

Abstract
Ganciclovir (GCV) and acyclovir (ACV) are guanine nucleoside analogues that inhibit lytic herpesvirus replication. GCV and ACV must be monophosphorylated by virally encoded enzymes to be converted into nucleotides and incorporated into viral DNA. However, whether GCV and/or ACV phosphorylation in Epstein-Barr virus (EBV)-infected cells is mediated primarily by the EBV-encoded protein kinase (EBV-PK), the EBV-encoded thymidine kinase (EBV-TK), or both is controversial. To examine this question, we constructed EBV mutants containing stop codons in either the EBV-PK or EBV-TK open reading frame and selected for stable 293T clones latently infected with wild-type EBV or each of the mutant viruses. Cells were induced to the lytic form of viral replication with a BZLF1 expression vector in the presence and absence of various doses of GCV and ACV, and infectious viral titers were determined by a green Raji cell assay. As expected, virus production in wild-type EBV-infected 293T cells was inhibited by both GCV (50% inhibitory concentration [IC50] = 1.5 μM) and ACV (IC50 = 4.1 μM). However, the EBV-PK mutant (which replicates as well as the wild-type (WT) virus in 293T cells) was resistant to both GCV (IC50 = 19.6 μM) and ACV (IC50 = 36.4 μM). Expression of the EBV-PK protein in trans restored GCV and ACV sensitivity in cells infected with the PK mutant virus. In contrast, in 293T cells infected with the TK mutant virus, viral replication remained sensitive to both GCV (IC50 = 1.2 μM) and ACV (IC50 = 2.8 μM), although susceptibility to the thymine nucleoside analogue, bromodeoxyuridine, was reduced. Thus, EBV-PK but not EBV-TK mediates ACV and GCV susceptibilities.
Epstein-Barr virus (EBV) is a human herpesvirus that causes infectious mononucleosis and is associated with a variety of different human tumors (1, 47, 81). Like all herpesviruses, EBV can infect cells in either the latent or lytic form (13). The lytic form of infection is required for horizontal spread of the virus from cell to cell and from host to host. During the lytic form of viral replication, EBV uses a virally encoded DNA polymerase and the oriLyt replication origin to duplicate its genome (24, 36, 51). The lytic form of EBV replication can be effectively inhibited in vitro by the guanine nucleoside analogues, acyclovir (ACV) and ganciclovir (GCV) (11, 16, 50, 56). Since acyclovir is significantly less toxic than ganciclovir in patients, acyclovir is generally used to treat diseases associated with lytic EBV infection, such as oral hairy leukoplakia (3).
GCV and ACV cannot be incorporated into viral or cellular DNA unless they are phosphorylated and converted into nucleotides (17, 29). Work in other herpesvirus systems has demonstrated that the first step in GCV or ACV phosphorylation is not performed efficiently by cellular nucleoside kinases but can be carried out by virally encoded enzymes in cells infected with various herpesviruses (6, 17, 20, 25, 29, 75). Human herpes simplex virus 1 (HSV-1) and HSV-2 encode a viral thymidine kinase (TK) which mediates the first step in GCV and ACV phosphorylation in virally infected cells (20, 21, 73), and ACV- and GCV-resistant HSV mutants isolated from patients commonly have mutations in the viral thymidine kinase gene (5, 14, 46, 64) and less commonly within the viral DNA polymerase gene (61). In contrast, the human cytomegalovirus (HCMV) does not encode a viral thymidine kinase protein. Instead, in HCMV-infected cells, the virally encoded protein kinase (UL97) mediates the first step of GCV phosphorylation (53, 75) and (albeit much less efficiently) ACV phosphorylation (76). Once made, the triphosphorylated form of ACV (and to a lesser extent GCV) is a much better substrate for herpesvirus-encoded DNA polymerases than cellular DNA polymerases (17, 29) and thus inhibits viral DNA replication more effectively than cellular DNA replication.
EBV encodes both a thymidine kinase (EBV-TK, the product of the BXLF1 gene) (18, 54) and a protein kinase (EBV-PK, the product of the BGLF4 gene) (74). EBV-PK is a serine/threonine protein kinase that shares many of the same substrates as the UL97 cytomegalovirus (CMV) kinase (28). Although it has been demonstrated that both GCV and ACV are activated and phosphorylated in lytically infected EBV-positive cells, it remains unclear whether GCV and/or ACV phosphorylation in EBV-infected cells is mediated primarily by the viral protein kinase, the viral thymidine kinase, or both.
All studies to date investigating this question have been performed outside the context of the viral genome, and different groups have reported conflicting findings, particularly in regard to the effects of EBV-TK. For example, one group reported that EBV-TK (expressed in bacterial lysates) phosphorylates GCV and ACV in vitro (GCV more than ACV) (52), and another group found that overexpression of EBV-TK in cells enhances GCV phosphorylation and sensitivity to the cytotoxic effects of GCV and ACV (GCV more than ACV) (59). In contrast, two other groups reported that EBV-TK purified from bacterial lysates has a highly restricted substrate specificity in comparison to HSV-TK and phosphorylates ACV and GCV extremely poorly if at all (33, 78). Furthermore, in one of these studies, overexpression of EBV-TK in cells did not result in GCV phosphorylation or GCV-mediated cell killing (33).
