quantified by phosphorimager analysis. Results were normalized to vector control activity (Gal4 alone), which was set as 1. (B) Cell lysates were analyzed by immunoblot analysis using a Z antibody to demonstrate equal expression levels of Z in the experiment shown in panel A. β-Actin was used as a loading control.
Sumoylation decreases Z-mediated viral reactivation.
In a previous study, the effect of SUMO-1 overexpression on Z function was found to be promoter dependent in reporter gene assays, and the results of the promoter reporter gene assays did not correlate well with the ability of Z to activate these same promoters in the context of the intact viral genome (1). To examine more specifically how the sumoylation of ZK12 affects its ability to activate lytic viral gene expression when SUMO proteins are expressed at normal levels, 293 cells latently infected with an EBV mutant lacking the Z open reading frame (293-ZKO) were cotransfected with limiting amounts of Z, ZK12A, or ZK12R expression plasmid. Immunoblot analysis was performed 48 h posttransfection to measure the amounts of Z and the activated lytic viral proteins, BMRF1 and R. As shown in Fig. 6A, wild-type Z induced less BMRF1 and R expression than did the Z sumoylation mutants ZK12A and ZK12R. Similar results were obtained in the EBV-positive, latently infected Hone-Akata nasopharyngeal carcinoma cell line (Fig. 6B). Of note, when larger amounts of each Z construct were transfected into 293-ZKO cells or Hone-Akata cells, we found that the wild-type and mutant Z proteins had similar effects on BMRF1 and R expression (data not shown), likely due to limiting amounts of SUMO proteins in the cells. Taken together, these results suggest that the sumoylation of Z decreases its ability to induce lytic viral gene expression in the context of the intact viral genome when SUMO proteins are expressed at endogenous levels and Z protein is limiting.
FIG. 6.
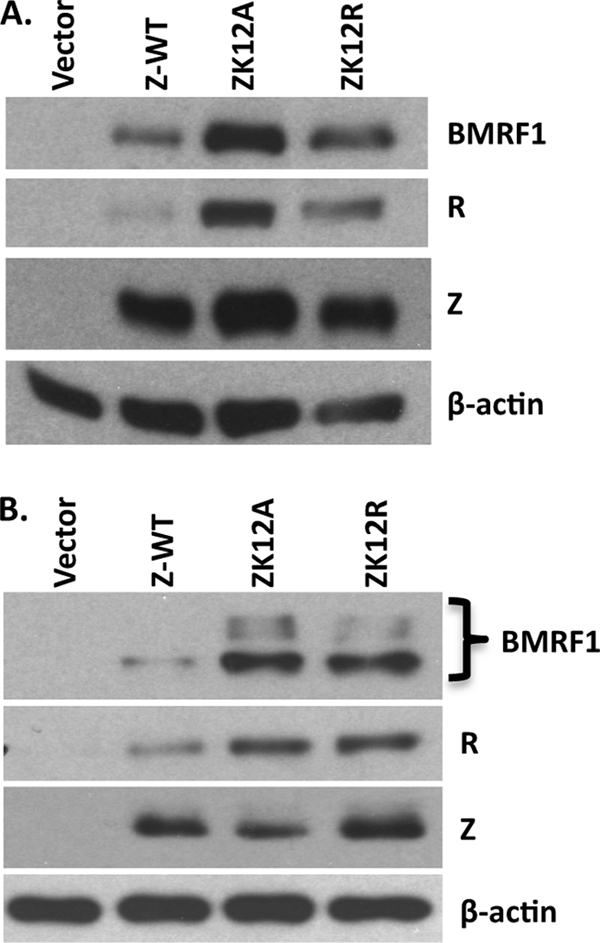
Sumoylation decreases Z-induced lytic gene expression. 293-ZKO cells (stably infected with an EBV mutant lacking the Z open reading frame) (A) or latently infected Hone-Akata cells (B) were transfected with expression plasmids encoding wild-type Z, ZK12A, ZK12R, or a vector control and examined by immunoblot analysis to quantitate BMRF1, R, and Z protein levels. β-Actin was used as a loading control.
Z sumoylation is not required for viral replication.
A Z mutant altered at both residues 12 and 13 (Zm12/13) previously was reported to be transcriptionally competent but unable to support plasmid-based lytic replication assays (66). To determine if the alteration of the K12 residue leads to a replication-defective phenotype in the context of the intact viral genome, 293-ZKO cells were cotransfected in parallel with wild-type Z, ZK12A, and ZK12R expression plasmids. Three days posttransfection, virus production was examined using the green Raji cell assay as previously described (41). As shown in Fig. 7A and B, the ZK12A mutant had a modest increase in virus production compared to that of wild-type Z, while replication mediated by the ZK12R mutant was similar to that of wild-type virus. Similar results were obtained in two separate experiments. In contrast, the Zm12/13 mutant, which previously was reported to be replication incompetent using a plasmid-based lytic replication assay, also was highly impaired for the ability to mediate virus production in 293-ZKO cells, although its ability to transcriptionally activate the early lytic viral protein BMRF1 was similar to that of wild-type Z (Fig. 7C and D).
