

Abstract
Viruses of the Paramyxoviridae family bind to their host cells by using hemagglutinin-neuraminidase (HN), which enhances fusion protein (F)-mediated membrane fusion. Although respiratory syncytial virus and parainfluenza virus 5 of this family are suggested to trigger host cell signaling during infection, the virus-induced intracellular signals dictating virus-cell fusion await elucidation. Using an F- or HN-F-containing reconstituted envelope of Sendai virus, another paramyxovirus, we revealed the role and regulation of AKT1 and Raf/MEK/ERK cascades during viral fusion with liver cells. Our observation that extracellular signal-regulated kinase (ERK) activation promotes viral fusion via ezrin-mediated cytoskeletal rearrangements, whereas AKT1 attenuates fusion by promoting phosphorylation of F protein, indicates a counteractive regulation of viral fusion by reciprocal activation of AKT1 and mitogen-activated protein kinase (MAPK) cascades, establishing a novel conceptual framework for a therapeutic strategy.
Infection by paramyxoviruses initiates with the binding of the viral glycoprotein, hemagglutinin-neuraminidase (HN), to the host cell surface sialoglycoconjugate. This is followed by HN-F interaction, which triggers a conformational change(s) in the viral F protein that subsequently completes the fusion process (39). Previous work from us and others showed that Sendai virus (SeV) envelopes devoid of HN protein (F-virosomes [FV]) can also bind and fuse with liver cells due to the high-affinity interaction between the exposed sugar residues on F protein and the asialoglycoprotein receptor (ASGPR) on hepatocytes (3-5, 30, 31, 35, 43, 50). However, a significant reduction in fusion potential of the virus occurs in the absence of HN protein. Recently, we showed that the histidine residue at position 247 (H247) in HN acts as a switch for triggering virus-cell fusion (27). The available data thus suggest that HN provides an activation signal to F protein (3, 10, 27) following the binding with sialoglycoconjugate and eventually accentuates the fusion potential of the virus, whereas ASGPR serves only as an alternate receptor for Sendai virus (5, 30, 35).
However, in addition to cell surface receptors for viral glycoproteins, viruses might also require some other cellular factors from their hosts for efficient fusion and entry. On the other hand, membrane fusion is a critical step in the course of viral infection in the case of enveloped animal viruses, so it is logical to think of host cell regulation at this level itself to modulate viral entry. While the importance of virus-cell surface receptor interaction for fusion and entry is established, the role of intracellular signaling in regulating this process is still not clear. It has been suggested that virus-cell surface receptor interactions can elicit two types of signals, i.e., conformational changes of viral particles and concomitant intracellular signals triggering specific cellular reactions (19). Indeed, cellular signal transduction pathways and associated protein kinases are implicated in retrovirus-induced cell-cell fusion (56). For instance, HIV-1 envelope interacts with the CCR5 coreceptor and activates the Gαq pathway to trigger HIV-1-induced cell-cell fusion (20). Recent studies with respiratory syncytial virus (RSV) and parainfluenza virus 5 (PIV5) also suggested a requirement for host cell signaling in infection (18, 26, 33). However, these reports failed to explain the effect of host cell signaling specifically on viral fusion-mediated entry. Nonetheless, these reports indicate the relative susceptibility or resistance of host cells to viral entry based on their signaling status.
Extracellular signal-regulated kinases 1 and 2 (ERK1/2), which mediate diverse specific responses to various stimuli, such as cytokines, growth factors, and hormones, have recently been implicated in the infection processes of many enveloped viruses, including paramyxoviruses (19, 26, 42). The infection processes for intact RSV and influenza virus activate the ERK1/2 pathway (26, 42), but the exact relationship between cell signaling and membrane fusion during infection, following interaction between the viral ligand and the host cell receptor, is still lacking. The involvement of several proteins during infection of such viruses remains one of the major hurdles in deciphering the fine interplay of cellular signaling and fusion. We have eliminated this limitation by using Sendai virus FV, which is free of viral genetic material (and HN) and thus can generate exclusive information regarding the role of host cell signaling in membrane fusion.
In the present study, we attempted to decipher the role of intracellular signaling pathways, if any, in the process of membrane fusion-mediated viral entry. We provide evidence for an interesting interplay between two distinct signaling pathways (AKT1 and Raf/MEK/ERK) in modulating Sendai virus F-induced FV-cell and cell-cell fusion. We show that whereas the Raf/MEK/ERK cascade supports fusion through host cell cytoskeletal rearrangement, AKT/PKB signaling acts as a host defense pathway which negatively regulates F protein-induced membrane fusion through phosphorylation of the fusion protein itself, which so far has not been reported for any viral infection. Additionally, the presence of HN in FV (HNFV) was seen to augment the fusion efficiency of FV, as described before. But here we demonstrate that the His247 residue of HN functions as a molecular switch to trigger F protein-mediated FV fusion, in part by regulating AKT1 phosphorylation, apart from its established role of inducing a conformational change in F. Based on our results, we propose that variations in virus-host cell interactions, which remain unexplained as yet, are due to differential activation of the host cell signaling pathways such that activation of one renders the host resistant to fusion/infection, whereas activation of the other makes that host susceptible. Considering the high incidences of drug resistance emanating from mutation-induced changes in viral genomes, these findings altogether point toward the need to develop efficient antiviral therapeutics involving the host cell response (58).
MATERIALS AND METHODS
Cells, virus, antibodies, and reagents.
Human liver cells (Huh-7 and HepG2) were obtained from the American Type Culture Collection and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) and antibiotics. SeV (Z strain) was grown in embryonated chicken eggs and purified according to standard procedures (3). Phospho-p44/42 mitogen-activated protein kinase (MAPK) antibody (pERK1/2; 9106S), phospho-p38 antibody (9211S), phospho-ezrin(Thr567)/radixin(Thr564)/moesin(Thr558) antibody (3141), ezrin/radixin/moesin antibody (3142), anti-phospho-Raf-1 (9421), anti-c-Raf (9422), anti-phospho-PKA C (Thr197) (4781), and anti-PKA C-α (4782) were purchased from Cell Signaling Technology. Anti-Sendai virus and anti-F antibodies were raised in mice (28). Rabbit anti-human ASGPR (hepatic) antibody was obtained from Martin Spiess, Basel, Switzerland. Anti-Flag M2 antibody (F1804), anti-ceramide (IgM; C8104) antibody (mouse monoclonal), anti-glutathione S-transferase (anti-GST) antibody (G7781), anti-lysozyme antibody, anti-rabbit IgG-tetramethyl rhodamine isocyanate (TRITC), and anti-mouse IgG-fluorescein isothiocyanate (FITC) were purchased from Sigma. Anti-ERK1 (sc-94), anti-p-AKT1(Ser 473) (sc7985), anti-AKT (1/2/3) (H-136; sc-8312), anti-hemagglutinin (anti-HA) antibody (HA probe F-7; sc-7392), anti-c-Jun N-terminal kinase 2 (anti-JNK2) antibody (sc-7345), anti-p38 (sc-535), and anti-phosphotyrosine antibody (sc-7020) were purchased from Santa Cruz Biotech. Anti-phospho-JNK antibody (V793A) was purchased from Promega. Anti-phosphothreonine antibody (KP3801) and anti-mouse IgM-horseradish peroxidase (HRP) antibody were obtained from Calbiochem. N-4-Nitrobenzo-2-oxa-1,3-diazole phosphatidylethanolamine (NBD-PE) was purchased from Avanti. SM2 Bio-Beads were obtained from Bio-Rad. Triton X-100 (TX-100) was obtained from Aldrich. Lysozyme (chicken egg white), trypsin (type III), dithiothreitol (DTT), wheat germ agglutinin (WGA), EDTA (disodium salt), rhodamine isothiocyanate (RITC), and cytochalasin B (cytoB; C6762) were purchased from Sigma. Bodipy FL phallacidin (B607) and jasplakinolide (JASP; J7473) were purchased from Molecular Probes. AKT inhibitor [1l-6-hydroxymethyl-chiro-inositol 2-(R)-2-O-methyl-3-O-octadecylcarbonate] (IAKT; 124005), SB202190 (559388), protein kinase A inhibitor 5-24 (IPKA; 116805), JNK inhibitor I (L) form (cell permeative) (IJNK; 420116), wortmannin (681675), okadaic acid (OKA) (495604), and tautomycin (TAU) (580551) were purchased from Calbiochem. PD98059 (specific MEK inhibitor) was purchased from New England Biolabs.
Conjugation of RITC with lysozyme and preparation of various types of RITC-L-loaded virosomes.
RITC was chemically conjugated to lysozyme (RITC-L), and reconstituted RITC-L-loaded Sendai viral envelopes, containing wild-type (wt) or mutant F alone [FV or (T234A)FV] or wt or mutant HN and F together [HNFV or (H247A)HNFV], were prepared as described earlier (55). Heat inactivation (heat control) of virosomes [HCFV, HCHNFV, and HC(H247A)HNFV] was performed according to the method in our previously published report (55).
Transmission electron microscopy (TEM).
HepG2 cells growing in DMEM were serum starved for 14 h and pretreated with cytoB (10 μM) for 1 h followed by incubation with 80 μg FV. Cells were washed with Dulbecco's phosphate-buffered saline (DPBS) and fixed in 1% glutaraldehyde in 0.01 M phosphate buffer, pH 7.3, for 1 h at 4°C. Cells were washed thrice, before a secondary fixation in 2% OsO4 (in the same buffer) for 1 h at room temperature, and then washed again thrice with phosphate buffer. Cells were then successively dehydrated in ethanol, infused with propylene oxide, and embedded in Epon 812. Ultrathin sections were obtained using a Reichert-Jung microtome and were double stained with uranyl acetate and lead citrate. Grids of specimens were incubated with anti-lysozyme antibody, washed with DPBS, and incubated with gold-labeled (15 nm) anti-rabbit IgG. Specimens were examined using a Philips CM-10 transmission electron microscope at 75 kV, with the magnification ranging from ×8,000 to ×100,000.
Immunocytochemistry and confocal microscopy.
HepG2 cells grown on polylysine-coated coverslips were incubated with 80 μg of various virosomes for 1 h, washed thrice with DPBS, fixed with 3.7% paraformaldehyde (20 min, room temperature), and permeabilized with 0.4% Tween 20 in DPBS (15 min, room temperature). For immunofluorescence, cells were incubated with specific primary and secondary antibodies after being blocked with 5% FCS. After being washed, cells were counterstained with DAPI (4′,6-diamidino-2-phenylindole; 100 ng/ml) for 5 min. The coverslips were then mounted with 0.1 M propyl gallate and 0.5% glycerol on slides, sealed, and viewed under a confocal laser scanning microscope (Aquastic TCS-SP2 optical beam splitter; Leica, Germany). For visualizing RITC-L in fusion assays, cells were directly processed for slide preparation after fixation, permeabilization, and DAPI counterstaining. Fixed HepG2 cells were incubated with rabbit anti-human hepatic ASGPR (polyclonal) antibody without permeabilization, followed by goat anti-rabbit IgG-FITC to locate the plasma membrane. In the case of actin staining, fixed cells were blocked with 1% bovine serum albumin (BSA) for 1 h, and then Bodipy FL phallacidin (1 unit, i.e., 5 μl in 200 μl DPBS) was overlaid on cells for 1 h at room temperature. All images were analyzed by standard software provided by the manufacturer of the confocal microscope, and no outshining of red and green fluorescence was observed.
Cloning and site-directed mutagenesis.
Full-length Sendai virus HN and F genes in pGEMT were obtained from D. Kolakofsky, Geneva, Switzerland. HN and F cDNAs were subcloned into the eukaryotic expression vector pcDNA3.1(+) (Clontech), using BamHI/EcoRI restriction sites. Site-directed mutagenesis to generate single amino acid substitutions in the F protein (T234A, T272A, and Y316A) was performed with a QuikChange kit (Stratagene). GST-MEK-S218E/S222E double mutants were generated using pEBG-MEK1 plasmid. pcDNA3.1-HN(H247A) was constructed earlier in our laboratory (27). The specific mutation was confirmed by DNA sequencing.