There is also some evidence that EBV-PK contributes to GCV activation. 293 cells overexpressing EBV-PK were reported to have enhanced GCV phosphorylation and to be sensitized to GCV-mediated cell killing (27, 59, 67). However, overexpression of EBV-TK, but not EBV-PK, was reported to sensitize cells to killing by penciclovir, a drug that is similar to acyclovir (59).
Identifying the EBV protein(s) that mediates ACV and/or GCV sensitivity during normal lytic EBV infection might have important clinical implications for the treatment of patients with EBV-associated illnesses. In addition, there is increasing interest in using the cytotoxic effect of GCV, in combination with lytic replication-inducing agents, to kill EBV-positive tumor cells (34, 40, 66, 83), since phosphorylated ganciclovir kills host cells when incorporated into the cellular DNA. The use of GCV as an anticancer agent requires that drugs be developed that precisely activate expression of the EBV-encoded protein(s) mediating GCV phosphorylation in tumor cells.
To address the question of whether EBV-TK and/or EBV-PK contributes to the ability of GCV and/or ACV to inhibit lytic EBV replication in the context of the intact EBV genome, we constructed EBV mutants defective in either EBV-PK or EBV-TK function and compared the ability of GCV and ACV to inhibit lytic replication of the mutant virus versus that of the wild-type (WT) virus in 293T cells. These studies were made possible by our recent discovery that WT and PK mutant viruses produce infectious virions with similar efficiencies in 293T cells (57), even though the PK mutant virus is highly impaired for virus production in most other cell types (31, 62). We show here that the EBV-PK mutant but not the EBV-TK mutant is resistant to the antiviral effects of both GCV and ACV. These results indicate that the EBV-encoded protein kinase and not the EBV-encoded thymidine kinase activates GCV and ACV phosphorylation in cells that are lytically infected with EBV.
MATERIALS AND METHODS
Cell lines.
The 293T cell line is derived from human kidney epithelial cells transformed with adenovirus E1A and E1B and subsequently infected with simian virus 40 (SV40) virus and expresses the SV40 large T and small t antigens (48, 84). 293T cells were cultured in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS), 100 U of penicillin/ml, and 100 μg of streptomycin/ml. Raji cells (ATCC) were maintained in RPMI 1640 with 10% FBS and penicillin-streptomycin. All cells were grown at 37°C in a humidified 5%-CO2 incubator.
Chemicals.
Ganciclovir (GCV), acyclovir (ACV), and 5-bromo-2′-deoxyuridine (BrdU) were purchased from Sigma (St. Louis, MO). GCV was dissolved in 0.1 N HCl at 10 mg/ml as a stock solution. ACV was dissolved in 1 M HCl at 50 mg/ml as a stock solution. BrdU was dissolved in water as a concentration of 10 mg/ml.
Construction of PK and TK mutant viruses.
The PK mutant virus was created by inserting stop codons at residues 1 and 5 in the EBV-PK open reading frame (in the EBV B95-8 strain of bacterial artificial chromosome [BAC]) as described in reference 85. The detailed phenotype of this mutant is described in reference 57. The pGS284 shuttle vector (60) and the EBV wild-type BACmid p2089 (19) were previously described and were gifts from W. Hammerschmidt (German Research Center for Environment and Health, Munich, Germany). EBV WT BACmid p2089 contains a hygromycin resistance cassette and expresses green fluorescent protein (GFP) (for visualization of infected cells). To create the TK mutant virus, which contains a stop codon inserted at residue 2 in the TK open reading frame, the wild-type EBV sequence 144208 to 145410, flanking the EBV TK start site, was PCR amplified using the primers BXLF1-144208 (5′-GCGAGCTCGTGTTCGAAGAGACCAGAAGGCTTACC-3′) and BXLF-1-145410 (5′-GCTCTAGACAGCGGGAGAGGAGGTTTAGCAGG-3′) and cloned into pSP65 to make pSP65-TKreg. Site-directed mutagenesis was performed according to the manufacturer's protocol (Stratagene) to convert the second amino acid of EBV-TK to a stop codon using the primers BXLF1-STOP-A-S (5′-CTCCTTTCCTGGAAATCCCTACATGGATCCCACCCGGGGTC-3′) and BXLF1-STOP-S (5′-GACCCCGGGTGGGATCCATGTAGGGATTTCCAGGAAAGGAG-3′). The mutated EBV-TK open reading frame was then cut out of pSP65 and ligated into the shuttle vector pGS284 to yield pGS284-TKStop. pGS284-TKStop in Escherichia coli S17λpir was conjugated with the wild-type EBV BACmid p2089 in E. coli G500, and cointegrates were selected in LB containing carbenicillin and chloramphenicol. Cultures were then recovered in LB containing chloramphenicol only and plated on LB plates containing 5% sucrose and chloramphenicol. Colonies containing the correct mutations were further screened by DNA sequencing of the EBV-TK region and restriction enzyme analysis comparing the TKStop BACmid with wild-type BACmid DNA using BamHI, HindIII, SalI, and EcoRI independent digestions to make certain that no deletions or rearrangements of the viral genome had occurred.
