

Abstract
Rabies virus infection of dorsal root ganglia (DRG) was studied in vitro with cultured adult mouse DRG neurons. Recent in vivo studies of transgenic mice that express the yellow fluorescent protein indicate that neuronal process degeneration, involving both dendrites and axons, occurs in mice infected with the challenge virus standard (CVS) strain of rabies virus by footpad inoculation. Because of the similarities of the morphological changes in experimental rabies and in diabetic neuropathy and other diseases, we hypothesize that neuronal process degeneration occurs as a result of oxidative stress. DRG neurons were cultured from adult ICR mice. Two days after plating, they were infected with CVS. Immunostaining was evaluated with CVS- and mock-infected cultures for neuron specific β-tubulin, rabies virus antigen, and amino acid adducts of 4-hydroxy-2-nonenal (4-HNE) (marker of lipid peroxidation and hence oxidative stress). Neuronal viability (by trypan blue exclusion), terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining, and axonal growth were also assessed with the cultures. CVS infected 33 to 54% of cultured DRG neurons. Levels of neuronal viability and TUNEL staining were similar in CVS- and mock-infected DRG neurons. There were significantly more 4-HNE-labeled puncta at 2 and 3 days postinfection in CVS-infected cultures than in mock-infected cultures, and axonal outgrowth was reduced at these time points in CVS infection. Axonal swellings with 4-HNE-labeled puncta were also associated with aggregations of actively respiring mitochondria. We have found evidence that rabies virus infection in vitro causes axonal injury of DRG neurons through oxidative stress. Oxidative stress may be important in vivo in rabies and may explain previous observations of the degeneration of neuronal processes.
Rabies is an acute viral infection of the central nervous system (CNS) that is usually fatal in humans and animals (10). Despite its lethality, under natural conditions only relatively mild histopathological lesions are typically found in association with a paucity of degenerative neuronal changes (9, 21). Li et al. (13) showed severe destruction and disorganization of axons and disruption of synaptic structures in silver-stained hippocampal sections from mice infected intracerebrally with the pathogenic N2C strain of rabies virus. Infection with the challenge virus standard (CVS) strain of rabies virus has recently been evaluated using hindlimb footpad inoculation in adult transgenic mice expressing yellow fluorescent protein (26). In this model, dendritic beading and axonal swellings were prominent, and neuronal process degeneration appeared to account for the severe clinical disease and fatal outcome. The axonal swellings exhibited striking morphological similarities to the neurodegenerative changes that occur in the axons of neurons in diabetic sensory and autonomic neuropathy and in human immunodeficiency virus (HIV) infection (4, 12, 24, 35). Studies of cultured adult dorsal root ganglion (DRG) sensory neurons from type 1 diabetic rats have revealed that glucose-induced oxidative stress is a key instigator of axonal swellings (35). In humans with sensory neuropathy, the nerve endings exhibit aberrant axonal structures that contain numerous accumulated mitochondria, vesicles, and neurofilaments (4). The axonal swellings in cultured diabetic neurons were characterized in more detail and exhibited immunofluorescent staining for amino acid adducts of 4-hydroxy-2-nonenal (4-HNE), which is a marker of oxidative stress-dependent lipid peroxidation (30). We have, therefore, hypothesized that oxidative stress may etiologically play an important role in axonal swelling formation and subsequent neuronal process degeneration in rabies virus infection. In order to evaluate this hypothesis, we have studied CVS versus mock infection of cultured adult mouse DRG neurons in order to determine the role of oxidative stress on rabies virus-infected DRG neurons and their neurites (axons). We have used cultured DRG neurons because these neurons are known to be relatively permissive to rabies virus infection (2, 14, 27-29), which facilitates evaluation of processes involving axons.
MATERIALS AND METHODS
Virus.
The CVS-11 strain of fixed rabies virus (CVS), which was obtained from William H. Wunner (The Wistar Institute, Philadelphia, PA), was used in these studies. Baby hamster kidney (BHK) cells (C13 clone) grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% newborn calf serum (NCS) (PAA Laboratories, Etobioke, ON) were used for virus propagation. Viral assays of stock virus were performed by counting fluorescent foci on BHK cell monolayers.
DRG neuron cultures.