Synthetic oligonucleotide primers for site-directed mutagenesis (MWG Company) were used to generate double mutations (S218E and S222E) in MEK1. The primer set used was as follows: forward primer, 5′-CAGCGGGCAGCTCATCGACGAAATGGCCAACGAATTCGTGGGCACAAGGTCC-3′; and reverse primer, 5′-GGACCTTGTGCCCACGAATTCGTTGGCCATTTCGTCGATGAGCTGCCCGCTG-3′. Synthetic oligonucleotide primers (Microsynth) were used to introduce single amino acid mutations (substituting alanine for amino acids T234, T272, and Y316) into the F gene, using the plasmid construct pCDNA3.1-F. Primer sets used for different mutations were as follows: for T234A mutation, forward primer 5′-GGAGAGAAGAGCCGCGCGCTGCAGGCGCTGTC-3′ and reverse primer 5′-GACAGCGCCTGCAGCGCGCGGCTCTTCTCTCC-3′; for T272A mutation, forward primer 5′-GAACAGATCAAAGGAGCGGTGATAGATGTGGAC-3′ and reverse primer 5′-GTCCACATCTATCACCGCTCCTTTGATCTGTTC-3′; and for Y316A mutation, forward primer 5′-GACGGGGAGGAATGGGCTGTGACTGTCCCCAGC-3′ and reverse primer 5-GCTGGGGACAGTCACAGCCCATTCCTCCCCGTC-3′.
Purification of wild-type F and mutant F(T234A) from HepG2 cells.
The wild-type F and mutant F(T234A) proteins were expressed on the HepG2 cell surface and purified as described earlier (27).
Transfections (plasmids and small interfering RNAs [siRNAs]).
Cells were grown either in 6-well plates at a density of 1 × 106 cells in 2 ml DMEM for cell fusion assays or on poly-l-lysine-coated coverslips in 12-well plates for other cDNA transfections. The expression plasmids for dominant-negative AKT1, HA-AKT1 (K179A, S473A), Flag-myr AKT1 (pcDNA3 m2-Myr-AKT), wild-type pEBG-MEK1, constitutively active pEBG-MEK1 (S218E/S222E) (48), dominant-negative ERK1 [DNERK1, pCEP 4, or ERK-1(K91R)], pcDNA 3.1-F (wild type and its mutants), pcDNA3.1-HN, and pcDNA3.1-HN (H247A) were transfected into HepG2 or Huh-7 cells by use of Lipofectamine and Plus reagents (Invitrogen). Subconfluent monolayers were transfected with 0.4 μg of the desired plasmid. In order to check surface F (wild-type and mutant) and F protein expression, immunofluorescence and flow cytometry were used as described earlier (27).
Human ezrin siRNA (40 pmol) (sc-35349; Santa Cruz) and nonspecific (NS) siRNA (specific against the ornithine decarboxylase [ODC] gene of Aspergillus nidulans; sense strand, 5′-GAUCGUUGGCGUGAGCUUCCATT-3′; and antisense strand, 5′-UGGAAGCUCACGCCAACGAUCTT-3′) (40 pmol; Microsynth) were transfected into HepG2 cells by use of Lipofectamine 2000 reagent (Invitrogen), using the diluent medium Opti-MEM I (Invitrogen) per the supplier's protocol.
Fusion assays. (i) Cytosolic delivery of RITC-L in HepG2 cells.
HepG2 cells which had been grown in DMEM, serum starved for 14 h, and pretreated with cytoB (10 μM for 1 h) were washed with DMEM (without serum) and then further incubated with FV, (T234A)FV, HNFV, or (H247A)HNFV for 1 h in the same medium. Cells treated with heat-inactivated virosomes were used as a binding control (no fusion). Cells were washed with DPBS and processed further for either electron or confocal microscopy as described above. The percent fusion was calculated by counting the RITC-positive cells (red) among the total cells in a given field and was expressed in the form of a graph for all microscopic evaluations of membrane fusion.
(ii) Kinetics of fusion of Sendai virus FV (lipid mixing) with HepG2 cells.
HepG2 cells pretreated with IAKT (AKT inhibitor), PD98059 (specific MEK inhibitor), or dimethyl sulfoxide (DMSO) (solvent control) for 1 h were incubated on ice with NBD-PE-labeled virosomes [NBD-PE-FV and NBD-PE-(H247A)HNFV], prepared as described previously (27). Kinetics of fusion was recorded online by a spectrofluorimeter (FL3-22; Horiba Jobin) at 37°C, and its value was expressed as % fluorescence dequenched (FDQ), using the formula [(F − F0)/(Ft − F0)] × 100 (3).
(iii) Content mixing based on green and red fluorescent proteins (cell-cell fusion assay).
The effects of wild-type and mutated F proteins in fusion promotion were evaluated using a novel and sensitive content mixing assay and quantification by scoring of the number of fused cells (yellow cells), as described earlier (27). Two populations of cells (Huh-7 and HepG2) were used. In one case, the first cell population (Huh-7) was cotransfected with the desired F wild-type (wt) or mutant cDNA along with DsRed-N1 (red fluorescent protein) plasmid DNA. In another case, cells were cotransfected with HN wt or mutant cDNA along with F (wt) and DsRed-N1 plasmid DNAs. The second population of cells (HepG2; target cells) was transfected with pEGFP-N1 (green fluorescent protein) plasmid DNA. HN (wt) and F (wt) cotransfection into Huh-7 cells served as a positive control. After 24 h of transfection, DsRed-N1-, HN-, and F-cotransfected cells were treated with 5 μg/ml of trypsin (for activation of F0 to F1 and F2) and 0.22 mg/ml of neuraminidase before addition of target cells. Enhanced green fluorescent protein (EGFP)-expressing HepG2 cells (serving as the target cell population) were lifted and overlaid on the first set of cells. Cell-cell fusion was assessed in cells that showed both green fluorescence (450- to 490-nm-wavelength excitation filter, ft 510 dichroic mirror, and lp 520 emission filter on a Nikon Eclipse TE 300 epifluorescence microscope; Nikon, Japan), with a barrier filter of 510 nm, and red fluorescence (BP 546 excitation filter, ft 580 dichroic mirror, and lp 590 emission filter), with a barrier filter of 590 nm and a 20×/0.40 CF Achro LWD DL objective lens. Images were captured with a digital camera (Digital Sight DS-5 M; Nikon) attached to the microscope that gave a yellow image upon merging, using the Image-Pro Plus, version 5.1 (Media Cybernetics), software package. No spectral overlap was observed under these conditions.
Inhibition experiments.
HepG2 cells grown in DMEM were serum starved for 14 h, pretreated, wherever indicated, with 20 μM IAKT, 70 μM PD98059, 2 μM SB202190, 4 μM IJNK (JNK inhibitor), 20 nM IPKA (PKA inhibitor), 100 nM wortmannin, 10 μM cytoB, 2 nM OKA (PP2A inhibitor), 3 nM TAU (PP1 inhibitor), 1% DMSO, or 1% ethanol, and incubated with 80 μg of FV, HNFV, HCFV, (T234A)FV, or (H247A)HNFV for 1 h. Cells were then washed, and fusion was examined by confocal microscopy. In parallel experiments, cells were washed and processed for Western blotting or phosphatidylinositol 3-kinase (PI3K) assay (with and without wortmannin) or PP2A phosphatase assay (with and without okadaic acid).
Preparation of cytosolic/nuclear extracts and EMSA.
Following FV (40 and 80 μg), HNFV (80 μg), and HN(H247A)FV (80 μg) interaction with HepG2 cells (1 × 106 cells) under various conditions, nuclear and cytosolic extracts were prepared as described previously (38). The protein concentration in the extracts was measured by the Bradford assay. Electrophoretic mobility shift assays (EMSAs) were carried out following a published method (38). Briefly, a synthetic oligonucleotide consisting of a double-stranded TRE sequence (Promega) was used as a probe in this assay. A reaction mixture containing gel shift assay (GSA) buffer (20 mM HEPES, 5% glycerol, 50 mM NaCl, 1.5 mM MgCl2, 1 mM DTT), 8 μg of nuclear extract, 1 μg of poly(dI-dC), and 32P-labeled probe was made. Binding reactions were carried out for 60 min at 4°C, followed by incubation for 5 min at 25°C, using 10,000 cpm of 32P-labeled probe and 1 μg of poly(dI-dC) (Sigma). Competition binding experiments were performed with unlabeled oligonucleotide (cold probe) in excess. The reaction mixture was resolved in a 4% nondenaturing PAGE gel, dried, and visualized by autoradiography or phosphorimaging analysis.
In vitro kinase assays. (i) MBP phosphorylation assay.
After incubation with FV, HNFV, or (H247A)HNFV, HepG2 cells were lysed in a defined lysis buffer (38). Assessment of ERK activity was carried out by in vitro phosphorylation of myelin basic protein (MBP; Gibco BRL) following published protocols (38). For each sample, 10 μg of protein lysate was used in a reaction mixture containing 5 μg of MBP in kinase buffer (20 mM Tris-HCl [pH 7.5], 40 mM MgCl2, 10 μM ATP [Sigma]) and 2 μCi of [γ-32P]ATP (Amersham Pharmacia Biotech) and was incubated for 30 min at 30°C as described earlier (38). The reaction was stopped by the addition of Laemmli sample buffer, and products were resolved by SDS-15% PAGE. The upper half of the gel was used for Western analysis with anti-ERK antibody (Santa Cruz), using an enhanced chemiluminescence (ECL) detection system. The lower half of the gel, containing MBP, was dried and exposed to X-ray films. Coomassie blue (CB) staining was done with the dried blot to check that equal amounts of MBP were present in the reaction mixtures. The stained MBP bands were excised, and incorporated radioactivity was measured by liquid scintillation counting for calculations of fold activation.
(ii) PI3K assay.
HepG2 cells pretreated with wortmannin (100 nM) were incubated with 80 μg FV or HCFV, followed by washing with DPBS, and cell lysates were immunoprecipitated with mouse monoclonal anti-phosphotyrosine antibody by use of an immunoprecipitation kit (Boehringer Mannheim). The reactions were carried out in a volume of 0.1 ml containing the immunoprecipitated complex in kinase assay buffer (25 mM HEPES [pH 7.4], 10 mM MgCl2, and 1 mM EDTA) with phosphatidyl inositol (0.25 mg/ml), 100 mM ATP, and 15 μCi of [γ-32P]ATP, and the reaction mixtures were incubated at 30°C for 10 min. The reaction was terminated by addition of 1 ml acidified chloroform-methanol-HCl (2:1:0.03 [vol/vol]). Lipids were extracted as reported earlier (45). Briefly, the phases were resolved by addition of 0.5 ml chloroform and 0.5 ml water. The samples were vortexed and centrifuged at 800 × g for 10 min to separate the phases. The organic phase was recovered and dried under nitrogen. Dried samples were dissolved in chloroform-methanol (2:1) and spotted onto oxalate-treated plastic thin-layer chromatography (TLC) plates, where they were resolved using a solvent system consisting of chloroform-methanol-20% methylamine (65:35:10 [vol/vol/vol]). The spots corresponding to the position of radioactive phosphatidylinositol phosphate (PIP) were visualized by autoradiography.
IP.
Serum-starved HepG2 cells pretreated with cytoB (10 μM) or IAKT (20 μM) were incubated with FV, (T234A)FV, HNFV, or (H247A)HNFV, and lysates were prepared as described before (38) and subjected to anti-F immunoprecipitation (IP). Immune complexes were washed with wash buffers and resolved by SDS-10% PAGE, and proteins were detected by Western blotting.