293T cell clones.
293T cells were transfected with EBV WT BAC, the EBV-PK stop codon mutant construct (PKmut), or the EBV-TK stop mutant construct (TKmut) using Lipofectamine 2000 (Invitrogen) as described in the manufacturer's protocol. Selection of stable cell clones carrying the WT, PKmut, and TKmut BACmids was performed in DMEM medium with 100 μg/ml hygromycin. Stable clones were transfected with the BZLF1 and BRLF1 expression vectors (pSG5-Z and pSG5-R, gifts from S. Diane Hayward) (70) plus a gp110 expression vector (pRK5-BALF4, a gift from H. J. Delecluse) (63) to induce lytic replication. The supernatant from these cells was then used to infect Raji cells. The GFP-positive, hygromycin-resistant cell clones that produced the highest infectious viral titer were chosen for further study; several independent clones were selected and frozen at early passage for the WT and mutant viruses.
Drug susceptibility assays.
293T-WT, 293T-PKmut, and 293T-TKmut cells were cotransfected in six-well plates with a BZLF1 expression plasmid (pSG5-Z; 0.5 μg/well), a BRLF1 expression plasmid (pSG5-R; 0.5 μg/well), and a gp110 expression plasmid (pRK5-BALF4; 0.5 μg/well) to induce lytic replication in the presence or absence of GCV or ACV. In some experiments, 0.5 μg of a C-terminal FLAG-tagged WT EBV-PK expression vector (a gift from Manfred Marschall) (55) was added to the transfection mix to complement the defect of 293T-PKmut cells. Virus supernatants were harvested and filtered through an 0.8-μm filter 2 days after transfection. Viral titers were determined by the green Raji cell assay (38). Raji cells (4 × 105 in 1 ml medium/well) were infected in 24-well plates with a serial dilution of virus supernatant. Phorbol-12-myristate-13-acetate (TPA) (20 ng/ml) and sodium butyrate (3 mM) were added 24 h later. The next day, the GFP-positive Raji cells were scored using a fluorescence microscope. The number of green Raji cells per milliliter (GRU/ml) was calculated to determine the concentration of infectious virus particles. All experiments were performed in duplicate.
Immunoblot analysis.
293T-WT, 293T-PKmut, or 293T-TKmut cells were transfected with a BZLF1 expression vector in the presence or absence of a cotransfected EBV-PK expression vector to induce lytic replication. Immunoblots were performed as previously described (57). Primary antibodies used were as follows: anti-Z (mouse; 1:250 [Argene]), anti-EBV diffuse early antigen (BMRF1; mouse; 1:250 [Vector Laboratories]), anti-BGLF4 (C-terminal; rabbit; 1:1,000 [Abgent]), anti-EBV-TK antibody (rabbit; 1:2,000) (32), and β-actin antibody (mouse; 1:5,000). After primary antibody incubation, membranes were washed in 1× phosphate-buffered saline-5% milk-0.1% Tween 20 (PBST) three times (5 min for each wash) and incubated in horseradish peroxidase secondary antibody (Thermo Scientific) at a 1:10,000 (antirabbit) or 1:5,000 (antimouse) dilution. Membranes were then washed with PBST three times (15 min for each wash) and visualized by enhanced chemiluminescence (ECL) treatment (Pierce) and exposure to film.
Drug IC50 determinations.
293T-WT, 293T-PKmut, or 293T-TKmut cells were cotransfected in six-well plates with pSG5-Z (0.5 μg/well), pSG5-R (0.5 μg/well), and pRK5-BALF4 (0.5 μg/well) to induce lytic replication in the presence or absence of different doses of GCV or ACV for 48 h. For 293T-WT and 293T-TKmut cells, the doses of GCV were 0, 0.25 μg/ml, 0.5 μg/ml, 1 μg/ml, 2.5 μg/ml, and 5 μg/ml; doses of ACV were 0, 0.5 μg/ml, 1 μg/ml, 2.5 μg/ml, 5 μg/ml, and 10 μg/ml. For 293T-PKmut cells, the doses of GCV were 0, 1, 2.5, 5, 8, and 10 μg/ml; doses of ACV were 0, 5, 10, 25, 30, and 50 μg/ml. The green Raji cell assay was performed as described above to determine the titer of virus in duplicate transfections. The experiments were repeated twice. The data were analyzed by using the CurveExpert 1.4 software program to determine the 50% inhibitory concentration (IC50).