DRG were isolated from 6- to 10-week-old ICR mice (University of Manitoba, Winnipeg, MB, Canada). Mice were killed by cervical dislocation, the complete vertebral column was removed, and DRG were removed by dissection. DRG were mechanically and enzymatically dissociated in 0.125% collagenase type 4 (no. 47C9497; Worthington, Lakewood, NJ) in Ham's F-12 medium (no. 11765-054; Invitrogen, Carlsbad, CA) for 45 min at 37°C. Collagenase was neutralized by adding NCS. Cells were collected and triturated using a glass pipette in 10% NCS in F-12. The cell suspension was filtered through a 70-μm nylon mesh filter to remove nondissociated cells and myelin debris and centrifuged at 300 × g for 10 min. The cells were resuspended in 1 ml F-12 and then spun through a column with 15% essentially fatty acid-free bovine serum albumin (BSA) (no. A9205; Sigma-Aldrich, St. Louis, MO) in F-12 at 900 × g for 10 min. Cell pellets were collected and resuspended in 2 ml F-12 medium with 2 mM l-glutamine and Bottenstein's N2 supplements without insulin (0.1 mg/ml transferrin, 20 nM progesterone, 100 μM putrescine, 30 nM sodium selenite, and 0.1 mg/ml fatty acid-free BSA). Cells were plated on 18-mm circle glass coverslips coated with 0.5 mg/ml poly-dl-ornithine (no. P8638; Sigma) and 2 μg/ml natural mouse laminin (no. 23017-015; Invitrogen). Cultures were maintained in N2/F-12 medium at 37°C in humidified air containing 5% CO2. The cultures were cultivated for 2 days prior to viral adsorption.
Determination of viability of infected DRG neuron cultures.
Two days after plating, viral adsorption of cultured DRG neurons was performed with CVS at a multiplicity of infection of 10 fluorescent focus-forming units per cell (in N2/F-12 with 2% NCS) for 1 h, or cultures were mock infected, and then fresh N2/F12 medium was added. At 24, 48, and 72 h postinfection (p.i.), cells were stained with trypan blue, and viability was determined using the trypan blue exclusion assay, which is based on the ability of viable cells to exclude the vital dye trypan blue, whereas nonviable cells fail to exclude the vital dye and show trypan blue staining in their cytoplasm.
Immunocytochemistry for tubulin, rabies virus antigen, and 4-hydroxy-2-nonenal adducts.
CVS- and mock-infected cultured DRG neurons at 24, 48, and 72 h postinfection (p.i.) were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (pH 7.4) for 15 min and permeabilized with 0.3% Triton X-100 in PBS for 3 min. The slides were blocked for 1 h at room temperature in blocking reagent (no. 11 096 176 001; Roche, Mannheim, Germany), NCS, and PBS in a ratio of 1:1:3. Cultures were evaluated with fluorescent staining after incubation overnight at 4°C with the following primary antibodies: mouse monoclonal anti-β-tubulin isotype III (no. T8660; Sigma-Aldrich) (1:300) as a pan-neuronal marker to label cell bodies and neurites, polyclonal rabbit anti-rabies virus antibody (obtained from K. Charlton, Canadian Food Inspection Agency, Ottawa, ON, Canada) (1:1,000) or mouse anti-rabies virus nucleocapsid protein monoclonal antibody (5DF12) (1:80) (obtained from A.I. Wandeler, Canadian Food Inspection Agency, Ottawa, ON, Canada), and polyclonal rabbit anti-4-hydroxy-2-nonenal (4-HNE) adducts (no. ALX-210-767-R100; Alexis Biochemicals, San Diego, CA) (1:100). Secondary antibodies included cyanine 3 (Cy3)-conjugated donkey anti-mouse IgG (no. 711-165-152; Jackson ImmunoResearch Laboratories, West Grove, PA) and fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit IgG (no. 711-095-152; Jackson ImmunoResearch Laboratories) (1:250), which were incubated in blocking buffer for 1 h at room temperature. Coverslips were mounted on slides with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (H-1200, Vector Laboratories, Burlingam, CA) and examined using a Carl Zeiss Axioskop 2 mot microscope equipped with fluorescein isocyanate, Cy3, and DAPI filters and coupled to an AxioCam camera with Axo-Vision 3 software. Quantitative assessments of 4-HNE puncta were performed with masked images, so that the evaluator was not aware if samples were from CVS- or mock-infected groups, and the number of labeled puncta was analyzed relative to axonal length (in μm).
TUNEL staining.
Apoptotic cell death in the CVS- and mock-infected DRG cultures was evaluated with terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining using a TMR red in situ cell death detection kit (no. 12156792910; Roche, Indianapolis, IN). Cells were stained after fixation for 1 h with 4% paraformaldehyde in PBS (pH 7.4) and permeabilization with 0.1% Triton X-100 in 0.1% sodium citrate. Cells were then stained with the TUNEL reaction mixture for 1 h in a humidified chamber in the dark at 37°C. Negative controls were evaluated by using all reagents except terminal transferase. Positive controls for TUNEL staining were pretreated with 1 μg/ml DNase I (Pharmacia Biotech, Baie d'Urfe, Quebec) for 15 min at 37°C in order to produce enzymatic DNA fragmentation. Double-staining was performed with TUNEL staining immediately followed by immunostaining for rabies virus antigen with an FITC-conjugated anti-rabies virus antibody (Millipore, Temecula, CA) (1:200). Stained DRG cultures were mounted with Vectashield mounting medium with DAPI and analyzed using fluorescence microscopy with green fluorescence for rabies virus antigen and red fluorescence for TUNEL staining. TUNEL-positive cells containing red fluorescent nuclei were quantitatively assessed with masked images.