Immune complexes obtained by anti-ceramide immunoprecipitation from lysates prepared from HepG2 cells which had been pretreated with cytoB and incubated with FV, HCFV, HNFV, (H247A)HNFV, or (T234A)FV were resuspended in 10 mM PBS, and 200 μl CHCl3-H2O (1:1) was added. The mixture was centrifuged at 800 × g for 10 min, and the organic phase was recovered, spotted onto oxalate-treated plastic TLC plates, and resolved by using a solvent system consisting of chloroform-methanol (95:5) and 20% methylamine (vol/vol). Spots corresponding to the position of ceramide were visualized by DAB (Sigma) staining after Western blotting.
Western blotting.
Cell lysates prepared as described before (38) were resolved by SDS-10% PAGE, and proteins were electrotransferred to a nitrocellulose membrane (Bio-Rad). The membrane was incubated with primary antibody (described elsewhere), washed, incubated with HRP-conjugated secondary antibody, and visualized by an ECL detection system (Sigma). The same blot was stripped and reprobed with another antibody as described elsewhere. Ponceau staining confirmed equal protein loading.
Reverse transcription-PCR (RT-PCR) amplification of ezrin gene-specific transcript.
Total RNAs from various virosome-treated samples of HepG2 cells were extracted using Trizol (Gibco BRL), and 5 μg was reverse transcribed using Superscript RNase H− reverse transcriptase (Gibco BRL) and gene-specific antisense primers per the manufacturer's protocol. The PCR primer set was obtained from Santa Cruz Biotech [ezrin (h)-PR; 565-bp product], and the product was amplified with the following PCR cycling profile: 94°C for 1 min, 58°C for 1 min (or 55°C for β-actin primers; Stratagene), and 72°C for 1 min, with a final extension of 72°C for 10 min. β-Actin was included for normalization.
PP2A phosphatase assay.
Serum-starved HepG2 cells pretreated with 10 μM cytoB, 2 nM okadaic acid, or 3 nM tautomycin were incubated with FV, HCFV, HNFV, (H247A)HNFV, or (T234A)FV for 1 h, washed thrice with DPBS, and lysed in 100 μl of phosphatase extraction buffer (containing 20 mM imidazole, 2 mM EDTA, 2 mM EGTA in 20 mM Tris-HCl, pH 7.0, with a protease inhibitor cocktail) to give a low endogenous phosphate level. Lysates (170 μg protein each) were subjected to immunoprecipitation with anti-PP2A antibody, followed by phosphatase assay according to the manufacturer's protocol (PP2A immunoprecipitation phosphatase assay kit; Millipore). Specific activities were calculated using a phosphate standard curve.
Statistical analysis.
All experiments were performed in triplicate, and the results were compared with appropriate controls by use of Student's t test. P values of <0.01 were considered significant.
RESULTS
Membrane fusion of FVs with HepG2 cells.
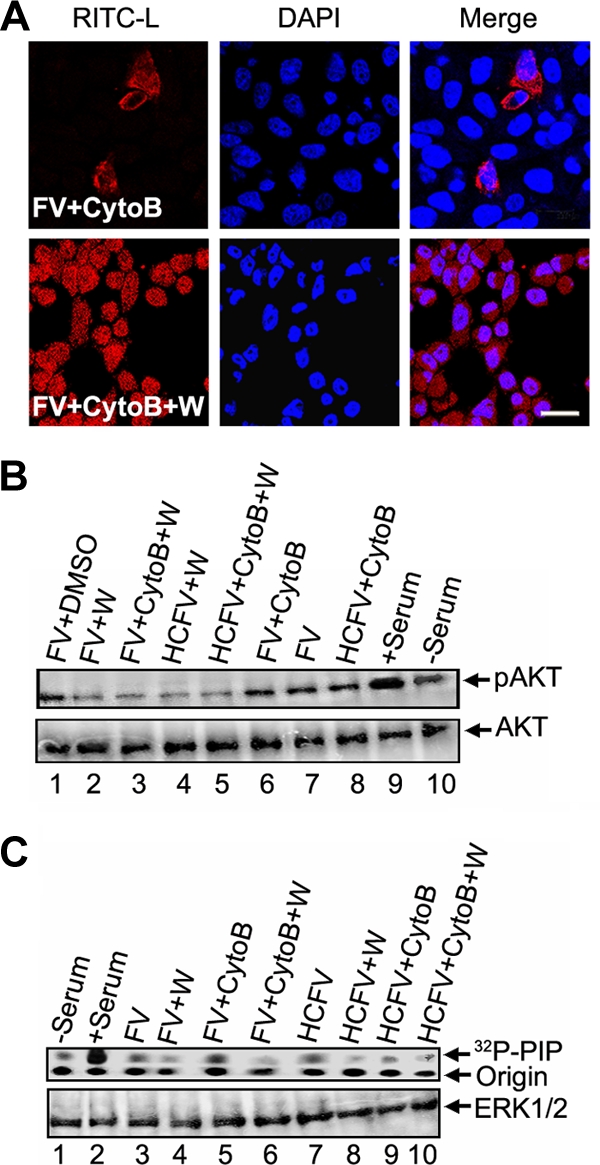
FVs can fuse with HepG2 cells at the surface or be endocytosed after binding to ASGPRs. Since endocytic entry of FVs can elicit cellular signals and that can interfere with the fusion-mediated cell signaling pathways, we used cytochalasin B (10 μM)-treated HepG2 cells throughout the study. CytoB effectively blocks endocytosis of FV/HCFV at this concentration (Fig. 1A, B, and C). In order to determine FV fusion with HepG2 cells and to differentiate it from binding followed by receptor-mediated endocytosis of FV, immunoconfocal (RITC-L probe and FITC-labeled anti-rabbit IgG antibody) and immunoelectron (anti-lysozyme antibody) microscopic analyses were carried out (Fig. 1A, B, and C). HCFVs in the presence of 10 μM cytoB (34) bound to HepG2 cells through interaction with ASGPRs but failed to be endocytosed, thus serving as a binding/negative control (punctate fluorescence), supporting the presence of intact FVs on the cell surface only in the presence of cytoB and both on the cell surface and in the cytosol in the absence of cytoB (Fig. 1A, compare rows 4 and 3), thus functioning as a control to negate the effect of F-ASGPR interaction on intracellular signaling. In order to differentiate the plasma membrane from the endocytic compartment of HepG2 cells, an immunolocalization experiment with specific anti-ASGPR (human) was carried out under nonpermeabilized conditions prior to confocal microscopy. Colocalization of RITC and FITC (FITC-labeled anti-rabbit IgG antibody) on the plasma membranes (yellow fluorescence) of HepG2 cells incubated with FV/HCFV in the presence of cytoB, but not in the case of endocytosed FV/HCFV (in the absence of cytoB), confirmed the role of cytoB as an inhibitor of endocytosis (Fig. 1A, compare rows 1 and 2 to rows 3 and 4). Similarly, it is known that Sendai virus enters its host cells both by fusion with the plasma membrane and via endocytosis (4), and fusion-active FVs failed to exhibit any punctate fluorescence pattern in the cytosol in the presence of cytoB (Fig. 1A, compare rows 2 and 1). The effect of 10 μM cytoB on endocytosis inhibition is also presented in a quantitative manner (Fig. 1B). This notion was also supported by TEM analysis (Fig. 1C, compare panels c and d), where binding of HCFV (particles in Fig. 1C, panel d, in the presence of cytoB) could easily be discerned with localized immunogold labeling in the absence of cytoB (Fig. 1C, panel c). On the other hand, interaction of FVs with HepG2 cells in the presence of cytoB in the cell cytosol, as monitored by RITC fluorescence (diffused) (Fig. 1A, row 2) and immunogold labeling (arrows in Fig. 1C, panel b), reflects the membrane fusion property (only) of the F protein. Altogether, our data also eliminate any interfering role of cytoB in membrane fusion per se. These results are in accordance with our earlier published work (3, 4) showing that heat-treated FV is capable of specifically binding with the liver cell receptor (ASGPR) but is not able to induce membrane fusion. Similar fusion profiles to that of FV were also observed in the case of HNFV and (H247A)HNFV (data not shown).
FIG. 1.

Fusion-mediated cytosolic delivery of RITC-L. (A) FV (80 μg)-HepG2 cell fusion was monitored by RITC-L delivery into the cytosol, using confocal microscopy as described in Materials and Methods. Cells appeared red (diffused fluorescence) after fusion with FV. Nuclei (blue) were visualized by staining with DAPI. The plasma membrane appeared green by development with a FITC-labeled secondary antibody. CytoB and HCFV stand for cytochalasin B-pretreated HepG2 cells and heat-inactivated FV, respectively. Arrowheads indicate representative areas of RITC-L localization (punctate fluorescence) as a result of endocytosis, while plasma membrane-bound FVs are shown by tailed arrows. Images shown are typical examples from more than three experiments. Bars, 23.81 μm. (B) Graphical representation (expressed as mean ± standard deviation [SD]) showing the percentage of RITC-L FVs inside the cytosol for FV and HCFV, with and without cytoB, as quantified after viewing 100 fields during acquisition of confocal images. (C) Immuno-TEM localization of RITC-L during interaction of FV with HepG2 cells. HepG2 cells fused with FV (80 μg) were labeled first with anti-lysozyme rabbit antibodies and then with 15-nm-gold-particle-labeled anti-rabbit IgG antibodies. (a) Cytosolic and vesicular labeling of gold particles to ensure fusion of FVs in the absence of cytoB, showing both fusion and endocytosis. (b) Localization of gold particles in the cytosol, depicting fusion of FVs in the presence of cytoB. (c) Vesicles showing the endocytosis of heat-inactivated FVs. (d) Heat-inactivated FVs (HCFVs) were bound (four particles) to the plasma membrane in the presence of cytoB. (e) Magnified view of the bound HCFVs in panel d, showing the labeling of gold particles. Images are typical of three experiments. The arrows indicate representative areas of RITC-L localization.
Membrane fusion is accompanied by ERK1/2 phosphorylation.
It has been shown that ERK1/2 plays a critical role in viral infection (19, 26, 37, 42). Therefore, we evaluated the activation of the MAPK signaling pathways in HepG2 cells during interaction with FV. Western blot analysis showed that FV-HepG2 cell fusion led to a 6-fold increase in ERK1/2 activation (Fig. 2A, top panel, compare lanes 3 to 6 with lane 1). Heat-treated FV (HCFV) failed to activate ERK1/2 in both the presence and absence of cyto B (Fig. 2A, lanes 7 to 9), thus making this response specific to membrane fusion. Furthermore, fusion with FV was specifically associated with activation of ERK1/2, in a dose-dependent manner, but not with activation of JNK and p38 (Fig. 2A). The presence of HN in the virosome (HNFV) augmented ERK1/2 activation further (Fig. 2A, lane 10), to the level of intact Sendai virus (unpublished observation). The heat-treated HNFV (HCHNFV) and mutant (H247A mutant of HN) H247AHNFV displayed similar ERK1/2 activation profiles to those of HCFV and FV, respectively (data not shown), suggesting an association between membrane fusion and ERK1/2 activation. Since FV-induced pERK peaked 1 h after stimulation (Fig. 2B), in all subsequent experiments the assessment of host cell signaling was carried out after incubation with FVs for 1 hour. Serum, a well-known stimulator of the MAPK cascade (7), served as a positive control for ERK activation (Fig. 2A, lane 2, and B, lane 7).
FIG. 2.