RESULTS
Establishment of 293T cell clones latently infected with WT, PKmut, and TKmut viruses.
EBV mutants containing stop codon insertions in residues 1 and 5 of the EBV-PK open reading frame or residue 2 of the EBV-TK open reading frame (in an EBV B958 strain BACmid construct) were constructed as described in Materials and Methods and transfected into 293T cells. Stable 293T cell clones with the latent form of EBV infection were then selected by treating cells with hygromycin (100 μg/ml) for 14 days. Thirty resistant clones for each virus were subsequently screened for the ability to produce high-titer virus following BZLF1 transfection as described in Materials and Methods. Clones which produced high-titer virus were kept for further experiments and frozen at early passage. Three different clones from the WT or mutant viruses in 293T cells were expanded and analyzed. Similar results were obtained with each clone.
To confirm that 293T-PKmut cells do not express EBV-PK, we examined the level of endogenous EBV-PK expression in BZLF1/BRLF1-transfected 293T-WT and 293T-PKmut cells. 293T cells with WT EBV infection but not PKmut infection had detectable EBV-PK expression (Fig. 1A). In addition, the hyperphosphorylated form of the viral BMRF1 protein, which is observed only in the presence of EBV-PK (10, 30, 31), was found in BZLF1/BRLF1-transfected 293T-WT cells but was not seen in BZLF1/BRLF1-transfected 293T-PKmut cells (Fig. 1B). Cotransfection with an EBV-PK expression vector restored the hyperphosphorylated form of BMRF1 in the PKmut virus-infected cells (Fig. 1B). This result confirms that 293T-PKmut cells do not have EBV-PK function.
FIG. 1.
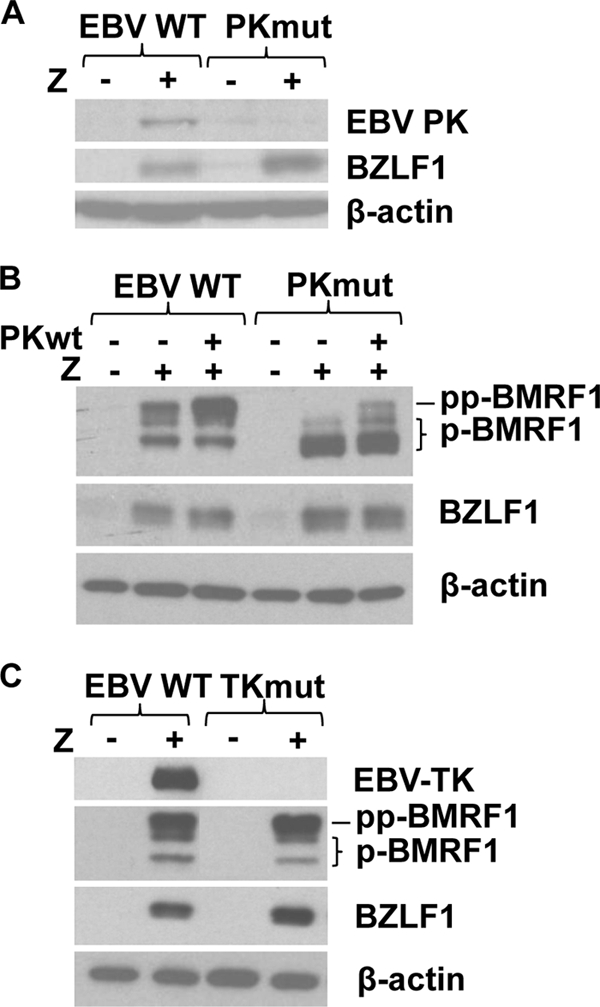
Creation of stable 293T cell clones latently infected with WT, PKmut, and TKmut viruses. (A) 293T cells latently infected with EBV WT or PKmut viruses were transfected with vector control or BZLF1 and BRLF1 expression vectors (“Z”) to induce lytic gene expression, and extracts were analyzed by immunoblotting for EBV-PK, BZLF1, and β-actin. (B) 293T cells latently infected with EBV WT or PKmut viruses were transfected with vector control or BZLF1 and BRLF1 expression vectors in the presence or absence of a cotransfected EBV-PK expression vector, as indicated. Extracts were analyzed by immunoblotting for BZLF1, BMRF1, and β-actin. The hyperphosphorylated (pp-BMRF1) and phosphorylated (p-BMRF1) forms of the BMRF1 protein are indicated; the hyperphosphorylated form of BMRF1 requires expression of EBV-PK (10, 30, 31). (C) 293T cells infected with WT or TKmut viruses were transfected with vector control or the BZLF1 (Z) and BRLF1 expression plasmids. Extracts were prepared and analyzed by immunoblotting for expression of the lytic EBV proteins, BMRF1, BZLF1, EBV-TK, and cellular β-actin.