FIG. 7.

ZK12 sumoylation is not required for lytic viral replication. (A) 293-ZKO cells were cotransfected in parallel with empty vector, wild-type Z, or ZK12A or ZK12R expression plasmid. Three days posttransfection, supernatants were harvested and the green Raji cell titration assay was performed to quantitate viral titers. The amount of infectious virus produced by each condition is shown relative to the amount produced by the wild-type Z (set as 100%). (B) The level of Z expression in the transfected 293-ZKO cells shown in panel A was examined by immunoblot analysis. (C) 293-ZKO cells were cotransfected with vector control, the combination of the wild-type Z plus R expression vectors, or the Zm12/13 mutant and R expression plasmids. The amount of infectious virus in each condition (relative to wild-type Z, set as 100%) was quantitated as described for panel A. (D) BMRF1 and Z expression levels were examined by immunoblot analysis. β-Actin was used as a loading control.
These results, while confirming that Zm12/13 is replication defective, show that the sumoylation of Z at lysine 12 is not required for EBV lytic replication. Instead, the Z phenylalanine residue 13 may be important for EBV replication. The relatively minor effect of Z sumoylation on lytic viral replication compared to that of lytic viral gene expression may reflect the partially impaired ability of the ZK12A and ZK12R mutants to disperse PML bodies, as PML dispersion is thought to contribute to the lytic viral replication of other herpesviruses (24).
EBV-PK reduces Z sumoylation.
The EBV-encoded protein kinase (EBV-PK) recently was reported to directly interact with and phosphorylate Z at residue 209 and to inhibit the ability of Z to activate its own transcription (6). Since the phosphorylation of proteins often affects their ability to be sumoylated and the KSHV homologue of EBV-PK has been shown to reduce the sumoylation of the KSHV homologue of BZLF1 (K-bZip) (43), we examined the effect of overexpressed EBV-PK on Z sumoylation. HeLa cells were transfected with wild-type Z or ZK12A in the presence of SUMO-1 (Fig. 8A), SUMO-2 (Fig. 8B), or SUMO-3 (Fig. 8C) with or without an EBV-PK expression plasmid. As shown in Fig. 8A to C, EBV-PK greatly reduced the amount of Z sumoylation mediated by any of the three different SUMO isoforms. In contrast, EBV-PK did not affect the expression of the SUMO proteins or SUMO-1 modification of the cellular RanGAP1 protein (Fig. 8D). These results suggest that EBV-PK disrupts the sumoylation of Z by SUMO-1, SUMO-2, and SUMO-3.
FIG. 8.

EBV-PK inhibits Z sumoylation. HeLa cells were transfected with various combinations of wild-type Z, ZK12A, EBV-PK, and myc-SUMO-1 (A), HA-SUMO-2 (B), or HA-SUMO-3 (C) expression plasmid, and cell lysates were prepared 48 h posttransfection for immunoblot analysis. Antibody against myc was used to detect myc-SUMO-1 protein, antibody against HA was used to detect the HA-tagged SUMO-2 and SUMO-3 proteins, and antibody against Z was used to examine Z expression. (D) HeLa cells were transfected with EBV-PK and myc-SUMO-1 expression plasmids, and immunoblot analysis was performed on the cell lysates to examine both free SUMO-1 and SUMO-1-modified RanGAP1 levels using an anti-myc antibody. β-Actin was used as a loading control for all immunoblots.
To determine if EBV-PK expression regulates Z sumoylation in the context of the intact viral genome, we compared the amount of Z sumoylation in 293 cells containing wild-type EBV to that of 293 cells stably infected with an EBV-PK mutant (containing inserted stop codons in the first and fifth residues of the EBV-PK open reading frame). The complete phenotype of the EBV-PK mutant will be described in another manuscript (unpublished data). 293 wild-type or EBV-PKmut cells were transfected with a Z expression plasmid in the presence or absence of a cotransfected EBV-PK expression vector, and the level of Z sumoylation was examined by immunoblot analysis (Fig. 9). Lytically induced 293 cells containing the EBV-PKmut virus had a higher level of the sumo-modified form of Z (11% of the total Z protein sumo-modified) compared to that of the 293 cells with wild-type EBV, in which the sumo-modified form of Z was difficult to detect. Furthermore, cotransfection with Z and EBV-PK expression plasmids abolished Z sumoylation in the EBV-PKmut cells. Interestingly, we also found that EBV-PK modestly increased the ability of wild-type Z to activate the expression of the R protein from the latent viral genome. These results demonstrate that EBV-PK reduces Z sumoylation in the context of the virus as well as in EBV-negative cells.