Membrane fusion-associated activation of ERK in HepG2 cells. (A) Probing of fusion-associated MAPK pathways. After FV-, HCFV-, or HNFV-HepG2 cell fusion (dose dependent) under the indicated conditions, lysates were probed with anti-pERK, -pJNK, or -pp38 in Western blots, followed by stripping and reprobing with anti-ERK, -JNK, or -p38, respectively. The amount of lysate loaded is indicated. Relative fold activation (expressed as mean ± SD for three independent experiments) is represented graphically (densitometric analysis). (B) Kinetic profile of ERK activation during FV-HepG2 cell fusion upon incubation with 80 μg FV. (C) Assessment of functional activity of activated ERKs. The indicated lysates (from panel A) were assayed for MBP phosphorylation. Reaction mixtures were resolved by SDS-15% PAGE. The lower half of the gel was autoradiographed (middle panel) after CB staining (lower panel), and the upper half was incubated with anti-ERK to ensure equal loading (upper panel). The graph indicates fold activation (expressed as mean ± SD for three independent experiments) of MBP phosphorylation (lanes 3 to 10). MBP bands were excised and radioactivity was measured. (D) Detection of pERK, pJNK, and pp38 levels during FV-HepG2 cell fusion. Fused cells were fixed, stained for pERK (rows 1 and 2), pJNK (row 3), or pp38 (row 4) with FITC-labeled secondary antibody (green), and merged with RITC-L (red) delivered to the cytosol after FV fusion. Nuclei were stained with DAPI (blue). Bar, 15.59 μm. (E) Raf-1 activation in cells after fusion with FV, HCFV, HNFV, or (H247A)HNFV was checked by anti-pRaf-1 Western analysis (upper panel), followed by stripping and reprobing with anti-cRaf (lower panel). (F) EMSA was performed with nuclear extracts to detect AP-1 activation. Lane 8 was loaded with a 50-fold molar excess of cold competitor during DNA-protein interaction.
In order to confirm the functional activation of ERKs in cellular extracts, an in vitro MBP phosphorylation assay was performed. Similar to elevated pERK levels ex vivo, we observed 6- and 8-fold elevations in MBP phosphorylation by the extracts from HepG2 cells after their fusion with FV and HNFV, respectively (Fig. 2C, middle panel, lanes 4, 5, and 8). Fusion-associated ERK1/2 activation was further confirmed microscopically by the merging of pERK staining (green) and FV-delivered cytosolic RITC-L (Fig. 2D, rows 1 and 2), as similar merging was not observed in the case of HCFV. Specific activation of pERK was further confirmed by the absence of such merging in the cases of pp38 and pJNK (Fig. 2D, rows 3 and 4). The Raf-1 activation profile, as evidenced by phosphorylation of Raf-1, matched with that of ERK1/2 activation, suggesting a role of Raf-1 in FV-induced ERK activation (Fig. 2E). Since MAPK activation leads to AP-1 activation (24), we tested the FV-induced AP-1 binding activity in the HepG2 cells. Fusion with HNFV was accompanied by 2-fold higher levels of AP-1 complex formation (as shown by EMSA) than those of FV- and (H247A)HNFV-induced complex formation (Fig. 2F, lane 10 versus lanes 5 and 9), suggesting a close association of fusion with the ERK/AP-1 axis.
Phosphorylation of ERK1 is required for fusion.
In order to understand the role of ERK activation in FV fusion, we attempted to examine their causal relationship both by pretreating HepG2 cells with PD98059 (MEK1 inhibitor) and by overexpressing the dominant-negative mutant of ERK1 (DN) in HepG2 cells. In both cases, there was a significant decrease in ERK1 phosphorylation (Fig. 3C). Fusion-mediated delivery of RITC-L to the target cells was also drastically impaired under both conditions (ca. 4-fold reduction of fusion) (Fig. 3A and B). The reduced fusion efficiencies of HNFV and (H247A)HNFV reflected a similar pattern to that of FV (data not shown). In contrast, overexpression of constitutively active MEK-1 (pEBG-MEK1S218E/S222E) (48), which resulted in increased ERK1/2 activation (Fig. 3F and G), markedly increased the transfer of RITC-L from FV to the target cells (Fig. 3D, middle and lower panels, and E). Similar results to those obtained with FV were obtained with HNFV and (H247A)HNFV (data not shown). These results were complemented by abrogation of infection of HepG2 cells by Sendai virus in the presence of PD98059 or DN (unpublished observation). Our results thus suggest that activation of ERK1 regulates FV-HepG2 fusion.
FIG. 3.

Activation of ERK1 is required for membrane fusion. (A to C) Inhibition of cellular ERKs abrogates fusion. (A) Fusion of 80 μg FV with HepG2 cells pretreated with the MEK-1 inhibitor PD98059 (PD) or transfected with the dominant-negative ERK-1 expression vector (DN) is shown. Note that RITC-L (red) in the cytosol indicates a fusion event. Results are graphically represented in panel B as percentages of fused cells. Error bars indicate SD values for three independent experiments. (C) Corresponding cell lysates were analyzed for pERK levels by Western blotting. The same blot was stripped and reprobed with anti-ERK to ensure equal loading. (D) Constitutively active ERK/MAPK pathway increases RITC-L transfer. The images show fusion of FVs with cells transfected with an expression vector for constitutively active MEK-1 (CA MEK-1, GST tagged). Immunocytochemical analysis shows fused cells stained for GST (green; FITC-labeled secondary antibody) and RITC-L (red) delivered to the cytosol (middle and lower panels). The upper panel represents fusion in nontransfected cells. Results are graphically represented in panel E as percentages of fused cells. Error bars indicate SD values for three independent experiments. (F and G) Effect of constitutively active MEK-1 expression on pERK levels in HepG2 cells. HepG2 cells growing in DMEM were transfected with either wt MEK-1 or CA MEK-1 by use of Lipofectamine reagent. Colocalization of CA MEK-1 (anti-GST and FITC-labeled secondary antibody; green) with increased cellular pERK levels (red) confirms constitutive ERK activation in these cells (lower panel). In the case of wt MEK-1 expression, no such colocalization was observed (upper panel). (G) An immunoblot of lysates from cells transfected with either wt or CA MEK-1 was probed with anti-GST, anti-pERK1/2, and anti-ERK1, as indicated. The same blot was stripped and reprobed. Images were captured by confocal microscopy. Bars, 23.81 μm (A), 15.59 μm (D), and 10 μm (F).
Inhibition of AKT activity enhances membrane fusion.
Next, we sought to investigate the involvement of any other signaling pathway in the event of fusion/entry. HepG2 cells were pretreated with chemical inhibitors of various signaling cascades. Interestingly, only pretreatment of HepG2 cells with a potent chemical inhibitor of AKT (IAKT) enhanced fusion with FV, by 4-fold, compared to that observed in cells without IAKT (Fig. 4A, row 5, and B). In contrast, inhibition of other pathways, viz, PKA, p38, and JNK pathways, or use of a solvent control (DMSO) had no effect on FV-cell fusion. It is important that the exposure times of virosomes to cells were the same in all pretreatments with inhibitors. The specificity of inhibition of these pathways was further confirmed by immunoblot analysis of the cell lysates used in Fig. 4A (Fig. 4C to F). AKT or protein kinase B (PKB) is a key regulator of the PI3K signaling pathway and plays a crucial role in many cellular processes (13) as well as in replication of negative-strand nonsegmented RNA viruses (33, 51). Therefore, we further examined the role of AKT activity in FV-HepG2 cell fusion. We subsequently observed a reciprocal relationship between the levels of pAKT and pERK, as shown in Fig. 4G. The negative effect of AKT on fusion was further substantiated by the observation that overexpression of myristoylated AKT1 (myrAKT1), which is membrane anchored and thus constitutively activates PKB signaling, resulted in 5-fold reduced fusion, whereas inhibition of AKT1 expression by its dominant-negative mutant, DNAKT1, enhanced fusion ∼4-fold in comparison to FV-HepG2 cell fusion without DNAKT1 (Fig. 5A and B). The effects of expression of myrAKT1 and DNAKT1 on the accumulation of pAKT were confirmed in immunoblots (Fig. 5C and D). Moreover, the effect of DNAKT1 on FV-cell fusion was validated microscopically by merging of the HA tag (green) and RITC-L fluorescence (Fig. 5E), based on comparison with its effect on HCFV-cell interaction. Another critical piece of evidence of the inhibitory effect of AKT1 on fusion was demonstrated by a sensitive online fluorescence dequenching assay of FV-cell fusion (Fig. 5F). Both IAKT-pretreated cells and DNAKT1-expressing cells showed faster kinetics and a greater extent of fusion than did untreated cells. Interestingly, PD98059 pretreatment did not inhibit the rate and extent of fusion.
FIG. 4.

Effects of various signaling inhibitors on membrane fusion. (A) FV-HepG2 cell fusion was evaluated in the presence or absence of p38, PKA, JNK, and AKT inhibitors. (B) Fusion was assayed by cytosolic RITC-L delivery, and the percentage of fused cells is represented graphically. Error bars indicate SD values for three independent experiments. (C to F) Effects of inhibitors were confirmed by Western analysis. (G) Assessment of pERK levels in IAKT-pretreated cells. Lysates from IAKT-pretreated HepG2 cells were probed with anti-pERK, -ERK, -pAKT, and -AKT in the same Western blot after stripping and reprobing, in the order given. Images were captured by confocal microscopy. Bars, 23.81 μm.
FIG. 5.

Inhibition of AKT activity enhances membrane fusion. (A to E) Effect of dominant-negative (DNAKT1) and constitutively active (myrAKT1) AKT1 expression on membrane fusion. (A) HepG2 cells transfected with HA-DNAKT1 or Flag-myrAKT1 (pcDNA m2-Myr-AKT) expression vector were allowed to fuse with FV. (B) Percentages of fused cells are represented graphically. Western analyses of lysates from DNAKT1 (C)- and myr-AKT1 (D)-transfected cells are shown. Reprobing with anti-AKT ensured equal loading. (E) Merging of DNAKT1 expression profile with fusion. Immunofluorescence analysis shows HepG2 cells fused with FV and stained for HA-DNAKT1 (HA tag, using FITC-labeled secondary antibodies; green) and RITC-L (red). (F) Effects of AKT and ERK inhibition on kinetics of FV-HepG2 cell fusion. Ten micrograms of NBD-PE-labeled virosomes was incubated with PD98059- or IAKT-pretreated cells (1 × 106) in 1 ml ice-cold DPBS containing 1.5 mM Ca2+. Virosome-cell fusion was recorded as described in the text. Images were captured by confocal microscopy. Bars, 23.81 μm (A) and 15.59 μm (E).
In order to understand the mechanism of regulation of AKT1 in HepG2 cells during fusion, we tested the effects of wortmannin, a potent inhibitor of PI3K, on fusion with FV. Wortmannin-treated cells exhibited complete inhibition of PI3K activity (Fig. 6B and C) and showed similar effects on fusion to those observed with IAKT- and DNAKT1-treated cells (Fig. 6A).
FIG. 6.
Effect of PI3K inhibition on fusion of FV. (A) Serum-starved HepG2 cells were pretreated with wortmannin (100 nM) and cytoB (10 μM), followed by 1 h of incubation with 80 μg FV (RITC-L loaded). Images were captured with a confocal microscope. Virosome-cell fusion was assessed by RITC-L delivery into the cytosol. (B) Corresponding cell lysates were subjected to SDS-10% PAGE, followed by Western blotting using anti-phospho-AKT antibody (upper panel). The same blot was stripped and reprobed with anti-AKT antibody (lower panel). (C) HepG2 cells were also assayed for PI3K activity after similar treatments. Cells were lysed and equal amounts of protein (lower panel) were used for immunoprecipitation with mouse monoclonal anti-phosphotyrosine antibodies, and the immunocomplexes were assayed for the ability to phosphorylate PI to PIP as described in the text. Bar, 23.81 μm.
HN affects fusion by inhibition of AKT-dependent F phosphorylation.