To confirm that 293T-TKmut cells are unable to express the EBV-TK protein, we performed immunoblot analysis on the lysates of BZLF1/BRLF1-transfected 293T-WT and 293T-TKmut cells (Fig. 1C) using an anti-EBV-TK antibody. BZLF1/BRLF1 transfection induced EBV-TK and BMRF1 expression in 293T-WT cells as expected, but the EBV-TK protein was not detectable in 293T-TKmut cells, although the BMRF1 protein was induced (Fig. 1C). These results indicate that the 293T-TKmut cells do not express the EBV-TK protein.
PKmut and TKmut viruses are not defective for lytic virus production in 293T cells.
The phenotype of the PKmut virus in 293 cells versus 293T cells is described in detail elsewhere (57). Similar to the results of previous studies, loss of EBV-PK expression was found to severely impair the ability of PKmut-infected 293 cells to release infectious viral particles, since EBV-PK is required for nuclear egress of the virus in this cell type (31, 62). Somewhat surprisingly, we found that PKmut was not impaired for release of infectious viral particles following BZLF1 transfection in 293T cells (Fig. 2). The TKmut virus also replicated at least as well as WT EBV in 293T cells (Fig. 2). The ability of TKmut to replicate in 293T cells was not unexpected, since EBV mutants containing foreign gene insertions that should inactivate viral EBV-TK function have already been described and shown to replicate in tumor cell lines (58, 72). The unexpected ability of PKmut to produce virus efficiently in 293T cells allowed us to ask whether PKmut or TKmut virus is more resistant to ACV or GCV treatment than WT virus.
FIG. 2.
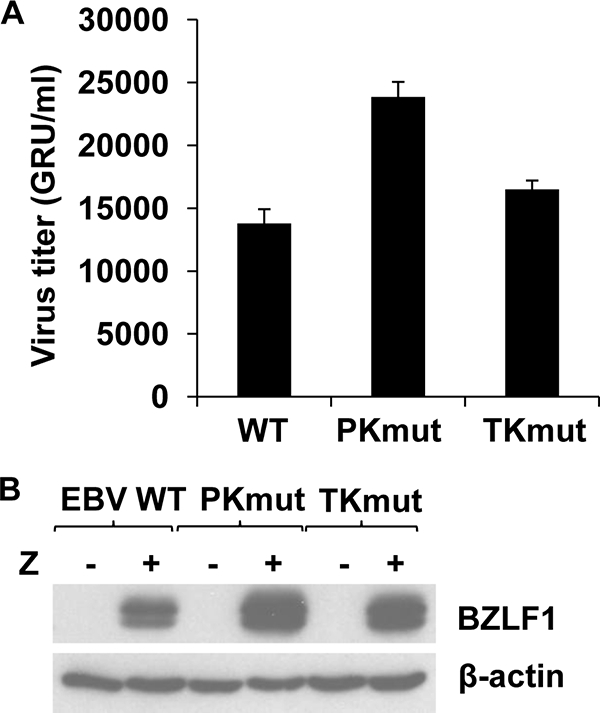
The PKmut and TKmut viruses are not defective for viral production in 293T cells. (A) 293T cells infected with WT, PKmut, or TKmut viruses were transfected with the BZLF1, BRLF1, and gp110 expression vectors to induce lytic infection. Viral titers were quantitated by infecting Raji cells with various amounts of the supernatant 72 h posttransfection and counting the number of GFP-positive Raji cells using a fluorescence microscope. The number of GFP+ Raji cells per ml (GRU/ml) is shown. Results from three independent experiments are shown. (B) Extracts from the 293T cells used to create virus for panel A were immunoblotted with antibodies against BZLF1 and β-actin.
TKmut virus remains highly sensitive to ACV.
HSV replication is efficiently inhibited by ACV (IC50 = 0.45 to 1.47 μM) (65), and the viral thymidine kinase protein is required for viral susceptibility to ACV (69). HCMV, which does not have a virally encoded thymidine kinase, is relatively resistant to the antiviral effect of ACV (IC50 = 77 ± 2.1 μM) (76). We therefore hypothesized that EBV-TK may be required for susceptibility of EBV to ACV. To determine if this is the case, 293T-WT and 293T-TKmut cells were transfected with BZLF1/BRLF1/gp110 expression vectors to induce lytic replication in the presence or absence of ACV (25 μg/ml). Viral titers were examined 2 days later using the green Raji cell assay as described in Methods. Duplicate transfections were performed for each condition, and each experiment was performed at least twice using independently derived WT and TKmut clones. As shown in Fig. 3A, ACV inhibited replication of both WT and TKmut virus by approximately 95%. These results suggest that EBV-TK does not mediate the antiviral effects of ACV in cells with lytic EBV infection, and they are consistent with the previous findings that neither highly purified EBV-TK nor EBV-TK expressed by itself in cells phosphorylates ACV (33).