Evaluation of neurite (axon) outgrowth.
The level of total neurite (axon) outgrowth of DRG neurons from CVS- and mock-infected cultures was analyzed after staining for β-tubulin. Images of cultures were taken from at least 10 random fields from each well. The total number of neuronal cell bodies and the number of intersects of their neurites with a vertical grid were counted using a morphometric approach using ImageJ 1.42 software (available at http://rsbweb.nih.gov/ij/) with masked images. The total number of intersects per neuron was taken as the parameter of total axon outgrowth as previously described (35).
Assessment of 4-HNE puncta and axonal outgrowth with antioxidant NAC.
N-acetyl cysteine (NAC) was used as an antioxidant to revert the oxidative damage phenotype. Preliminary studies showed that 1 mM NAC was well tolerated by the DRG cultures. Two days after plating, DRG cultures were infected with CVS or mock infected (see above), and at 48 h p.i. 1 mM NAC was added to the media. At 72 h p.i., cells were fixed with buffered 4% paraformaldehyde and immunostaining was performed for β-tubulin and 4-HNE (see above).
Mitochondrial localization.
CVS- and mock-infected DRG cultures at 72 h p.i. were treated with 1 μM MitoTracker Deep Red FM dye (no. M22426; Molecular Probes, Eugene, OR), which is a carbocyanine-based mitochondrian-selective probe, for 15 min for localization of actively respiring (“live”) mitochondria. Subsequently, the cells were fixed, immunostaining was performed for 4-HNE (see above), and mounting media were added. The fluorescent signals were examined using a Carl Zeiss LSM510 confocal inverted microscope.
Statistical analyses.
Standard two-tailed unpaired Student t tests with Welch's correction, which did not assume equal variances, were used for evaluating the significance of the difference between the means from assays comparing CVS- and mock-infected cultures. A value of P of <0.05 was considered statistically significant.
RESULTS
Characterization of primary neuron cultures.
At 3 days after plating, 65 to 87% of the cells isolated from DRG expressed the neuronal marker β-tubulin III as assessed using immunofluorescent staining, indicating that they were DRG neurons (Fig. 1C and 2A). These neurons also exhibited well-developed axonal projections, and the high level of growth was indicative of a healthy and viable culture (Fig. 1C and 2A). CVS infection resulted in the appearance of multiple axonal swellings of different shapes and sizes by 72 h p.i. (Fig. 1D, F, and H, arrowheads) but not at 24 h p.i. (Fig. 1C).
FIG. 1.

CVS infection causes formation of axonal swellings in DRG cultures. Fluorescence microscopy showing CVS-infected DRG neurons at 24 h (A, C, E, and G) and at 72 h (B, D, F, and H) p.i. Staining with DAPI shows neuronal nuclei (A and B). Staining for β-tubulin III shows two neuronal cell bodies at 24 h p.i. (C) and one (large spherical body) at 72 h p.i. (D). (E) There is strong rabies virus antigen staining of one of the two neuronal cell bodies at 24 h p.i., but not of the other, demonstrating that CVS infects only a subpopulation of DRG neurons. Definite axonal swellings are not yet present at 24 h p.i. (C, E, and G), but axonal swellings are well established at 72 h p.i. (D, F, and H, arrowheads). Rabies virus antigen is strongly expressed in the neuronal cell bodies and axons at 24 h and 72 h p.i. (E to H) and also in axonal swellings at 72 h p.i. (F and H).
FIG. 2.

CVS infection but not mock infection induces formation of 4-HNE-labeled axonal swellings. Fluorescence microscopy showing mock-infected (A, C, and E) and CVS-infected (B, D, and F) DRG neurons at 72 h p.i. β-Tubulin is a marker of DRG neuronal cell bodies and axons (red), and expression of β-tubulin in CVS-infected neurons (B) showed multiple axonal swellings, but a lack of axonal swellings in mock-infected neurons (A). 4-HNE (green) was poorly expressed in the axons of mock-infected DRG neurons (C), but showed greater expression in the axons of CVS-infected neurons and accumulation in regions with axonal swellings (D). In CVS-infected neurons, the merging of signals for β-tubulin and 4-HNE (yellow) showed there was strong expression of these elements in axons, both with axonal swellings (arrowheads) and without axonal swellings (arrow) (F) but not in mock-infected neurons (E).
CVS infection was associated with immunostaining for rabies virus antigen along axons and in axonal swellings.
At 24 h p.i., 33% of neuronal cell bodies (identified by positive β-tubulin immunostaining) expressed rabies virus antigen versus 54% at 48 h and 52% at 72 h p.i., with no significant increase at the later two time points (P > 0.4). There was low background staining in mock-infected cultures. There was strong staining for rabies virus antigen in a subpopulation of neuronal cell bodies with associated staining in axons and intense signals in axonal swellings that increased in size from 48 h to 72 h p.i. (Fig. 1F and H).
CVS infection did not result in loss of viability in DRG neurons.