Our results support the notion that successful membrane fusion of FV with HepG2 cells requires AKT inhibition. To study the mechanism of regulation, we used a bioinformatic approach to screen for potential AKT-interacting domains in F protein. A motif search by the Motif Scan program (http://scansite.mit.edu) under high-stringency conditions resulted in three potential kinase sites in the F protein (Fig. 7), among which Thr 234 was identified as a putative strong AKT phosphorylation site. The Motif Scan graphic results (Fig. 7), though, indicated T234 as the only potential AKT kinase site in F protein; it also displayed T272 (in PKC epsilon) and Y316 (in Itl kinase, a tyrosine kinase present in T cells) as other non-AKT phosphorylation sites. In order to establish the role of the T234 residue in fusion, we constructed three different mutants of F protein (F-T234A, F-T272A, and F-Y316A). Residues T272 and Y316 were mutated to serve as nonspecific controls. Cell surface expression of all three mutants and wt F was found to be similar (Fig. 8 and Table 1). Subsequently, F protein (wt or mutant), either alone or together with HN (wt or mutant), was coexpressed with DsRed-N1 in Huh-7 cells, which were then used for fusion with HepG2 cells expressing EGFP (target cells), followed by scoring of the fused yellow cells (Fig. 9A and B). A remarkable enhancement of F-T234A fusion with untreated cells (ca. 2.5-fold more than that of wt F and the other two mutants) (Fig. 9A and C) or of wt F fusion with IAKT-pretreated cells (Fig. 9B, row 3) suggested an inhibitory effect of T234 phosphorylation on fusion. Despite the fact that HN- and F-coexpressing cells served as a positive control (Fig. 9A, top panels), the H247AHN- and F-coexpressing cells behaved like those expressing wt F alone (reduced fusion activity) (compare second row of Fig. 9A with bottom row of Fig. 9B), thereby confirming our earlier observation regarding the role of the H247 residue of HN protein in the activation of F-induced virosome-cell and cell-cell fusion (27). However, this effect was abrogated in the presence of IAKT, which significantly increased fusion with mutant HN (Fig. 9B, fourth and fifth rows). Importantly, inhibition of cell-cell fusion mediated by HN and wt F coexpression or by wt F alone in the presence of PD98059 (Fig. 9B, first and second rows, respectively) further supported the role of ERK1 in virosome-cell fusion, as discussed above.
FIG. 7.

Motif Scan results for F protein. The F protein sequence (GenBank accession no. BAD 74228) was included in a Motif Scan (http://scansite.mit.edu) search for a candidate AKT kinase site(s). As shown, one site (threonine 234) was identified as a potential site for AKT phosphorylation.
FIG. 8.
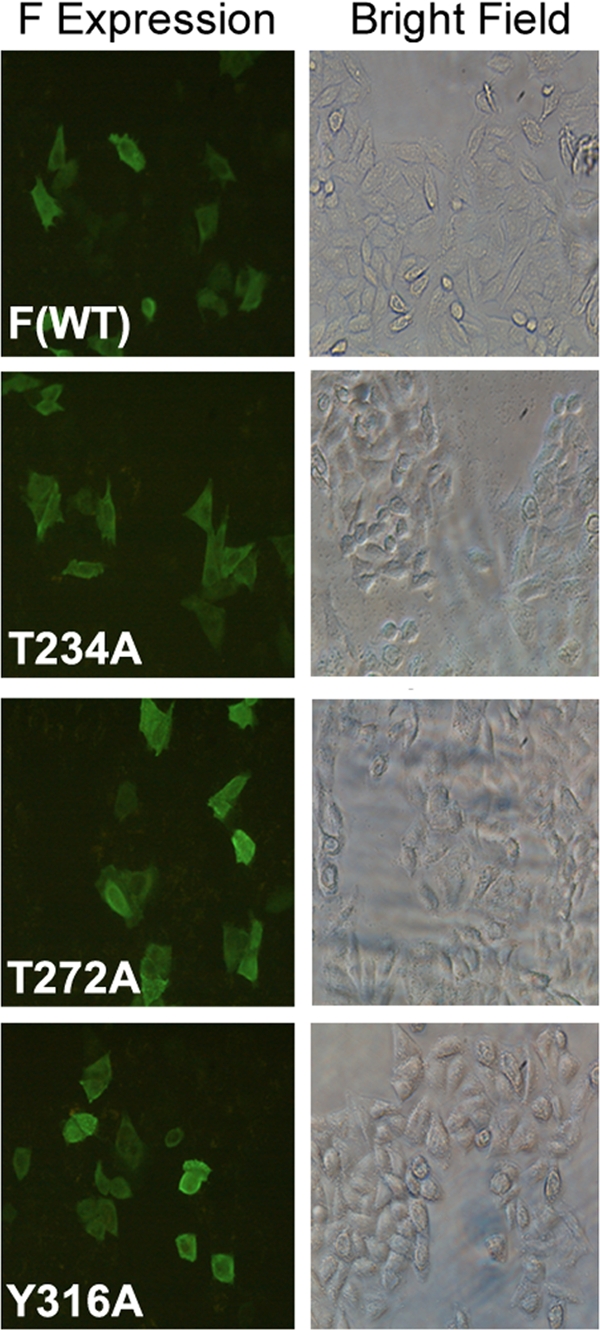
Expression of different F mutants. Cells were transfected with pcDNA 3.1-F plasmid encoding wild-type F or different F mutants (T272A, T234A, or Y316A) as described in Materials and Methods, and F expression was evaluated by immunocytochemistry using anti-F antibody followed by FITC-labeled secondary antibody.
TABLE 1.
Surface expression of F proteins
| Protein | Relative expressiona | Relative MFIb |
|---|---|---|
| F (wt) | 1.0 | 1.0 |
| T272A | 0.95 | 0.87 |
| T234A | 0.92 | 0.84 |
| Y316A | 0.98 | 0.85 |
Relative expression refers to the number of positive cells in each sample, normalized with respect to wild-type F.
Relative mean fluorescence intensity (MFI) denotes the amount of fluorescence a given particle population carries, normalized to wild-type F.
FIG. 9.

Mutation of F and HN proteins affects fusion. (A) Cell-cell fusion of different F mutant-expressing cells was monitored by content mixing. Images were captured with an epifluorescence microscope. Cell-cell fusion of cells with HN and wt F cotransfection served as a positive control. (B) Effect of PD98059 or IAKT on cell-cell fusion of cells with both HN and F proteins (first row) and only F protein (second and third rows), determined by overlaying target cells as described in the text. The last two rows indicate the effect of mutation in HN (H247A) on cell-cell fusion in the absence or presence of IAKT pretreatment of overlaid cells. (C) Percent fusion activity was scored relative to wild-type HN fusion activity. Error bars indicate SD values for three independent experiments. (D) Effect of mutation in HN or F on virosome-cell fusion. Virosomes (80 μg) with the indicated mutations in F or HN were tested for fusion with HepG2 cells. The third and fourth rows indicate the effect of mutation in HN (H247A) on virosome-cell fusion in the absence or presence of IAKT pretreatment. (E) Cell lysates (from panel D) were probed with anti-pAKT and anti-AKT in Western blots and graphically represented to show the effect of HN contact on AKT activity. (F) AKT phosphorylates F protein. The indicated virosomes were fused with cytoB- and/or IAKT-pretreated HepG2 cells. Anti-F (lanes 1 to 7 and 9) and anti-Sendai virus (lane 8) immunoprecipitates were probed with anti-F, followed by stripping and anti-phosphothreonine Western analysis to detect F phosphorylation (confirmed by a band corresponding to the 45-kDa F1 fragment). The same band showed threonine phosphorylation after immunoblotting with anti-phosphothreonine. The third panel indicates equal loading, and the bottom panel shows Ponceau staining. (G) Phosphorylation of F is fusion dependent. Cells expressing F (wt and T234A mutant; lanes 1 and 2, respectively) were immunoprecipitated with anti-F (middle panel), followed by stripping and immunoblotting by anti-phosphothreonine (top panel) to detect F phosphorylation. FV fused with HepG2 cells served as a positive control for F phosphorylation (lane 3). ERK1/2 served as a loading control (bottom 3). Bars, 100 μm (A and B) and 23.81 μm (D).
Additionally, we also observed a similar effect of mutant F and HN on virosome-cell fusion by using corresponding RITC-L-loaded FV and HNFV (Fig. 9D). Similar to the case for cell-cell fusion, a significant enhancement of FV-HepG2 cell fusion was observed in the case of T234A (but not T272A) mutant F in the absence of HN (Fig. 9D, compare rows 5 and 7), as well as by inhibition of AKT1 in the case of (H247A)HNFV (Fig. 9D, row 4), supporting the crucial role of T234 and AKT1 in regulating fusion. Importantly, enhanced phosphothreonine signals of F in the case of either (H247A)HNFV or FV alone (Fig. 9F, topmost blot, lanes 2 and 7) further confirmed phosphorylation of F during fusion. These results were also supported by corresponding reductions (∼3-fold) in pAKT levels (Fig. 9E, bars 4 and 5) and phosphothreonine signals of F (Fig. 9F, topmost blot, lanes 3 and 8) in the presence of (H247A)HNFV and IAKT together or of HNFV alone. It may be noted here that (T234A)F (fused with HepG2 cells) and nonfused wild-type F (both expressed in chick embryonic cells) were not phosphorylated (Fig. 9F, lanes 1 and 10). Furthermore, the HepG2 cell-expressed F proteins (both wt and T234A mutant) were immunoprecipitated with anti-F antibody, followed by Western blotting with anti-phosphothreonine antibody. Neither of the two proteins showed any phosphorylation signal (compared to the FV fused to HepG2 cells), thus indicating that the phosphorylation of F (wt) protein is a fusion-associated phenomenon (Fig. 9G, compare lanes 1 and 2 with lane 3). This also rules out the possibility of host cell-expressed wild-type F protein being phosphorylated by AKT1.
Notably, a significant reduction in the phosphorylation of AKT1 as well as of F in the case of HNFV but not (H247A)HNFV suggests that HN protein (specifically the H247 residue in HN) regulates F-induced virosome-cell and cell-cell fusion, mainly by diminishing pAKT levels and subsequent F activation. Therefore, we envisage the reduction of pAKT levels by HN contact to be the crucial event in the case of replication-competent intact Sendai virus during entry into the cell.
Transient actin depolymerization supports membrane fusion and is regulated by MAPKs.