FIG. 3.
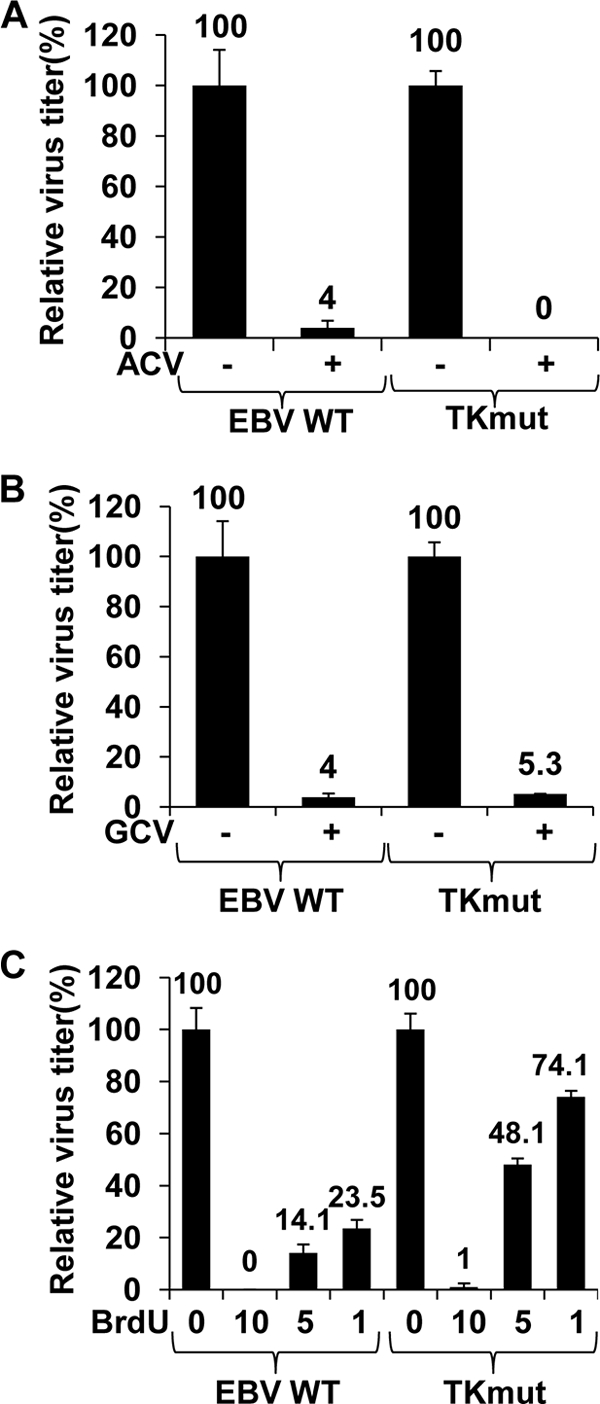
TKmut has sensitivities to GCV and ACV similar to those of WT EBV. 293T cells infected with WT EBV or TKmut EBV were transfected with BZLF1/BRLF1/gp110 expression vectors to induce lytic replication in the presence or absence of acyclovir (25 μg/ml) (A) or GCV (5 μg/ml) (B). Viral titer and viral replication were examined 48 h later as described in the legend for Fig. 2. The average viral titer produced by the 293T WT and TKmut cells in the absence of drug is set as 100% for each cell line (± standard deviation). (C) 293T cells infected with WT EBV or TKmut virus were transfected with BZLF1/BRLF1/gp110 expression vectors to induce lytic replication in the presence or absence of different concentrations of BrdU (10, 5, or 1 μg/ml) for 48 h. Viral titers were quantitated as described in the legend for Fig. 2. The average viral titer produced by the 293T WT and TKmut cells in the absence of drug is set as 100% for each cell line (± standard deviation).
TKmut virus is also highly sensitive to GCV.
HSV is susceptible to both ACV (IC50 = 0.45 to 1.47 μM) (65) and GCV (IC50 = 0.45 to 1.3 μM) (9), and in both cases this susceptibility is mediated by the virally encoded TK (15, 69). In contrast, HCMV is much more susceptible to GCV (IC50 = 5.1 μM) (76) than ACV (IC50 = 77 ± 2.1 μM) (76), and in HCMV, susceptibility to GCV is mediated by the virally encoded UL97 PK (which is a less active enzyme than HSV-TK). To determine if EBV susceptibility to the antiviral effects of GCV requires EBV-TK, 293T-WT and 293T-TKmut cells were transfected with the BZLF1/BRLF1/gp110 expression vectors to induce lytic replication in the presence or absence of GCV (5 μg/ml), and the virus titer was determined. As shown in Fig. 3B, GCV (5 μg/ml) inhibited replication of WT and TK mutant virus to similar degrees (approximately 95% at the dosage used) in 293T-WT and 293T-TKmut cells. These results indicate that EBV-TK is not required for GCV to inhibit the lytic form of EBV replication.