Z sumoylation is not regulated by phosphorylation of Z residue S209.
Since EBV-PK has been reported to phosphorylate ZS209 (6), we next determined if the ZS209 residue modulates Z sumoylation. 293 PKmut cells were transfected with expression plasmids encoding wild-type Z, the ZK12A mutant, or a ZS209A mutant in the presence or absence of an EBV-PK expression plasmid, and the amount of Z sumoylation was examined by immunoblotting. As shown in Fig. 10, ZS209A was sumoylated similarly to wild-type Z in the absence of cotransfected EBV-PK, and the presence of EBV-PK decreased the level of Z sumoylation to a similar extent in the wild-type and mutant Z constructs. These results suggest that the effect of EBV-PK on Z sumoylation is not directly mediated by phosphorylation at residue S209.
FIG. 10.
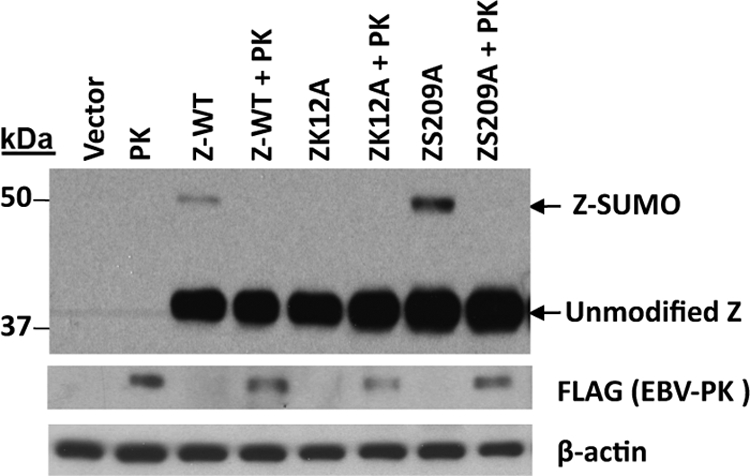
ZS209 phosphorylation is not required for the EBV-PK inhibition of Z sumoylation. 293-EBV-PKmut cells were transfected with wild-type Z, ZK12A, or ZS209A expression plasmid with or without an EBV-PK expression plasmid. Immunoblot analysis was performed to determine Z and EBV-PK protein levels. Antibody against FLAG was used to detect FLAG-tagged EBV-PK. β-Actin was used as a loading control.
DISCUSSION
The EBV immediate-early Z protein plays an essential role in initiating the switch between latent and lytic EBV infection. Z serves as a transcriptional activator of early lytic viral genes and is required for OriLyt-mediated virus replication. In addition, Z has multiple effects on the host cell environment that are predicted to be advantageous for lytic viral replication, including inhibiting p53 function (75), regulating cell cycle progression (11), and dispersing PML nuclear bodies (2). Although it has been shown previously that Z is sumoylated at lysine residue 12 (2), the effect of this sumoylation on Z transcriptional and replication functions has not been well studied. In this report, we show that Z sumoylation inhibits its function as a transcriptional activator (Fig. 5) and is not required for lytic viral replication (Fig. 7). Furthermore, we show that the EBV-encoded protein kinase (EBV-PK) reduces Z sumoylation (Fig. 8) and that this effect does not involve the EBV-PK-mediated phosphorylation of ZS209 (Fig. 10). Taken together, our results suggest that Z sumoylation helps to promote viral latency, and that one function of EBV-PK during viral reactivation is to prevent or reverse Z sumoylation.
Sumoylation has been shown to affect the function of various transcription factors in both positive and negative ways, but most commonly sumoylation represses transcriptional function. In this study, we found that the transcriptional activation domain of wild-type Z (fused to the Gal4 DNA binding domain) has less transcriptional activity than the Z sumoylation mutants ZK12A and ZK12R (Fig. 5). In addition, we found that the ability of limiting amounts of Z to induce lytic reactivation in EBV-positive cells was enhanced by the mutation of the ZK12 residue (Fig. 6). These data all are consistent with the interpretation that the sumoylation of the K12 residue acts to inhibit Z function as a transcriptional activator.