While standardizing the concentration of cytoB to be used to specifically inhibit endocytosis and not have any pleiotropic effects on the cells, we repeatedly observed increased fusion of FVs with HepG2 cells in the presence of 20 μM cytoB, even though the fusogenic potential of the batch of FVs used was no different from that of earlier batches, as assessed by their hemolytic activity (data not shown). CytoB (20 μM) is also known to act as an actin-depolymerizing agent (36), in addition to being an endocytosis inhibitor. However, no significant difference in binding of FV and HCFV with HepG2 cells in the presence or absence of cytoB (20 μM) was observed (data not shown). This may support the notion that cytoB pretreatment could not affect the binding process. In fact, phalloidin staining showed extensive actin depolymerization in HepG2 cells at this concentration of cytoB (Fig. 10A, rows 5 and 6). Thus, we hypothesized that actin depolymerization caused by cytoB (20 μM) might support fusion by removing the barrier of the actin network and may be controlled by MAPK signaling during FV-HepG2 cell fusion. It is known that cytoskeletal remodeling is the final step of membrane fusion (15). Moreover, cell-cell fusion induced by Sendai virus has also been shown to be associated with cytoskeletal remodeling (21). Using established actin-remodeling agents (cytoB and JASP), we attempted to unfold the molecular mechanism of ERK/MAPK-mediated enhancement of F-induced fusion of the Sendai viral envelope with HepG2 cells. As shown in Fig. 10, pretreatment of cells with 20 μM cytoB resulted in extensive actin depolymerization, with concomitant enhancement of FV-cell fusion (ca. 4-fold) (Fig. 10A, row 5, and B), compared to that in untreated cells (Fig. 10A and B, rows with control, FV alone, and FV in the presence of 10 μM cytoB, which inhibits endocytosis only). Notably, pretreatment of cells with 20 μM cytoB reversed the partial inhibition of fusion by 70 μM PD98059 (Fig. 10A, rows 4 and 6). On the other hand, inhibition of pAKT (also a modulator of actin [54]) again resulted in enhanced fusion (Fig. 10A, row 7) that was neutralized in the presence of PD98059 (Fig. 10A, row 8). A state of polymerized actin by pretreatment of HepG2 cells with 0.3 μM JASP was established. This observation was based on our standardization by a microscopic analysis following a published protocol (9). Treatment with 0.3 μM JASP showed a complete inhibition of fusion, with a concomitant increase in the polymerized form of actin (Fig. 10A, row 9). To confirm this relationship between actin depolymerization and fusion, we also used latrunculin A (another known actin-depolymerizing agent) in a parallel experiment and obtained similar results (data not shown). MAPK activation is known to regulate actin remodeling through an AP-1-induced target gene product, ezrin (40, 53). We thus studied the possibility of any effect of activation of ERK1 and AP-1, followed by fusion, on ezrin activity (Fig. 10C to E). Fusion of FVs clearly indicated an increase in ezrin expression (∼3-fold) (Fig. 10E) and its phosphorylation (∼3-fold) (Fig. 10D) over basal levels. Also, suppression of endogenous ezrin by using ezrin-specific siRNA in the target cells completely abolished FV-cell fusion (Fig. 10C, upper panels) compared to that in the presence of the nonspecific siRNA control (Fig. 10C, lower panels). Similarly, (T234A)FV, HNFV, and (H247A)HNFV could not fuse with the ezrin siRNA-expressing target cells (data not shown).
FIG. 10.

Effect of actin depolymerization on FV-cell fusion. (A) FVs (80 μg) were fused with HepG2 cells pretreated with the indicated doses of cytoB, IAKT, PD98059, and JASP alone or in combination. Fluorescence analysis is shown for fused cells stained for RITC-L (red) and F-actin (green; Bodipy FL-phallacidin). Nuclei were counterstained with DAPI. Tailed arrows and arrowheads represent actin depolymerization and polymerization, respectively, in the case of cytoB (20 μM) and JASP pretreatments. Arrowheads in the red channel in the last row show virosomes bound to the plasma membrane. The phallacidin (M) panels denote magnified images. (B) Percentages of fused cells are represented graphically. Error bars indicate SD values for three independent experiments. (C to E) Ezrin profile during membrane fusion. (C) Ablation of endogenous ezrin inhibits fusion. HepG2 cells were transfected with ezrin-specific siRNA (ezrin siRNA) or control siRNA (NS siRNA), and fusion with FV was analyzed. (D) Levels of phospho-ezrin, ezrin, and ERK (equal loading) were checked in lysates from HepG2 cells with the indicated treatments by Western blotting. (E) Fusion enhances ezrin mRNA levels. RT-PCR analysis of ezrin mRNA from HepG2 cells undergoing the indicated treatments and fusion with FV showed a product of 565 bp for the ezrin gene, which was visualized by ethidium bromide staining. β-Actin was included for normalization (specific 650-bp product). Densitometric quantitation of the bands depicts relative amounts of ezrin mRNA among different samples, with the NS siRNA-treated sample value set at 1. Error bars indicate SD values for three independent experiments. (F) Effect of JASP or PD98059 treatment on kinetics of FV-HepG2 cell fusion. Ten micrograms of NBD-PE-labeled virosomes was incubated with JASP (0.3 μM)- or PD98059 (70 μM)-pretreated HepG2 cells (1 × 106) in 1 ml ice-cold DPBS containing 1.5 mM Ca2+. Virosome-cell fusion was recorded as described in the text. (G) Western profiles (for the cells used for panel F) showing pERK levels in JASP- and PD98059-pretreated HepG2 cells after fusion with FV. Images were captured by confocal microscopy. (H) Effect of JASP on F phosphorylation. Following fusion (in the absence or presence of JASP), FV-HepG2 cell lysates were immunoprecipitated with anti-F (middle panel), followed by immunoblotting with anti-phosphothreonine antibody (top panel) to detect phosphorylation of F protein (lanes 1 and 2). ERK1/2 served as a loading control (bottom panel). Bars, 23.81 μm.
Figure 10A, row 9, shows that inhibition of depolymerization of actin by JASP (a specific inhibitor of actin depolymerization) does not allow the establishment of an aqueous fusion pore (core mixing). Having this information, we performed a membrane-mixing (hemifusion) assay under such conditions. It was apparent that kinetics of lipid mixing were more or less similar and were not affected by the presence of JASP. As shown in Fig. 10A, row 9, JASP inhibited core mixing, but its failure to affect lipid mixing (Fig. 10F) clearly indicates the establishment of a hemifusion state of the JASP-pretreated target cells (HepG2). The binding of FV and HCFV with HepG2 cells in the presence of 0.3 μM JASP was found to be unaffected (data not shown). Furthermore, these hemifused cells were evaluated for their pERK levels, and it was concluded that the hemifusion state potentially activated similar pERK levels to those with complete membrane fusion (Fig. 10G, compare lanes 2 and 3). Altogether, these results show that elevation of pERK levels in response to hemifusion leads to activation of ezrin through AP-1, which supports viral entry by bringing about actin remodeling at the cell cortex. Nevertheless, a significant phosphorylation of F in the absence of JASP (but not in its presence) indicated a possibility that pore opening is a prerequisite for phosphorylation of F protein (Fig. 10H, compare lanes 1 and 2).
Membrane fusion triggers ceramide synthesis and activates PP2A.
Ceramides are a novel class of lipid second messengers that originate from membrane sphingomyelin (produced by the hydrolysis of sphingomyelin in response to a variety of stress-related stimuli) and regulate Ras-Raf-MAPK cascades (22). Therefore, to address the molecular mechanism underlying the upstream activation of ERK1 by F-protein-induced membrane fusion, we studied the cellular levels of ceramide and the activity of its immediate downstream effector PP2A during FV-HepG2 cell fusion (Fig. 11A to C). An appreciable, 2.5-fold activation of ceramide synthesis over the basal level (Fig. 11A) corroborated with a 2-fold increase in PP2A activity (during fusion) (Fig. 11B). Interestingly, inhibition of PP2A activity by OKA (a PP2A-specific inhibitor when used at low concentrations) completely inhibited FV-HepG2 cell fusion (Fig. 11C, top panels) compared to that of the respective controls (Fig. 11C, second and bottom rows). This effect was not observed with TAU, a PP1 inhibitor, and indicated an important role of PP2A in regulating fusion. The fusion of HNFV, (H247A)HNFV, and (T234A)FV with target cells was similar to that of FV in inducing ceramide production and PP2A activity (data not shown).
FIG. 11.

Effect of fusion on ceramide synthesis and phosphatases. (A) FV or HCFV was incubated with HepG2 cells pretreated with cytoB (10 μM). Lysed cells were subjected to immunoprecipitation with anti-ceramide antibody. Ceramide immunocomplexes were resolved by TLC and visualized by DAB staining. The aligned bar diagram shows fold activation of ceramide synthesis. Error bars indicate SD values for three independent experiments. (B) Lysates prepared from HepG2 cells pretreated with OKA or TAU and fused with FV were used to assess PP2A activity as described in the text, which is expressed as specific activity in the form of a bar diagram, with SD values shown for three independent experiments. (C) Serum-starved HepG2 cells were incubated with 2 nM OKA, 3 nM TAU, or ethanol (EtOH; solvent control) for 1 h, followed by incubation with 80 μg FV. Cells were washed and images were captured by confocal microscopy. Bars, 23.81 μm.
DISCUSSION
Viral infection is known to modulate host cell signaling (19, 26, 33, 37, 42, 51), but the initial signaling arising from viral fusion that eventuates in either a successful or abortive entry remains to be established. This study was aimed at addressing this issue by employing reconstituted Sendai viral envelope having only F protein (FV). Since it is derived from intact virus, FV represents the best physiological model with which to study host-virus interaction. However, we also retained the HN protein throughout our studies to see (and compare) the status of signaling during fusion with the natural fusion machinery (F-HN complex). We demonstrated that specific activation of a host mitogenic cascade, the Raf/ERK/MAPK axis, occurs in response to membrane fusion. The fusion of HNFV with the host cell and infection of HepG2 cells with Sendai virus also resulted in a similar activation of ERK. Moreover, inhibition of fusion as well as underlying signaling events in response to heat-treated FVs (known to retain their binding to the ASGPR) (4) further supported the observation that besides undergoing efficient recycling, ASGPR-carbohydrate interactions failed to exert any signal transduction in hepatocytes (25). Mitogenic stimulation appears to be regulatory for effective fusion, as preactivation of ERK1 augmented, whereas ERK1 inhibition substantially reduced, the fusion efficiency. In contrast, both fluorescence dequenching and transfer of RITC-L to assay lipid merging and cytosolic core mixing as respective markers of fusion revealed an unexpected inhibitory role of AKT kinase in membrane fusion. However, pretreatment with PD98059 alone could not inhibit the lipid mixing, suggesting an intermediate “hemifusion” state which, acting as a stress factor, induces the MAPK pathway. This speculation may be supported by the notion that complete fusion takes place through several metastable stages of activation of F protein (31). The fact that JASP inhibits core mixing but not lipid mixing clearly indicates the establishment of a hemifusion state of the JASP-pretreated target cells (HepG2 cells). Furthermore, these hemifused cells were evaluated for pERK levels, and it was concluded that the hemifusion state potentially activated similar pERK levels to those with complete membrane fusion. Altogether, these data may be taken as additional support for our proposition that a hemifusion state induced by F protein in HepG2 cells acts as a stress factor leading to the induction of pERK.