TKmut virus has reduced susceptibility to BrdU.
5-Bromodeoxyuridine (BrdU) is a nucleoside analog of thymidine that has antiviral properties and is a known substrate of EBV-TK (33). To study the sensitivity of the EBV-TK mutant to BrdU, 293T-WT and 293T-TKmut cells were induced into lytic infection in the presence or absence of different doses of BrdU. As shown in Fig. 3C, BrdU inhibited virus production in 293T-WT cells in a dose-dependent manner, and TKmut was less susceptible than the WT virus to the antiviral effect of low-dose BrdU. For example, BrdU at a dose of 1 μg/ml inhibited WT virus replication by 76% but inhibited TKmut virus replication by only 26%. These results indicate that the EBV TKmut virus is partially resistant to BrdU-inhibited virus replication. The ability of BrdU to prevent viral replication of TKmut at higher doses likely reflects the cellular toxicity of this drug, which is also a substrate for human TK1.
PKmut virus has reduced susceptibility to ACV and GCV.
To determine if PKmut virus has reduced susceptibility to either GCV or ACV, 293T cells infected with WT or PKmut viruses were induced into lytic infection as described previously and treated with either no drug or increasing amounts of GCV and ACV. As shown in Fig. 4A, the PKmut virus was much more resistant to GCV than the WT virus. For example, while GCV at the 5-μg/ml dose inhibited WT virus replication by 95%, it inhibited replication of the PKmut virus by only 38% (Fig. 4A). PKmut was also found to be much less susceptible than the WT virus to the antiviral effects of ACV (Fig. 4B). While the 25-μg/ml dose of ACV inhibited WT virus replication by 93%, it inhibited the replication of the PKmut virus by only 52%. Similar results were obtained using independently derived clones of the PKmut virus (data not shown).
FIG. 4.
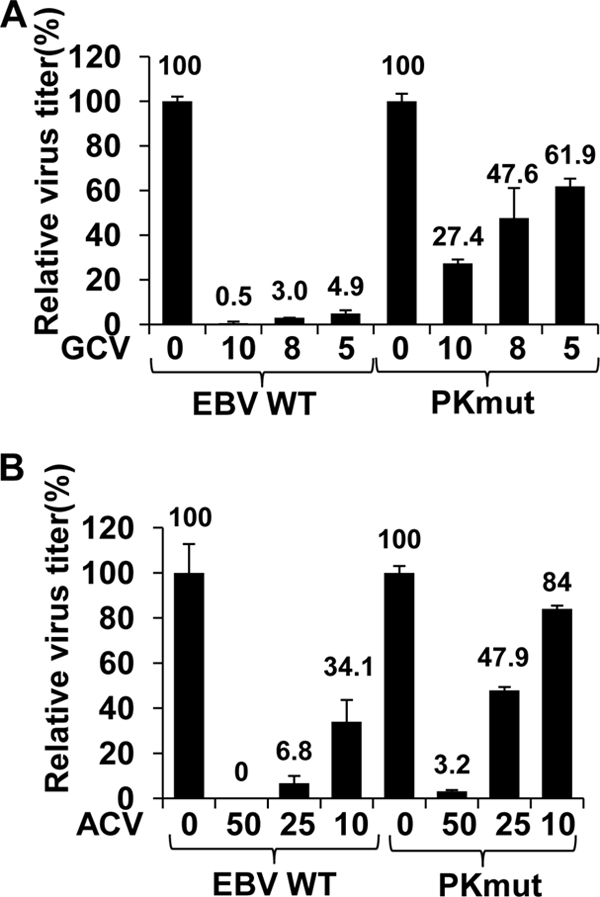
PKmut has reduced susceptibility to acyclovir and ganciclovir compared to WT EBV. 293T cells infected with WT EBV or PKmut EBV were transfected with BZLF1/BRLF1/gp110 expression vectors to induce lytic replication. Following transfection, cells were treated with no drug or different doses of GCV (10, 8, or 5 μg/ml) (A) or ACV (50, 25, or 10 μg/ml) (B) for 48 h. Viral titers were examined 48 h later. The average viral titer produced by the 293T WT and PKmut cells in the absence of drug is set as 100% for each cell line (± standard deviation). Results from two independent experiments are shown.
GCV and ACV IC50s for PKmut virus are much higher than IC50s for WT EBV.