Cells were infected with CVS or mock infected and analyzed using trypan blue exclusion for neuronal viability at 24, 48, and 72 h p.i. (Fig. 3). CVS-infected cultures did not show a significant change (loss) of viability in comparison with the mock-infected cultures (P > 0.5).
FIG. 3.

CVS infection does not affect viability of DRG neurons. Viability of CVS-infected (black bars) and mock-infected (white bars) DRG cultures was assessed with the trypan blue exclusion method, which revealed nonviable neurons in the culture at 24, 48, and 72 h p.i. (n = 500 cells for each value, with a total of 3,000 cells evaluated). There was no significant change in viability at 24 (P = 0.7), 48 (P = 0.5), or 72 (P = 0.8) h p.i. Error bars represent standard error of the mean (SEM).
TUNEL staining did not show CVS-induced neuronal apoptosis of DRG neurons.
TUNEL staining was performed on CVS- and mock-infected cultures at 24, 48, and 72 h p.i. as a marker of neuronal apoptosis in order to determine whether rabies virus infection induced neuronal apoptotic death in the cultures, which would be expected to have associated effects on neurites (axons) (Fig. 4 and 5). Cultures pretreated with DNase (positive control) showed strong TUNEL staining in nuclei (Fig. 4D), and there was low background staining in cultures with an omission of the terminal transferase enzyme (data not shown). The CVS-infected cultures did not show a significant change (increase) in TUNEL staining in comparison with the mock-infected cultures (P > 0.4) (Fig. 5).
FIG. 4.

CVS infection does not trigger apoptosis in DRG neurons. Mock-infected (A to D) and CVS-infected (E and F) DRG neurons at 72 h p.i with DAPI staining (A and C) showing neuronal nuclei, immunostaining for rabies virus antigen showing strong expression in the cytoplasm of infected neurons (E), and TUNEL staining (B, D, and F). TUNEL staining shows a lack of signal in mock-infected (B) and CVS-infected (F) neurons but shows strong signals in neurons pretreated with DNase as a positive control (D).
FIG. 5.
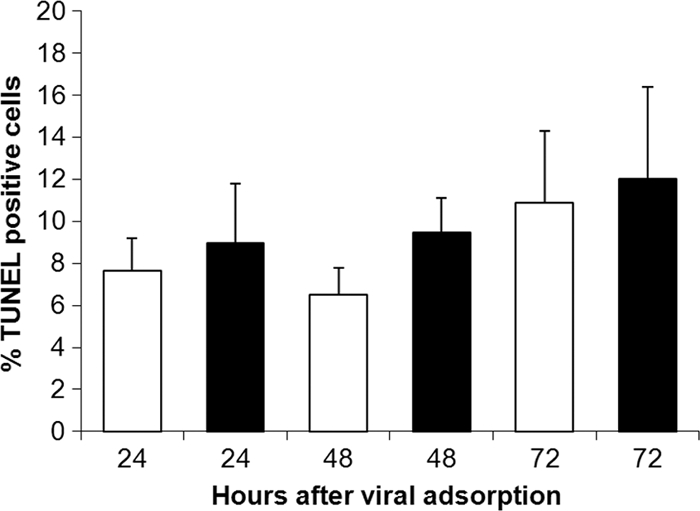
TUNEL staining of CVS-infected (black bars) and mock-infected (white bars) DRG cultures at 24, 48, and 72 h p.i. (n = 200 to 300 cells evaluated for each value). There was no significant change in the percentage of TUNEL-positive cells at 24 (P = 0.7), 48 (P = 0.4), or 72 (P = 0.8) h p.i. Error bars represent SEM.
Positive immunofluorescent staining for amino acid adducts of 4-HNE of CVS-infected DRG neurons indicated the presence of oxidative stress.
Fluorescence microscopy showed strong immunostaining for amino acid adducts of 4-HNE, a consequence of ongoing lipid peroxidation and hence a marker of oxidative stress; axons of CVS-infected DRG neurons; and the presence of axonal swellings with intense immunostaining for 4-HNE (Fig. 2). Some 4-HNE-immunostained axons demonstrated swellings (puncta), whereas others did not (Fig. 2D and F). CVS-infected neurons demonstrated relatively prominent axonal swellings and HNE-labeled puncta in comparison with uninfected neurons in the CVS-infected cultures. There was a progressive enlargement of the axonal swellings from 48 h to 72 h p.i., with a variety of shapes being formed (e.g., kidney-shaped and a range of circular profiles) (Fig. 2). Quantitative evaluation of the number of 4-HNE-immunostained puncta per 100 μm axon in mock- versus CVS-infected cultures showed no significant change at 24 h p.i. but a 143% increase at 48 h p.i. (P = 0.006) and a 109% increase at 72 h p.i. (P = 0.0008) in CVS infection (Fig. 6).
FIG. 6.