We investigated the mechanisms of the counteracting roles of pAKT1 and MAPKs in the fusion process. Using a series of cell-cell and virosome-cell fusion experiments, we concluded that pAKT1 significantly reduces fusion efficiency by enhancing phosphorylation of F protein, most likely at the T234 residue. Our claim is further supported by the enhancement of F (alone)-induced cell-cell fusion by IAKT and its inhibition by PD98059. Interestingly, the failure of H247AHN to activate F-induced cell-cell fusion (27) could be rectified by the presence of IAKT, suggesting an alternate and novel role of HN in supporting fusion through AKT inhibition, in addition to inducing a conformational change (10) in F protein. Although H247AHN could bind the target cells through the sialic acid receptor and was enzymatically active (27), it failed to inactivate pAKT. In contrast, binding of HN impaired the pAKT level, indicating that the contact of HN with target cells deactivates AKT, as previously observed with measles virus (2). This observation confirmed the hypothesis that a histidine residue of HN acts as a trigger for F-induced fusion (27) by inhibiting AKT signaling. Thus, we predicted that the contact of HN with host cells during viral infection diminishes the pAKT level and might prevent phosphorylation of F protein. An indirect proof of phosphorylation of F protein in the fused cells in the absence of active HN contact and IAKT pretreatment endorses our interpretation of pAKT-induced F inactivation and subsequent inhibition of both cell-cell and virosome-cell fusion. However, direct biochemical/biophysical evidence(s) in support of phosphorylation of F residue T234 and its effect on fusion and data showing that the mutation of the T234 residue does not change the overall conformation of the protein, inhibiting the phosphorylation of another residue, have yet to be investigated. Yet the Motif Scan search detected T234 as the only potential phosphorylation site (Fig. 7) and our results showed that mutation at the 234th residue in F protein did not change the fusion activity of FVs (in comparison to FVs containing wild-type F protein), as confirmed by hemolytic assay of fusion (unpublished observation). Nevertheless, such a motif search failed to pick up any probable phosphorylation site in the cytoplasmic domain of F protein. Therefore, the possibility that a conformational change in F protein at the 234th residue is associated with enhanced fusion and decreased phosphorylation at another residue is less likely but needs to be ascertained clearly. The current working model of membrane fusion induced by viral class I fusion glycoproteins includes membrane contact, membrane merger (hemifusion), and the opening of an aqueous fusion pore (complete fusion; specifically, a transition between the fusion pore [FP] and a solute-permissive fusion pore [FPs]) (6, 23). It was recently demonstrated that the fusion peptide of parainfluenza virus 5 F (positioned at the immediate vicinity of heptad repeat A [HRA]) is sandwiched between two subunits of the trimer DII and DIII domains in its metastable/prefusion conformation (59). Furthermore, it is known that the DIII domain (containing the T234 residue) gives the core structure of the globular head in the prefusion conformation around which HRA folds and thus functions in a prefusion/metastable coiled-coil form of the HRA-DIII complex. Moreover, in general, viral fusion proteins such as Sendai virus F and gp41 of HIV, which trigger fusion at neutral pH, remain resistant to proteases during fusion (14, 31). This protease-resistant property of the six-helix bundle form is indicative of insertion of the HRA-DIII complex into a nonbilayer lipid intermediate during hemifusion (prefusion state). Additional support for this notion may be drawn from a model proposed by us for influenza virus HA (analogous to Sendai virus F protein [47])-mediated fusion in which the central trimeric coiled-coil domain establishes close contacts with the nonbilayer lipid phase of the viral membrane and target cell membrane during hemifusion (6). Keeping with this view and the fact that activated AKT (pAKT) remains associated with the lipid bilayer of the plasma membrane of host cells (12), our speculation that T234 of F protein is phosphorylated by membrane-associated pAKT (rather than cytosolic pAKT) may fit well. However, it may be important that none of these models of membrane fusion by type I fusion protein predicts exposure of T234 to the cytoplasmic surface of the plasma membrane, where pAKT is located. The signaling via AKT might confer a host defense response through phosphorylation and inactivation of F protein, which is counterbalanced by a contemporaneous activation of MAPK cascades. Thus, the reciprocity between these two signaling pathways plays a decisive role in viral fusion with the host cells (Fig. 12). These observations imply that paramyxovirus-resistant host cells have an elevated pAKT level but less ERK activation and that differential Ras activation modulates the permissibility to viral fusion (16, 17, 49). Thus, our observations establish a new framework for redefining the host cell susceptibility or resistance to a given virus and thereby reveal a novel therapeutic strategy. In that process, the Sendai virosomes may serve as an efficient tool for targeted delivery of drugs.
FIG. 12.

Model for membrane fusion-induced host cell signaling. The reciprocal relationship between AKT1 and the ERK/MAPK pathway during HNFV-HepG2 cell fusion is simplified in this schematic model. The impairment of AKT activation by HN protein contact with target cells (through terminal sialic acid [TSA] receptors of HepG2 cells) prevents the phosphorylation of F protein and allows complete fusion (hemifusion and core mixing) catalyzed by F protein (with F bound to ASGPR of HepG2 cells). Meanwhile, stress during the hemifusion state (lipid mixing) induces the ERK/MAPK pathway through ceramide synthesis, which in turn supports membrane fusion through ezrin-induced actin depolymerization. Ceramide-activated PP2A seems to lie at the crossing of two opposing pathways, with one supporting the fusion event (Raf/ERK/MAPK) and the other inhibiting it (AKT).
Actin depolymerization is implicated in HIV fusion with its host cell and in secretory granule fusion with the cell membrane (60, 61). Therefore, we tested the association between cytoskeletal rearrangement and membrane fusion in our model of viral entry. The enhanced fusion of FV with HepG2 cells and its concomitant actin depolymerization in the presence of 20 μM cytoB, but inhibition by ERK1 inhibitors, support the role of ERK1-dependent cytoskeletal rearrangement in virus-cell fusion. We proposed that upon ERK1-independent lipid mixing, the slow process of core mixing required the transient removal of the actin barrier, which is regulated by AP-1/ezrin-directed cytoskeletal rearrangements. Ezrin, a known AP-1-induced target gene product, regulates membrane fusion via MAPK-regulated actin rearrangements (15, 40, 53). Ezrin association with the membrane and the associated actin network leads to remodeling of the actin barrier for productive fusion (8). The enhanced ezrin expression, concurrent increase in pEzrin level during successful progression of membrane fusion, and reduced fusion with the host cells expressing ezrin-specific siRNA validated this concept. The influence of HN and H247AHN in affecting ezrin upregulation and its activation provided more evidence of fusion-associated ezrin induction. Moreover, the role of H247 of HN in transmitting the signal emanating from HN contact with cell surface receptors and culminating in AKT inactivation supports the efficient fusion of intact Sendai virus and HNFV with liver cells.
Lastly, we attempted to delineate the events responsible for activation of Raf/MEK/ERK required for efficient viral fusion. Ceramide, a bioactive lipid produced under stress, is known to regulate cytoskeletal rearrangements and also to bind to and activate c-Raf, leading to the activation of MAPKs (22, 62). Moreover, PP2A is a ubiquitously expressed and ceramide-regulated phosphatase (11, 29) involved in various cellular processes, such as modulation of insulin signaling by direct dephosphorylation of AKT (1, 41, 44, 52). Thus, assuming fusion to be a “stressful” event, we monitored ceramide synthesis, and our results clearly indicated its synthesis, with a concomitant increase in PP2A activity, under lipid mixing stress. Complete inhibition of FV-cell fusion in the presence of OKA suggested a role for ceramide-activated PP2A in membrane fusion apart from ceramide-mediated Raf activation. It may be proposed that PP2A lies at the juncture of two opposing pathways, one supporting the fusion event (Raf/ERK/MAPK) and the other inhibiting it (AKT), where PP2A might play a critical role in regulating fusion by dephosphorylating pAKT. Additionally, these results also validated our earlier proposition of factor “X” (46), a protein or lipid factor associated with host cells which is required for efficient and biologically significant fusion of Sendai virus. Therefore, ceramide synthesis could be a potential candidate for the proposed factor “X,” showing a differential activation of Raf/MEK/ERK signaling during fusion.
Taken together, the results led to a proposed model (Fig. 12) to summarize our overall findings. Our study thus provides a unique discernment of host-virus interaction at the initial stage and demonstrates the involvement of cellular signaling machineries in the regulation of membrane fusion-mediated entry. This knowledge may help in developing safe and effective antiviral strategies, especially with the emergence of drug resistance due to structural alteration of viral components (19, 32, 58). Although we have successfully completed a preclinical study on bilirubin gene therapy in jaundiced rats by use of FV (57), an exploration of the present knowledge of modulation of host cell signaling in fusion enhancement in such a model is awaited.
Acknowledgments
This study was supported by the Department of Biotechnology, Government of India. N.R.S. thanks the Council of Scientific and Industrial Research, Government of India, for a senior research fellowship. Special financial support from the Delhi University is also gratefully acknowledged.
We thank Bhaskar Saha, Jayanta Roy-Chowdhury, Anu Puri, Jaharul Haque, Siddhartha Jana, Nisheeth Agarwal, Debashis Mitra, Suman Kundu, Siddhartha Ghosh, Priyanka Sharma, Anuja Krishnan, Suman Dhar, Neelam Sarkar, and Shyamal Goswami for many helpful discussions and critical reviews of the manuscript. We also thank Subhra Chakraborty, Confocal Microscope Facility, NIPGR, and Kanury V. Subba Rao, ICGEB, New Delhi, for help with image capturing.
Footnotes
Published ahead of print on 17 February 2010.
REFERENCES
- 1.Andjelkovic, N., S. Zolnierowicz, C. Van Hoof, J. Goris, and B. A. Hemmings. 1996. The catalytic subunit of protein phosphatase 2A associates with the translation termination factor eRF1. EMBO J. 15:7156-7167. [PMC free article] [PubMed] [Google Scholar]
- 2.Avota, E., A. Avots, S. Niewiesk, L. P. Kane, U. Bommhardt, V. ter Meulen, and S. Schneider-Schaulies. 2001. Disruption of Akt kinase activation is important for immunosuppression induced by measles virus. Nat. Med. 7:725-731. [DOI] [PubMed] [Google Scholar]
- 3.Bagai, S., A. Puri, R. Blumenthal, and D. P. Sarkar. 1993. Hemagglutinin-neuraminidase enhances F protein-mediated membrane fusion of reconstituted Sendai virus envelopes with cells. J. Virol. 67:3312-3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bagai, S., and D. P. Sarkar. 1994. Fusion-mediated microinjection of lysozyme into HepG2 cells through hemagglutinin neuraminidase-depleted Sendai virus envelopes. J. Biol. Chem. 269:1966-1972. [PubMed] [Google Scholar]
- 5.Bitzer, M., U. Lauer, C. Baumann, M. Spiegel, M. Gregor, and W. J. Neubert. 1997. Sendai virus efficiently infects cells via the asialoglycoprotein receptor and requires the presence of cleaved F0 precursor proteins for this alternative route of cell entry. J. Virol. 71:5481-5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blumenthal, R., D. P. Sarkar, S. Durell, D. E. Howard, and S. J. Morris. 1996. Dilation of the influenza hemagglutinin fusion pore revealed by the kinetics of individual cell-cell fusion events. J. Cell Biol. 135:63-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulton, T. G., S. H. Nye, D. J. Robbins, N. Y. Ip, E. Radziejewska, S. D. Morgenbesser, R. A. DePinho, N. Panayotatos, M. H. Cobb, and G. D. Yancopoulos. 1991. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65:663-675. [DOI] [PubMed] [Google Scholar]