Previous studies examining Z sumoylation were conducted using a Z mutant in which both amino acids 12 and 13 were mutated (Zm12/13). Since this double mutant was reported to be replication incompetent in a transient plasmid-based replication assay (66), it remained unclear whether the sumoylation of ZK12 is required for lytic viral replication in the context of the intact viral genome. Our results here clearly show that ZK12 sumoylation is not required for viral replication (Fig. 7). In fact, we found that the sumoylation-deficient ZK12A mutant is modestly increased (compared to wild-type Z) for the ability to induce virus production, which is consistent with its enhanced transcriptional activity (Fig. 5). Additionally, we have confirmed that the Zm12/13 mutant cannot produce infectious viral particles (Fig. 7). These results indicate that Z sumoylation at lysine 12 is not required for viral replication, and that amino acid residue 13 plays an important role in Z-induced replication but not in transcriptional activation.
We previously showed that wild-type Z disperses PML bodies (2). Here, we demonstrate that the Z sumoylation mutants ZK12A, ZK12R, and Zm12/13 are partially defective for the ability to disperse PML bodies when the amount of Z protein is limiting compared to the amount of PML protein (Fig. 4). PML bodies function as an intrinsic antiviral response to infection, and the disruption of PML bodies is thought to be important for herpesvirus replication (24). The relatively modest increase in viral production mediated by the Z sumo-defective K12A and K12R mutants, given the ability of these mutants to increase Z transcriptional function, may reflect their partially impaired ability to disperse PML bodies.
The three members of the SUMO family, SUMO-1, SUMO-2, and SUMO-3, all are able to covalently modify lysine residues on a variety of proteins. The results in this paper indicate that Z can be sumoylated by all three SUMO members, and that lysine 12 is the only lysine required for Z sumoylation (Fig. 1 and 2). Since multiple different SUMO-2/3 proteins can be attached to a single lysine residue, we speculate that the larger forms of sumo-modified Z reflect the attachment of more than one SUMO-2/3 to Z; these larger forms also may contain SUMO-1. Alternatively, since some proteins have been shown to be modified simultaneously by both sumoylation and ubiquitination at a single lysine (71), the larger sumo-modified forms of Z could be ubiquitinated as well. Nevertheless, our finding that the overexpression of the sentrin protease 1 (which specifically cleaves SUMO moieties from proteins) prevents all of the various different forms of sumo-modified Z (Fig. 1) suggests that each of the higher-weight forms of sumo-modified Z depends upon at least one SUMO moiety being attached to K12. At present, it is unclear whether SUMO-1 has a different effect on Z function than does SUMO-2/3.
The sumoylation of Z is unusual in a number of respects. Although Z sumoylation is relatively efficient in some cell types (including HeLa), sumoylation usually occurs at the consensus sequence ΨKXE, with Ψ representing a hydrophobic amino acid followed by a lysine (K), any amino acid (X), and a glutamate (E) (5). The amino acid region of Z that is sumoylated encodes VKFT and does not conform to the ΨKXE sumoylation consensus sequence. Although most sumoylated proteins also contain a SUMO-interacting domain (SID), allowing them to interact directly with SUMO proteins, the Z sequence does not have an obvious SID. It currently remains unknown if a direct interaction between Z and SUMO proteins (mediated though some other part of Z) is required for the sumoylation of ZK12. In addition, the SUMO E3 ligase responsible for sumoylating Z has not yet been identified.
Similarly to the sumo-mediated repression of cellular transcription factors, in which the steady-state sumoylation level usually is less than 5% (30), we found that only a small portion of Z is sumo-modified on immunoblot analysis, yet this modification appears to inhibit Z transcriptional function. It has been hypothesized that sumoylated transcription factors recruit repressive factors to promoters and induce chromatin remodeling that results in decreased gene expression even when the transcription factor is no longer sumoylated (30, 38). Therefore, only a small percentage of Z may need to be sumoylated to inhibit Z function and promote viral latency.
Although sumoylation affects the cellular localization of some proteins, in this study we did not find that the wild-type Z has a different cellular localization from that of the sumo-deficient ZK12 mutants (Fig. 3). Interestingly, we found that the vast majority of Z in HeLa cells is localized to the nuclear matrix. The nuclear matrix has been shown to be a site for DNA replication as well as transcription, and the localization of transcription factors in the nuclear matrix can, in some cases, enhance their function by promoting access to transcriptional machineries (19). Given that the vast majority of Z resides within the nuclear matrix, it will be interesting to determine if any of the EBV promoters activated by Z contain matrix localization sequences that allow them to preferentially locate in the nuclear matrix as well. The simultaneous localization of the Z protein and Z-responsive EBV promoters to the nuclear matrix might allow limiting amounts of Z to preferentially interact with key viral target promoters rather than irrelevant ZREs in the cellular genome.