CVS infection caused a significant elevation in 4-HNE staining within axons. Evaluation of 4-HNE puncta in CVS-infected (black) and mock-infected (white) DRG neurons at 24, 48, and 72 h p.i. (n = total of 126 neurons for CVS infection and 99 neurons for mock infection). There was no significant change in the number of 4-HNE puncta per 100-μm axon at 24 h p.i. (P = 0.4), but there was a highly significant (**) 143% increase (2.3 ± 0.7 versus 5.6 ± 1.0) at 48 h p.i. (P = 0.006) and 109% increase (4.0 ± 0.6 versus 8.3 ± 1.1) at 72 h p.i. (P = 0.0008) in mock-infected neurons versus CVS-infected neurons.
Neurite (axonal) outgrowth of DRG neurons was reduced in CVS infection.
Total axonal outgrowth was assessed in mock- and CVS-infected cultures at 24, 48, and 72 h p.i. At 24 h, there was no significant change in axonal outgrowth (P = 0.10), but at 48 h and 72 h p.i., there were 42% and 60% reductions in axonal growth in CVS infection, respectively, with high statistical significance (P = 0.008 and P = 0.003, respectively) (Fig. 7).
FIG. 7.

CVS infection diminishes total axonal outgrowth in DRG cultures. Assessment of axonal outgrowth in CVS-infected (black bars) and mock-infected (white bars) DRG cultures assessed by using the number of intersects per neuron (n = total of 98 neurons for CVS infection and 66 neurons for mock infection). There was no significant difference at 24 h p.i. (P = 0.10), but there was a highly significant (**) 42% reduction (167 ± 22 versus 97 ± 9) at 48 h p.i. (P = 0.008) and 60% reduction (149 ± 26 versus 60 ± 5) in axonal growth at 72 h p.i. (P = 0.003) in mock-infected neurons versus CVS-infected neurons. Error bars represent SEM.
NAC treatment reduced 4-HNE immunostaining in DRG neurons.
Therapy for 24 h with NAC strongly reduced the 4-HNE immunostaining in CVS-infected neurons evaluated at 72 h p.i. versus mock-infected neurons (Fig. 8). Axonal swellings remained present in cultures treated with NAC, indicating that 24 h of NAC therapy was not sufficient to completely reverse the axonal injury in CVS infection.
FIG. 8.

Fluorescent staining of CVS-infected DRG cultures without NAC treatment (A, C, and E) and with NAC treatment (B, D, and F) with staining for β-tubulin (red) (A and B), 4-HNE (green) (C and D), and a merging (yellow) (E and F). In the NAC-treated culture (D and F), there was a marked reduction in the expression of 4-HNE versus the untreated culture (C and E). Axonal swellings (arrowheads) remained present in both the untreated (E) and NAC-treated (F) cultures, but coexpression of β-tubulin and 4-HNE was much greater in the untreated culture (E).
Mitochondrial localization of axonal swellings and 4-HNE immunostaining.
Fluorescence microscopy showed colocalization of actively respiring mitochondria and immunostaining for 4-HNE in many axonal swellings of CVS-infected cultures at 72 h p.i. and a normal appearance of signals without axonal swellings in mock-infected cultures (Fig. 9).
FIG. 9.

Fluorescent staining of mock-infected (A, C, and E) and CVS-infected (B, D, and F) cultures 72 h p.i. with MitoTracker red dye (for actively respiring mitochondria) (red) (A and B), 4-HNE (green) (C and D), and a merging of MitoTracker red and 4-HNE (yellow) (E and F). There is localization of signals for both actively respiring mitochondria and 4-HNE in many axonal swellings (arrowheads) in the CVS-infected culture (F) but not in the mock-infected culture (E).
DISCUSSION
Although rabies is a highly lethal infectious disease involving the CNS, under natural conditions there are usually a paucity of degenerative neuronal changes in the CNS observed by routine methods (9, 21). Recently, detailed studies of experimental rabies using a transgenic mouse model that expresses the yellow fluorescent protein in a subpopulation of neurons have shown extensive degenerative changes involving neuronal processes, whereas conventional histopathology showed inflammatory changes without apparent degenerative neuronal changes (26). With the development of severe clinical neurological disease, fluorescence microscopy showed marked structural abnormalities, especially beading and/or swelling, in dendrites and axons of layer V cortical pyramidal neurons; severe involvement of axons in the brainstem and inferior cerebellar peduncle; and severe abnormalities affecting axons of cerebellar mossy fibers. Toluidine blue-stained resin sections and electron microscopy showed vacuolation in cortical neurons that corresponded to swollen mitochondria and vacuolation in the neuropil of the cerebral cortex. Axonal swellings were observed. Vacuolation was also observed ultrastructurally with axons and presynaptic nerve endings. The involvement of axons has a striking morphological similarity to the neurodegenerative changes that occur in diabetic sensory and autonomic neuropathy, in which a key feature is the presence of axonal swellings that are composed of accumulations of mitochondria and cytoskeletal proteins (e.g., neurofilaments) (12, 24).