- 8.Bretscher, A., K. Edwards, and R. G. Fehon. 2002. ERM proteins and merlin: integrators at the cell cortex. Nat. Rev. Mol. Cell. Biol. 3:586-599. [DOI] [PubMed] [Google Scholar]
- 9.Bubb, M. R., I. Spector, B. B. Beyer, and K. M. Fosen. 2000. Effects of jasplakinolide on the kinetics of actin polymerization. An explanation for certain in vivo observations. J. Biol. Chem. 275:5163-5170. [DOI] [PubMed] [Google Scholar]
- 10.Dallocchio, F., M. Tomasi, and T. Bellini. 1995. Activation of the Sendai virus fusion protein by receptor binding. Biochem. Biophys. Res. Commun. 208:36-41. [DOI] [PubMed] [Google Scholar]
- 11.Dobrowsky, R. T., and Y. A. Hannun. 1992. Ceramide stimulates a cytosolic protein phosphatase. J. Biol. Chem. 267:5048-5051. [PubMed] [Google Scholar]
- 12.Downward, J. 1998. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 10:262-267. [DOI] [PubMed] [Google Scholar]
- 13.Du, K., and P. N. Tsichlis. 2005. Regulation of the Akt kinase by interacting proteins. Oncogene 24:7401-7409. [DOI] [PubMed] [Google Scholar]
- 14.Dutch, R. E., R. N. Hagglund, M. A. Nagel, R. G. Paterson, and R. A. Lamb. 2001. Paramyxovirus fusion (F) protein: a conformational change on cleavage activation. Virology 281:138-150. [DOI] [PubMed] [Google Scholar]
- 15.Eitzen, G. 2003. Actin remodeling to facilitate membrane fusion. Biochim. Biophys. Acta 1641:175-181. [DOI] [PubMed] [Google Scholar]
- 16.Farassati, F., W. Pan, F. Yamoutpour, S. Henke, M. Piedra, S. Frahm, S. Al-Tawil, W. I. Mangrum, L. F. Parada, S. D. Rabkin, R. L. Martuza, and A. Kurtz. 2008. Ras signaling influences permissiveness of malignant peripheral nerve sheath tumor cells to oncolytic herpes. Am. J. Pathol. 173:1861-1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farassati, F., A. D. Yang, and P. W. Lee. 2001. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat. Cell Biol. 3:745-750. [DOI] [PubMed] [Google Scholar]
- 18.Gower, T. L., M. K. Pastey, M. E. Peeples, P. L. Collins, L. H. McCurdy, T. K. Hart, A. Guth, T. R. Johnson, and B. S. Graham. 2005. RhoA signaling is required for respiratory syncytial virus-induced syncytium formation and filamentous virion morphology. J. Virol. 79:5326-5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greber, U. F. 2002. Signalling in viral entry. Cell. Mol. Life Sci. 59:608-626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harmon, B., and L. Ratner. 2008. Induction of the Galpha(q) signaling cascade by the human immunodeficiency virus envelope is required for virus entry. J. Virol. 82:9191-9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirayama, E., M. Ama, K. Takahashi, A. Hiraki, and J. Kim. 2002. Changes in fused cells induced by hvj (Sendai virus): redistribution of cytoplasmic organelles and cytoskeletal reorganization. Cell Biol. Int. 26:347-353. [DOI] [PubMed] [Google Scholar]
- 22.Huwiler, A., J. Brunner, R. Hummel, M. Vervoordeldonk, S. Stabel, H. van den Bosch, and J. Pfeilschifter. 1996. Ceramide-binding and activation defines protein kinase c-Raf as a ceramide-activated protein kinase. Proc. Natl. Acad. Sci. USA 93:6959-6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jahn, R., T. Lang, and T. C. Sudhof. 2003. Membrane fusion. Cell 112:519-533. [DOI] [PubMed] [Google Scholar]
- 24.Karin, M. 1995. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270:16483-16486. [DOI] [PubMed] [Google Scholar]
- 25.Kim, S. H., J. H. Kim, and T. Akaike. 2003. Regulation of cell adhesion signaling by synthetic glycopolymer matrix in primary cultured hepatocyte. FEBS Lett. 553:433-439. [DOI] [PubMed] [Google Scholar]
- 26.Kong, X., H. San Juan, A. Behera, M. E. Peeples, J. Wu, R. F. Lockey, and S. S. Mohapatra. 2004. ERK-1/2 activity is required for efficient RSV infection. FEBS Lett. 559:33-38. [DOI] [PubMed] [Google Scholar]
- 27.Krishnan, A., S. K. Verma, P. Mani, R. Gupta, S. Kundu, and D. P. Sarkar. 2009. A histidine switch in hemagglutinin-neuraminidase triggers paramyxovirus-cell membrane fusion. J. Virol. 83:1727-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar, M., M. Q. Hassan, S. K. Tyagi, and D. P. Sarkar. 1997. A 45,000-M(r) glycoprotein in the Sendai virus envelope triggers virus-cell fusion. J. Virol. 71:6398-6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leoni, L. M., H. C. Shih, L. Deng, C. Tuey, G. Walter, D. A. Carson, and H. B. Cottam. 1998. Modulation of ceramide-activated protein phosphatase 2A activity by low molecular weight aromatic compounds. Biochem. Pharmacol. 55:1105-1111. [DOI] [PubMed] [Google Scholar]
- 30.Leyrer, S., M. Bitzer, U. Lauer, J. Kramer, W. J. Neubert, and R. Sedlmeier. 1998. Sendai virus-like particles devoid of haemagglutinin-neuraminidase protein infect cells via the human asialoglycoprotein receptor. J. Gen. Virol. 79:683-687. [DOI] [PubMed] [Google Scholar]
- 31.Ludwig, K., B. Baljinnyam, A. Herrmann, and C. Bottcher. 2003. The 3D structure of the fusion primed Sendai F-protein determined by electron cryomicroscopy. EMBO J. 22:3761-3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ludwig, S., O. Planz, S. Pleschka, and T. Wolff. 2003. Influenza-virus-induced signaling cascades: targets for antiviral therapy? Trends Mol. Med. 9:46-52. [DOI] [PubMed] [Google Scholar]
- 33.Luthra, P., D. Sun, M. Wolfgang, and B. He. 2008. AKT1-dependent activation of NF-kappaB by the L protein of parainfluenza virus 5. J. Virol. 82:10887-10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacLean-Fletcher, S., and T. D. Pollard. 1980. Mechanism of action of cytochalasin B on actin. Cell 20:329-341. [DOI] [PubMed] [Google Scholar]
- 35.Markwell, M. A., A. Portner, and A. L. Schwartz. 1985. An alternative route of infection for viruses: entry by means of the asialoglycoprotein receptor of a Sendai virus mutant lacking its attachment protein. Proc. Natl. Acad. Sci. USA 82:978-982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyake, K., P. L. McNeil, K. Suzuki, R. Tsunoda, and N. Sugai. 2001. An actin barrier to resealing. J. Cell Sci. 114:3487-3494. [DOI] [PubMed] [Google Scholar]
- 37.Naranatt, P. P., H. H. Krishnan, M. S. Smith, and B. Chandran. 2005. Kaposi's sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus. J. Virol. 79:1191-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nijhara, R., S. S. Jana, S. K. Goswami, A. Rana, S. S. Majumdar, V. Kumar, and D. P. Sarkar. 2001. Sustained activation of mitogen-activated protein kinases and activator protein 1 by the hepatitis B virus X protein in mouse hepatocytes in vivo. J. Virol. 75:10348-10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okada, Y. 1988. Sendai virus-mediated cell fusion. Curr. Top. Membr. Transp. 32:297-336. [Google Scholar]
- 40.Ozanne, B. W., H. J. Spence, L. C. McGarry, and R. F. Hennigan. 2007. Transcription factors control invasion: AP-1 the first among equals. Oncogene 26:1-10. [DOI] [PubMed] [Google Scholar]
- 41.Padmanabhan, S., A. Mukhopadhyay, S. D. Narasimhan, G. Tesz, M. P. Czech, and H. A. Tissenbaum. 2009. A PP2A regulatory subunit regulates C. elegans insulin/IGF-1 signaling by modulating AKT-1 phosphorylation. Cell 136:939-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pleschka, S., T. Wolff, C. Ehrhardt, G. Hobom, O. Planz, U. R. Rapp, and S. Ludwig. 2001. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 3:301-305. [DOI] [PubMed] [Google Scholar]
- 43.Ramani, K., Q. Hassan, B. Venkaiah, S. E. Hasnain, and D. P. Sarkar. 1998. Site-specific gene delivery in vivo through engineered Sendai viral envelopes. Proc. Natl. Acad. Sci. USA 95:11886-11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Resjo, S., O. Goransson, L. Harndahl, S. Zolnierowicz, V. Manganiello, and E. Degerman. 2002. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 14:231-238. [DOI] [PubMed] [Google Scholar]
- 45.Rizzo, M. T., and G. Weber. 1994. 1-Phosphatidylinositol 4-kinase: an enzyme linked with proliferation and malignancy. Cancer Res. 54:2611-2614. [PubMed] [Google Scholar]
- 46.Sarkar, D. P., and R. Blumenthal. 1987. The role of the target membrane structure in fusion with Sendai virus. Membr. Biochem. 7:231-247. [DOI] [PubMed] [Google Scholar]
- 47.Sarkar, D. P., S. J. Morris, O. Eidelman, J. Zimmerberg, and R. Blumenthal. 1989. Initial stages of influenza hemagglutinin-induced cell fusion monitored simultaneously by two fluorescent events: cytoplasmic continuity and lipid mixing. J. Cell Biol. 109:113-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schramek, H., E. Feifel, E. Healy, and V. Pollack. 1997. Constitutively active mutant of the mitogen-activated protein kinase kinase MEK1 induces epithelial dedifferentiation and growth inhibition in Madin-Darby canine kidney-C7 cells. J. Biol. Chem. 272:11426-11433. [DOI] [PubMed] [Google Scholar]
- 49.Strong, J. E., G. Wong, S. E. Jones, A. Grolla, S. Theriault, G. P. Kobinger, and H. Feldmann. 2008. Stimulation of Ebola virus production from persistent infection through activation of the Ras/MAPK pathway. Proc. Natl. Acad. Sci. USA 105:17982-17987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Subramanian, N., P. Mani, S. Roy, S. V. Gnanasundram, D. P. Sarkar, and S. Das. 2009. Targeted delivery of hepatitis C virus specific shRNA in mouse liver using Sendai virosomes. J. Gen. Virol. 90:1812-1819. [DOI] [PubMed] [Google Scholar]
- 51.Sun, M., S. M. Fuentes, K. Timani, D. Sun, C. Murphy, Y. Lin, A. August, M. N. Teng, and B. He. 2008. Akt plays a critical role in replication of nonsegmented negative-stranded RNA viruses. J. Virol. 82:105-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ugi, S., T. Imamura, H. Maegawa, K. Egawa, T. Yoshizaki, K. Shi, T. Obata, Y. Ebina, A. Kashiwagi, and J. M. Olefsky. 2004. Protein phosphatase 2A negatively regulates insulin's metabolic signaling pathway by inhibiting Akt (protein kinase B) activity in 3T3-L1 adipocytes. Mol. Cell. Biol. 24:8778-8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Usatyuk, P. V., and V. Natarajan. 2004. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J. Biol. Chem. 279:11789-11797. [DOI] [PubMed] [Google Scholar]
- 54.Vandermoere, F., I. El Yazidi-Belkoura, Y. Demont, C. Slomianny, J. Antol, J. Lemoine, and H. Hondermarck. 2007. Proteomics exploration reveals that actin is a signaling target of the kinase Akt. Mol. Cell. Proteomics 6:114-124. [DOI] [PubMed] [Google Scholar]
- 55.Verma, S. K., P. Mani, N. R. Sharma, A. Krishnan, V. V. Kumar, B. S. Reddy, A. Chaudhuri, R. P. Roy, and D. P. Sarkar. 2005. Histidylated lipid-modified Sendai viral envelopes mediate enhanced membrane fusion and potentiate targeted gene delivery. J. Biol. Chem. 280:35399-35409. [DOI] [PubMed] [Google Scholar]
- 56.Wang, W., Z. Jobbagy, T. H. Bird, M. V. Eiden, and W. B. Anderson. 2005. Cell signaling through the protein kinases cAMP-dependent protein kinase, protein kinase Cepsilon, and RAF-1 regulates amphotropic murine leukemia virus envelope protein-induced syncytium formation. J. Biol. Chem. 280:16772-16783. [DOI] [PubMed] [Google Scholar]
- 57.Wang, X., D. P. Sarkar, P. Mani, C. J. Steer, Y. Chen, C. Guha, V. Chandrasekhar, A. Chaudhuri, N. Roy-Chowdhury, B. T. Kren, and J. Roy-Chowdhury. 2009. Long-term reduction of jaundice in Gunn rats by nonviral liver-targeted delivery of Sleeping Beauty transposon. Hepatology 50:815-824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang, H., S. K. Kim, M. Kim, P. A. Reche, T. J. Morehead, I. K. Damon, R. M. Welsh, and E. L. Reinherz. 2005. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J. Clin. Invest. 115:379-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yin, H. S., X. Wen, R. G. Paterson, R. A. Lamb, and T. S. Jardetzky. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoder, A., D. Yu, L. Dong, S. R. Iyer, X. Xu, J. Kelly, J. Liu, W. Wang, P. J. Vorster, L. Agulto, D. A. Stephany, J. N. Cooper, J. W. Marsh, and Y. Wu. 2008. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 134:782-792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu, H. Y., and W. M. Bement. 2007. Control of local actin assembly by membrane fusion-dependent compartment mixing. Nat. Cell Biol. 9:149-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeidan, Y. H., R. W. Jenkins, and Y. A. Hannun. 2008. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J. Cell Biol. 181:335-350. [DOI] [PMC free article] [PubMed] [Google Scholar]