Finally, we also show here that Z sumoylation is inhibited by the virally encoded kinase EBV-PK. EBV-PK previously has been shown to bind to and phosphorylate the Z protein (6). We show that overexpressed EBV-PK rather dramatically reduces Z sumoylation mediated by SUMO-1, SUMO-2, or SUMO-3 (Fig. 8), but that this effect does not require the one residue in Z previously shown to be phosphorylated by EBV-PK, residue 209 (Fig. 10). EBV-PK does not inhibit the sumoylation of the cellular RanGAP1 protein, indicating that the effect is specific to Z. Importantly, we show that Z sumoylation is greatly enhanced in 293 cells infected with an EBV mutant missing the EBV-PK protein compared to cells infected with the wild-type virus, thus confirming that EBV-PK functions to inhibit Z sumoylation in the context of the intact viral genome (Fig. 9). Thus, EBV-PK likely alters Z function through more than one mechanism. In addition, our results suggest that EBV-PK regulates one or more cellular proteins involved in Z sumoylation, including the proteins that mediate Z sumoylation (such as Ubc9 and the as-yet-unknown E3 ligase) or the protein(s) that reverses Z sumoylation (such as the SUMO isopeptidases). Alternatively, the EBV-PK phosphorylation of a Z residue other than 209 may contribute to the loss of Z sumoylation. Although the mechanism by which EBV-PK decreases Z sumoylation currently remains unclear, our results suggest that one of the roles of EBV-PK during lytic viral reactivation is to prevent Z sumoylation and, thus, enhance its function as a transcriptional activator.
Acknowledgments
This work was supported by grants T32 CA009681, T32 AI078985, R01-CA58853, R01-CA66519, P01-CA022443, R01-H6064851, and R01-107034 from the National Institutes of Health.
We thank Lance Rodenkirch at the W. M. Keck Laboratory for Biological Imaging, University of Wisconsin, for helping with the confocal microscopy.
Footnotes
Published ahead of print on 24 February 2010.
REFERENCES
- 1.Adamson, A. L. 2005. Effects of SUMO-1 upon Epstein-Barr virus BZLF1 function and BMRF1 expression. Biochem. Biophys. Res. Commun. 336:22-28. [DOI] [PubMed] [Google Scholar]
- 2.Adamson, A. L., and S. Kenney. 2001. Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J. Virol. 75:2388-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adamson, A. L., and S. C. Kenney. 1998. Rescue of the Epstein-Barr virus BZLF1 mutant, Z(S186A), early gene activation defect by the BRLF1 gene product. Virology 251:187-197. [DOI] [PubMed] [Google Scholar]
- 4.Ahn, J. H., Y. Xu, W. J. Jang, M. J. Matunis, and G. S. Hayward. 2001. Evaluation of interactions of human cytomegalovirus immediate-early IE2 regulatory protein with small ubiquitin-like modifiers and their conjugation enzyme Ubc9. J. Virol. 75:3859-3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anckar, J., and L. Sistonen. 2007. SUMO: getting it on. Biochem. Soc. Trans. 35:1409-1413. [DOI] [PubMed] [Google Scholar]
- 6.Asai, R., A. Kato, and Y. Kawaguchi. 2009. Epstein-Barr virus protein kinase BGLF4 interacts with viral transactivator BZLF1 and regulates its transactivation activity. J. Gen. Virol. 90:1575-1581. [DOI] [PubMed] [Google Scholar]
- 7.Beech, S. J., K. J. Lethbridge, N. Killick, N. McGlincy, and K. N. Leppard. 2005. Isoforms of the promyelocytic leukemia protein differ in their effects on ND10 organization. Exp. Cell Res. 307:109-117. [DOI] [PubMed] [Google Scholar]
- 8.Bhende, P. M., W. T. Seaman, H. J. Delecluse, and S. C. Kenney. 2005. BZLF1 activation of the methylated form of the BRLF1 immediate-early promoter is regulated by BZLF1 residue 186. J. Virol. 79:7338-7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhende, P. M., W. T. Seaman, H. J. Delecluse, and S. C. Kenney. 2004. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat. Genet. 36:1099-1104. [DOI] [PubMed] [Google Scholar]
- 10.Calderwood, M. A., A. M. Holthaus, and E. Johannsen. 2008. The Epstein-Barr virus LF2 protein inhibits viral replication. J. Virol. 82:8509-8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cayrol, C., and E. K. Flemington. 1996. The Epstein-Barr virus bZIP transcription factor Zta causes G0/G1 cell cycle arrest through induction of cyclin-dependent kinase inhibitors. EMBO J. 15:2748-2759. [PMC free article] [PubMed] [Google Scholar]
- 12.Chang, L. K., Y. H. Lee, T. S. Cheng, Y. R. Hong, P. J. Lu, J. J. Wang, W. H. Wang, C. W. Kuo, S. S. Li, and S. T. Liu. 2004. Post-translational modification of Rta of Epstein-Barr virus by SUMO-1. J. Biol. Chem. 279:38803-38812. [DOI] [PubMed] [Google Scholar]