Diabetes-induced oxidative stress in sensory neurons and peripheral nerves is demonstrated by increased production of reactive oxygen species (16, 22, 35), lipid peroxidation (19, 35), and protein nitrosylation (18). It is believed the neurodegenerative outcome is energy failure in the nerve, observed as a decrease in high energy intermediates (e.g., phosphocreatine) (17, 32), impaired axonal transport of proteins (5), and suboptimal Ca2+ ion pumping (8, 11), and it is believed that these factors contribute to the sensory neuropathy. Because of morphological similarities with diabetic neuropathy, we have hypothesized that oxidative stress may also play a key role in degeneration of neuronal processes in rabies virus infection. The importance of oxidative stress during viral infections has been recognized for over a decade (25), and oxidative injury has been shown to be an important component in HIV infection (7, 34), particularly in HIV dementia (34), and also in experimental acute encephalitis caused by herpes simplex virus type 1 in mice (31).
We have studied CVS infection in cultured DRG sensory neurons because they are known to be relatively permissive to in vitro infection with rabies virus (2, 14, 27-29), with survival of rabies virus-infected DRG neurons for more than 20 days without cytopathic effects observed with the cultures (27). Hence, DRG neurons are an amenable neuronal cell type for an assessment of changes in morphology and/or growth of their neurites (axons) in rabies virus infection. In contrast, rabies virus infection of embryonic primary neurons from the cerebral cortex and hippocampus results in apoptotic cell death (15, 33).
Similar to previous reports of DRG neurons in cultures (14, 29), we have found that CVS infects only a subpopulation (33 to 54%) of DRG neurons. CVS infection did not cause a loss of neuronal viability over 72 h in CVS-infected DRG neurons above baseline levels observed with the mock-infected cultures. Similarly, CVS infection did not produce neuronal apoptosis of the DRG neurons over 72 h p.i. However, rabies virus infection did result in markedly reduced growth of axonal processes at 48 and 72 h p.i. in the infected cultures, indicating a functional abnormality with impairment of normal axonal growth.
The striking morphological abnormality in CVS-infected DRG neurons is the presence of multiple axonal swellings. These swellings are associated with intense immunostaining for both rabies virus antigen and amino adducts of 4-HNE; 4-HNE is a marker of lipid peroxidation that occurs in association with oxidative stress. Axonal swellings (as well as dendritic beading) were also observed with CVS-infected transgenic mice that expressed the yellow fluorescent protein (26), indicating a strong correlation between our in vitro observations and the findings in a mouse model of rabies. The major sources of reactive oxygen species in DRG could involve rabies virus-induced aberrant mitochondrial function, impaired calcium homeostasis, and/or enhanced NAD(P)H oxidase activity. We hypothesize that rabies virus infection, through these pathways, can result in enhanced production of reactive oxygen species and that this oxidative stress causes abnormal mitochondrial protein function and suboptimal axoplasmic structural protein function that combine to trigger impaired axonal transport and axonal degeneration. This oxidative stress results in lipid peroxidation and the formation of amino acid adducts of 4-HNE, which contributes to the modification in the function of key mitochondrial and cytoskeletal proteins, leading to the formation of axonal swellings. The axonal swellings are likely sites of mitochondrial and structural protein accumulation due to local blockade of axonal transport (6). In support of this hypothesis, we have found evidence of colocalization of 4-HNE immunostaining and staining of “live” mitochondria in axonal swellings of CVS-infected DRG neurons. Recently, it has been demonstrated that 4-HNE directly impairs mitochondrial function in cultured DRG neurons (1). Mitochondria likely play a central role as a mediator of neurodegeneration. Impaired mitochondrial function leads to impaired axonal transport and accumulation of organelles (including mitochondria) and structural proteins, resulting in axonal swellings. The absence of structural proteins and ATP-generating organelles leads to axonal degeneration and destruction of axon terminals, which was observed in detailed morphological studies in vivo with CVS-infected mice (26). Amino acid adducts triggered by 4-HNE have been shown previously to significantly impair axonal outgrowth (1), which may explain this finding for the CVS-infected cultures. We have found that treatment for 24 h with the antioxidant NAC markedly reduced 4-HNE immunostaining in CVS-infected neurons. Although axonal swellings persisted in the cultures, the time of initiation of treatment, the duration of treatment, and the concentration of NAC may all be important factors in reducing axonal injury related to oxidative stress in CVS infection. Further studies are needed in order to demonstrate what structural changes are reversible.
Clearly, axons of cultured DRG neurons are more vulnerable to the effects of oxidative stress than neuronal cell bodies. This was also observed to be the case in streptozotocin-induced type 1 diabetes in rats, in which there is no significant loss of DRG neurons for up to 12 months duration (3, 36). There are also similar findings in human postmortem studies of type 2 diabetic patients (23, 24). With HIV infection of cultured human DRG neurons, Hahn et al. (7) also observed a differential effect on axons and cell bodies. Axonal retraction was observed without neuronal cell death, there were reactive oxygen species produced in neuronal cell bodies, and the mitochondrial membrane potential was reduced, indicating mitochondrial dysfunction. However, the situation in DRG in vivo is quite different in rabies in both humans and animals, in which neuronal death is quite prominent without the typical features of necrosis or apoptosis (20); likely, this neuronal death is due to autophagy.