- 13.Chang, Y. N., D. L. Dong, G. S. Hayward, and S. D. Hayward. 1990. The Epstein-Barr virus Zta transactivator: a member of the bZIP family with unique DNA-binding specificity and a dimerization domain that lacks the characteristic heptad leucine zipper motif. J. Virol. 64:3358-3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, M. R., S. J. Chang, H. Huang, and J. Y. Chen. 2000. A protein kinase activity associated with Epstein-Barr virus BGLF4 phosphorylates the viral early antigen EA-D in vitro. J. Virol. 74:3093-3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chevallier-Greco, A., E. Manet, P. Chavrier, C. Mosnier, J. Daillie, and A. Sergeant. 1986. Both Epstein-Barr virus (EBV)-encoded trans-acting factors, EB1 and EB2, are required to activate transcription from an EBV early promoter. EMBO J. 5:3243-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Countryman, J., H. Jenson, R. Seibl, H. Wolf, and G. Miller. 1987. Polymorphic proteins encoded within BZLF1 of defective and standard Epstein-Barr viruses disrupt latency. J. Virol. 61:3672-3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Countryman, J., and G. Miller. 1985. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc. Natl. Acad. Sci. USA 82:4085-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darr, C. D., A. Mauser, and S. Kenney. 2001. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. J. Virol. 75:6135-6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davie, J. R., S. He, L. Li, A. Sekhavat, P. Espino, B. Drobic, K. L. Dunn, J. M. Sun, H. Y. Chen, J. Yu, S. Pritchard, and X. Wang. 2008. Nuclear organization and chromatin dynamics-Sp1, Sp3 and histone deacetylases. Adv. Enzyme Regul. 48:189-208. [DOI] [PubMed] [Google Scholar]
- 20.Delecluse, H. J., T. Hilsendegen, D. Pich, R. Zeidler, and W. Hammerschmidt. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 95:8245-8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickerson, S. J., Y. Xing, A. R. Robinson, W. T. Seaman, H. Gruffat, and S. C. Kenney. 2009. Methylation-dependent binding of the Epstein-Barr virus BZLF1 protein to viral promoters. PLoS Pathog. 5:e1000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Guindy, A. S., and G. Miller. 2004. Phosphorylation of Epstein-Barr virus ZEBRA protein at its casein kinase 2 sites mediates its ability to repress activation of a viral lytic cycle late gene by Rta. J. Virol. 78:7634-7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Guindy, A. S., S. Y. Paek, J. Countryman, and G. Miller. 2006. Identification of constitutive phosphorylation sites on the Epstein-Barr virus ZEBRA protein. J. Biol. Chem. 281:3085-3095. [DOI] [PubMed] [Google Scholar]
- 24.Everett, R. D., and M. K. Chelbi-Alix. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819-830. [DOI] [PubMed] [Google Scholar]
- 25.Farrell, P. J., D. T. Rowe, C. M. Rooney, and T. Kouzarides. 1989. Epstein-Barr virus BZLF1 trans-activator specifically binds to a consensus AP-1 site and is related to c-fos. EMBO J. 8:127-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feederle, R., M. Kost, M. Baumann, A. Janz, E. Drouet, W. Hammerschmidt, and H. J. Delecluse. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19:3080-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng, W. H., R. J. Kraus, S. J. Dickerson, H. J. Lim, R. J. Jones, X. Yu, J. E. Mertz, and S. C. Kenney. 2007. ZEB1 and c-Jun levels contribute to the establishment of highly lytic Epstein-Barr virus infection in gastric AGS cells. J. Virol. 81:10113-10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flemington, E., and S. H. Speck. 1990. Autoregulation of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 64:1227-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flemington, E., and S. H. Speck. 1990. Evidence for coiled-coil dimer formation by an Epstein-Barr virus transactivator that lacks a heptad repeat of leucine residues. Proc. Natl. Acad. Sci. USA 87:9459-9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geiss-Friedlander, R., and F. Melchior. 2007. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8:947-956. [DOI] [PubMed] [Google Scholar]
- 31.Gershburg, E., M. Marschall, K. Hong, and J. S. Pagano. 2004. Expression and localization of the Epstein-Barr virus-encoded protein kinase. J. Virol. 78:12140-12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gershburg, E., and J. S. Pagano. 2002. Phosphorylation of the Epstein-Barr virus (EBV) DNA polymerase processivity factor EA-D by the EBV-encoded protein kinase and effects of the L-riboside benzimidazole 1263W94. J. Virol. 76:998-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol. 2:1044-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gruffat, H., E. Manet, A. Rigolet, and A. Sergeant. 1990. The enhancer factor R of Epstein-Barr virus (EBV) is a sequence-specific DNA binding protein. Nucleic Acids Res. 18:6835-6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hacques, M. F., S. Muller, G. De Murcia, M. H. Van Regenmortel, and C. Marion. 1990. Use of an immobilized enzyme and specific antibodies to analyse the accessibility and role of histone tails in chromatin structure. Biochem. Biophys. Res. Commun. 168:637-643. [DOI] [PubMed] [Google Scholar]
- 36.Hammerschmidt, W., and B. Sugden. 1988. Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 55:427-433. [DOI] [PubMed] [Google Scholar]
- 37.Hardwick, J. M., P. M. Lieberman, and S. D. Hayward. 1988. A new Epstein-Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J. Virol. 62:2274-2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hay, R. T. 2005. SUMO: a history of modification. Mol. Cell 18:1-12. [DOI] [PubMed] [Google Scholar]
- 39.Heather, J., K. Flower, S. Isaac, and A. J. Sinclair. 2009. The Epstein-Barr virus lytic cycle activator Zta interacts with methylated ZRE in the promoter of host target gene egr1. J. Gen. Virol. 90:1450-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofmann, H., S. Floss, and T. Stamminger. 2000. Covalent modification of the transactivator protein IE2-p86 of human cytomegalovirus by conjugation to the ubiquitin-homologous proteins SUMO-1 and hSMT3b. J. Virol. 74:2510-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong, G. K., H. J. Delecluse, H. Gruffat, T. E. Morrison, W. H. Feng, A. Sergeant, and S. C. Kenney. 2004. The BRRF1 early gene of Epstein-Barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. J. Virol. 78:4983-4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Izumiya, Y., T. J. Ellison, E. T. Yeh, J. U. Jung, P. A. Luciw, and H. J. Kung. 2005. Kaposi's sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J. Virol. 79:9912-9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Izumiya, Y., C. Izumiya, A. Van Geelen, D. H. Wang, K. S. Lam, P. A. Luciw, and H. J. Kung. 2007. Kaposi's sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J. Virol. 81:1072-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karlsson, Q. H., C. Schelcher, E. Verrall, C. Petosa, and A. J. Sinclair. 2008. The reversal of epigenetic silencing of the EBV genome is regulated by viral bZIP protein. Biochem. Soc. Trans. 36:637-639. [DOI] [PubMed] [Google Scholar]
- 45.Kenney, S., J. Kamine, E. Holley-Guthrie, J. C. Lin, E. C. Mar, and J. Pagano. 1989. The Epstein-Barr virus (EBV) BZLF1 immediate-early gene product differentially affects latent versus productive EBV promoters. J. Virol. 63:1729-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kieff, E., and A. B. Rickinson. 2007. Epstein-Barr virus and its replication, p. 2603-2654. Fields virology, 5th ed. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 47.Laichalk, L. L., and D. A. Thorley-Lawson. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79:1296-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee, H. R., D. J. Kim, J. M. Lee, C. Y. Choi, B. Y. Ahn, G. S. Hayward, and J. H. Ahn. 2004. Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. J. Virol. 78:6527-6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao, G., F. Y. Wu, and S. D. Hayward. 2001. Interaction with the Epstein-Barr virus helicase targets Zta to DNA replication compartments. J. Virol. 75:8792-8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lieberman, P. M., and A. J. Berk. 1990. In vitro transcriptional activation, dimerization, and DNA-binding specificity of the Epstein-Barr virus Zta protein. J. Virol. 64:2560-2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lieberman, P. M., J. M. Hardwick, J. Sample, G. S. Hayward, and S. D. Hayward. 1990. The zta transactivator involved in induction of lytic cycle gene expression in Epstein-Barr virus-infected lymphocytes binds to both AP-1 and ZRE sites in target promoter and enhancer regions. J. Virol. 64:1143-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mahajan, R., C. Delphin, T. Guan, L. Gerace, and F. Melchior. 1997. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 88:97-107. [DOI] [PubMed] [Google Scholar]
- 53.Marschall, M., M. Stein-Gerlach, M. Freitag, R. Kupfer, M. van den Bogaard, and T. Stamminger. 2002. Direct targeting of human cytomegalovirus protein kinase pUL97 by kinase inhibitors is a novel principle for antiviral therapy. J. Gen. Virol. 83:1013-1023. [DOI] [PubMed] [Google Scholar]