In summary, we have found evidence that oxidative stress occurred in rabies virus-infected cultured DRG. These findings help explain the extensive degeneration of neuronal processes in a mouse model of rabies. More information is needed on the mechanisms involved in the production of oxidative stress in rabies virus infection and, ultimately, on whether therapeutic approaches can reduce oxidative damage and promote recovery. Oxidative stress may also play an important role in other viral infections of the nervous system.
Acknowledgments
This work was supported by Canadian Institutes of Health Research operating grant III-94590 (to A. C. Jackson and P. Fernyhough) and the St. Boniface General Hospital Research Foundation.
We thank Gordon Glazner for permitting access to the Carl Zeiss LSM510 confocal microscope.
Footnotes
Published ahead of print on 24 February 2010.
REFERENCES
- 1.Akude, E., E. Zherebitskaya, S. K. Roy Chowdhury, K. Girling, and P. Fernyhough. 2010. 4-Hydroxy-2-nonenal induces mitochondrial dysfunction and aberrant axonal outgrowth in adult sensory neurons that mimics features of diabetic neuropathy. Neurotox. Res. 17:28-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castellanos, J. E., M. Martinez, O. Acosta, and H. Hurtado. 2000. Nerve growth factor and neurotrophin-3 modulate the rabies infection of adult sensory neurons in primary cultures. Brain Res. 871:120-126. [DOI] [PubMed] [Google Scholar]
- 3.Cheng, C., and D. W. Zochodne. 2003. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes 52:2363-2371. [DOI] [PubMed] [Google Scholar]
- 4.Ebenezer, G. J., J. C. McArthur, D. Thomas, B. Murinson, P. Hauer, M. Polydefkis, and J. W. Griffin. 2007. Denervation of skin in neuropathies: the sequence of axonal and Schwann cell changes in skin biopsies. Brain 130:2703-2714. [DOI] [PubMed] [Google Scholar]
- 5.Fernyhough, P., and R. E. Schmidt. 2002. Neurofilaments in diabetic neuropathy. Int. Rev. Neurobiol. 50:115-144. [DOI] [PubMed] [Google Scholar]
- 6.Ferreirinha, F., A. Quattrini, M. Pirozzi, V. Valsecchi, G. Dina, V. Broccoli, A. Auricchio, F. Piemonte, G. Tozzi, L. Gaeta, G. Casari, A. Ballabio, and E. I. Rugarli. 2004. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J. Clin. Invest. 113:231-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn, K., B. Robinson, C. Anderson, W. Li, C. A. Pardo, S. Morgello, D. Simpson, and A. Nath. 2008. Differential effects of HIV infected macrophages on dorsal root ganglia neurons and axons. Exp. Neurol. 210:30-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang, T. J., N. M. Sayers, P. Fernyhough, and A. Verkhratsky. 2002. Diabetes-induced alterations in calcium homeostasis in sensory neurones of streptozotocin-diabetic rats are restricted to lumbar ganglia and are prevented by neurotrophin-3. Diabetologia 45:560-570. [DOI] [PubMed] [Google Scholar]
- 9.Iwasaki, Y., and M. Tobita. 2002. Pathology, p. 283-306. In A. C. Jackson and W. H. Wunner (ed.), Rabies. Academic Press, San Diego, CA.
- 10.Jackson, A. C., and W. H. Wunner. 2007. Rabies. Elsevier Academic Press, London, United Kingdom.