- 54.Matunis, M. J., E. Coutavas, and G. Blobel. 1996. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 135:1457-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McDonald, C. M., C. Petosa, and P. J. Farrell. 2009. Interaction of Epstein-Barr virus BZLF1 C-terminal tail structure and core zipper is required for DNA replication but not for promoter transactivation. J. Virol. 83:3397-3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moorman, N. J., D. O. Willer, and S. H. Speck. 2003. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J. Virol. 77:10295-10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Müller, S., and A. Dejean. 1999. Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J. Virol. 73:5137-5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Müller, S., M. J. Matunis, and A. Dejean. 1998. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 17:61-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neuhierl, B., R. Feederle, W. Hammerschmidt, and H. J. Delecluse. 2002. Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and efficiency of infection. Proc. Natl. Acad. Sci. USA 99:15036-15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nevels, M., W. Brune, and T. Shenk. 2004. SUMOylation of the human cytomegalovirus 72-kilodalton IE1 protein facilitates expression of the 86-kilodalton IE2 protein and promotes viral replication. J. Virol. 78:7803-7812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prieur, A., K. Nacerddine, M. van Lohuizen, and D. S. Peeper. 2009. SUMOylation of DRIL1 directs its transcriptional activity towards leukocyte lineage-specific genes. PLoS One 4:e5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ragoczy, T., L. Heston, and G. Miller. 1998. The Epstein-Barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. J. Virol. 72:7978-7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reyes, J. C., C. Muchardt, and M. Yaniv. 1997. Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J. Cell Biol. 137:263-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rickinson, A. B., and E. Kieff. 2006. Epstein-Barr virus, p. 2655-2700. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 65.Rooney, C. M., D. T. Rowe, T. Ragot, and P. J. Farrell. 1989. The spliced BZLF1 gene of Epstein-Barr virus (EBV) transactivates an early EBV promoter and induces the virus productive cycle. J. Virol. 63:3109-3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sarisky, R. T., Z. Gao, P. M. Lieberman, E. D. Fixman, G. S. Hayward, and S. D. Hayward. 1996. A replication function associated with the activation domain of the Epstein-Barr virus Zta transactivator. J. Virol. 70:8340-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schepers, A., D. Pich, and W. Hammerschmidt. 1993. A transcription factor with homology to the AP-1 family links RNA transcription and DNA replication in the lytic cycle of Epstein-Barr virus. EMBO J. 12:3921-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schepers, A., D. Pich, J. Mankertz, and W. Hammerschmidt. 1993. cis-acting elements in the lytic origin of DNA replication of Epstein-Barr virus. J. Virol. 67:4237-4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith, G. A., and L. W. Enquist. 1999. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J. Virol. 73:6405-6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takada, K., N. Shimizu, S. Sakuma, and Y. Ono. 1986. Trans-activation of the latent Epstein-Barr virus (EBV) genome after transfection of the EBV DNA fragment. J. Virol. 57:1016-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tatham, M. H., M. C. Geoffroy, L. Shen, A. Plechanovova, N. Hattersley, E. G. Jaffray, J. J. Palvimo, and R. T. Hay. 2008. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 10:538-546. [DOI] [PubMed] [Google Scholar]
- 72.Yeh, E. T., L. Gong, and T. Kamitani. 2000. Ubiquitin-like proteins: new wines in new bottles. Gene 248:1-14. [DOI] [PubMed] [Google Scholar]
- 73.Yu, X., Z. Wang, and J. E. Mertz. 2007. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog. 3:e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zalani, S., E. Holley-Guthrie, and S. Kenney. 1996. Epstein-Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proc. Natl. Acad. Sci. USA 93:9194-9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang, Q., D. Gutsch, and S. Kenney. 1994. Functional and physical interaction between p53 and BZLF1: implications for Epstein-Barr virus latency. Mol. Cell. Biol. 14:1929-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang, Q., Y. Hong, D. Dorsky, E. Holley-Guthrie, S. Zalani, N. A. Elshiekh, A. Kiehl, T. Le, and S. Kenney. 1996. Functional and physical interactions between the Epstein-Barr virus (EBV) proteins BZLF1 and BMRF1: effects on EBV transcription and lytic replication. J. Virol. 70:5131-5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhong, S., S. Muller, S. Ronchetti, P. S. Freemont, A. Dejean, and P. P. Pandolfi. 2000. Role of SUMO-1-modified PML in nuclear body formation. Blood 95:2748-2752. [PubMed] [Google Scholar]
- 78.zur Hausen, H., H. Schulte-Holthausen, G. Klein, W. Henle, G. Henle, P. Clifford, and L. Santesson. 1970. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 228:1056-1058. [DOI] [PubMed] [Google Scholar]