- 11.Kruglikov, I., O. Gryshchenko, L. Shutov, E. Kostyuk, P. Kostyuk, and N. Voitenko. 2004. Diabetes-induced abnormalities in ER calcium mobilization in primary and secondary nociceptive neurons. Pflugers Arch. 448:395-401. [DOI] [PubMed] [Google Scholar]
- 12.Lauria, G., M. Morbin, R. Lombardi, M. Borgna, G. Mazzoleni, A. Sghirlanzoni, and D. Pareyson. 2003. Axonal swellings predict the degeneration of epidermal nerve fibers in painful neuropathies. Neurology 61:631-636. [DOI] [PubMed] [Google Scholar]
- 13.Li, X. Q., L. Sarmento, and Z. F. Fu. 2005. Degeneration of neuronal processes after infection with pathogenic, but not attenuated, rabies viruses. J. Virol. 79:10063-10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez-Gutierrez, M., and J. E. Castellanos. 2007. Morphological and biochemical characterisation of sensory neurons infected in vitro with rabies virus. Acta Neuropathol. 114:263-269. [DOI] [PubMed] [Google Scholar]
- 15.Morimoto, K., D. C. Hooper, S. Spitsin, H. Koprowski, and B. Dietzschold. 1999. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J. Virol. 73:510-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikawa, T., D. Edelstein, X. L. Du, S. Yamagishi, T. Matsumura, Y. Kaneda, M. A. Yorek, D. Beebe, P. J. Oates, H. P. Hammes, I. Giardino, and M. Brownlee. 2000. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787-790. [DOI] [PubMed] [Google Scholar]
- 17.Obrosova, I. G. 2002. How does glucose generate oxidative stress in peripheral nerve? Int. Rev. Neurobiol. 50:3-35. [DOI] [PubMed] [Google Scholar]
- 18.Obrosova, I. G., P. Pacher, C. Szabo, Z. Zsengeller, H. Hirooka, M. J. Stevens, and M. A. Yorek. 2005. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes 54:234-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obrosova, I. G., H. C. Van, L. Fathallah, X. C. Cao, D. A. Greene, and M. J. Stevens. 2002. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 16:123-125. [DOI] [PubMed] [Google Scholar]
- 20.Rossiter, J. P., L. Hsu, and A. C. Jackson. 2009. Selective vulnerability of dorsal root ganglia neurons in experimental rabies after peripheral inoculation of CVS-11 in adult mice. Acta Neuropathol. 118:249-259. [DOI] [PubMed] [Google Scholar]
- 21.Rossiter, J. P., and A. C. Jackson. 2007. Pathology, p. 383-409. In A. C. Jackson and W. H. Wunner (ed.), Rabies. Elsevier Academic Press, London, United Kingdom.
- 22.Russell, J. W., D. Golovoy, A. M. Vincent, P. Mahendru, J. A. Olzmann, A. Mentzer, and E. L. Feldman. 2002. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 16:1738-1748. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt, R. E., L. N. Beaudet, S. B. Plurad, and D. A. Dorsey. 1997. Axonal cytoskeletal pathology in aged and diabetic human sympathetic autonomic ganglia. Brain Res. 769:375-383. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt, R. E., D. Dorsey, C. A. Parvin, L. N. Beaudet, S. B. Plurad, and K. A. Roth. 1997. Dystrophic axonal swellings develop as a function of age and diabetes in human dorsal root ganglia. J. Neuropathol. Exp. Neurol. 56:1028-1043. [DOI] [PubMed] [Google Scholar]
- 25.Schwarz, K. B. 1996. Oxidative stress during viral infection: a review. Free Radic. Biol. Med. 21:641-649. [DOI] [PubMed] [Google Scholar]
- 26.Scott, C. A., J. P. Rossiter, R. D. Andrew, and A. C. Jackson. 2008. Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome in experimental rabies in yellow fluorescent protein-expressing transgenic mice. J. Virol. 82:513-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsiang, H., P. E. Ceccaldi, and E. Lycke. 1991. Rabies virus infection and transport in human sensory dorsal root ganglia neurons. J. Gen. Virol. 72:1191-1194. [DOI] [PubMed] [Google Scholar]
- 28.Tsiang, H., E. Lycke, P.-E. Ceccaldi, A. Ermine, and X. Hirardot. 1989. The anterograde transport of rabies virus in rat sensory dorsal root ganglia neurons. J. Gen. Virol. 70:2075-2085. [DOI] [PubMed] [Google Scholar]
- 29.Tuffereau, C., K. Schmidt, C. Langevin, F. Lafay, G. Dechant, and M. Koltzenburg. 2007. The rabies virus glycoprotein receptor p75NTR is not essential for rabies virus infection. J. Virol. 81:13622-13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uchida, K. 2003. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog. Lipid Res. 42:318-343. [DOI] [PubMed] [Google Scholar]
- 31.Valyi-Nagy, T., S. J. Olson, K. Valyi-Nagy, T. J. Montine, and T. S. Dermody. 2000. Herpes simplex virus type 1 latency in the murine nervous system is associated with oxidative damage to neurons. Virology 278:309-321. [DOI] [PubMed] [Google Scholar]
- 32.Vincent, A. M., J. W. Russell, P. Low, and E. L. Feldman. 2004. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 25:612-628. [DOI] [PubMed] [Google Scholar]
- 33.Weli, S. C., C. A. Scott, C. A. Ward, and A. C. Jackson. 2006. Rabies virus infection of primary neuronal cultures and adult mice: failure to demonstrate evidence of excitotoxicity. J. Virol. 80:10270-10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams, R., H. Yao, F. Peng, Y. Yang, C. Bethel-Brown, and S. Buch. 2010. Cooperative induction of CXCL10 involves NADPH oxidase: implications for HIV dementia. Glia 58:611-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zherebitskaya, E., E. Akude, D. R. Smith, and P. Fernyhough. 2009. Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes 58:1356-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zochodne, D. W., V. M. Verge, C. Cheng, H. Sun, and J. Johnston. 2001. Does diabetes target ganglion neurones? Progressive sensory neurone involvement in long-term experimental diabetes. Brain 124:2319-2334. [DOI] [PubMed] [Google Scholar]