To quantitate more precisely the relative sensitivities of the WT versus PKmut viruses to ACV and GCV, we repeated the viral titer studies (in duplicate) using a wider spectrum of drug concentrations and calculated the concentration for each drug that inhibited viral replication by 50% (IC50) as measured by the green Raji cell assay in cells infected with the WT, PKmut, and TKmut viruses. The results of these studies are shown in Table 1. In similarity to previously reported values (11, 56, 71, 86), we found that ACV has an IC50 for WT EBV of 4.1 μM (0.92 μg/ml), while GCV is more potent, with an IC50 of 1.5 μM (0.39 μg/ml). In contrast, we found that ACV has an IC50 of 36.4 μM (8.19 μg/ml) for the PKmut virus, and GCV has an IC50 of 19.6 μM (5 μg/ml). The IC50s of ACV and GCV were actually lower for the TKmut virus than for the WT virus, further confirming that EBV-TK is not required for antiviral susceptibility of EBV to either ACV or GCV.
TABLE 1.
Antiviral activities of GCV and ACV against EBV in different cell lines
| Infection of cells | IC50 for GCV, μM (μg/ml) | IC50 for ACV, μM (μg/ml) |
|---|---|---|
| EBV WT | 1.5 (0.4) | 4.1 (0.9) |
| EBV PKmut | 19.6 (5.0) | 36.4 (8.2) |
| EBV TKmut | 1.2 (0.3) | 2.8 (0.6) |
Expression of the EBV-PK protein in trans rescues antiviral susceptibility of the PKmut virus to ACV and GCV.
The finding that PKmut-infected 293T cells are much less susceptible to both GCV- and ACV-mediated inhibition of EBV replication strongly implies that EBV-PK is required for activation of these nucleoside analogues. To further confirm this, 293T-PKmut cells were transfected with BZLF1/BRLF1/gp110 expression vectors in the presence or absence of an EBV-PK expression vector. Cells were treated with GCV or ACV or left untreated, and viral titers were determined. As shown in Fig. 5A, when the EBV-PK protein was expressed in trans, suppression of PKmut viral replication by GCV (5 μg/ml) was increased from 40% to 98% (similar to the level occurring in cells infected with WT EBV). Likewise, suppression of PKmut viral replication by ACV (25 μg/ml) was increased from 73% to 99% in the presence of cotransfected EBV-PK. These results confirm that the inability of ACV and GCV to efficiently suppress viral replication of the PKmut virus is due specifically to the loss of EBV-PK expression and not some other (unintentional) alteration in the EBV genome or 293T cell clones.
FIG. 5.
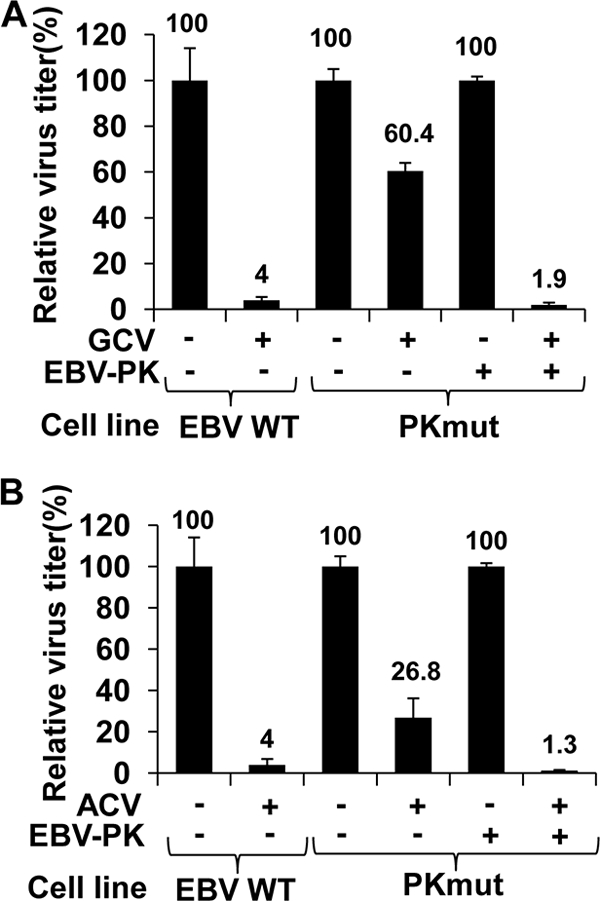
Expression of EBV-PK in trans restores susceptibility of PKmut to ganciclovir and acyclovir. 293T cells infected with WT or PKmut viruses were transfected with BZLF1/BRLF1/gp110 expression vectors to induce lytic replication in the presence or absence of a cotransfected EBV-PK expression vector, as indicated. Following transfection, cells were treated with no drug or GCV (5 μg/ml) (A) or ACV (25 μg/ml) (B) for 48 h, and viral titers were quantitated. The average viral titer produced for each transfection condition in the absence of drug is set as 100% for each cell line (± standard deviation). The average viral titer of cells treated with GCV or ACV is normalized to the average viral titer of cells in the absence of drug.
ACV and GCV have little cellular toxicity in 293T cells at doses which efficiently inhibit WT virus replication.
Phosphorylated GCV (and to a much lesser extent phosphorylated ACV) can be incorporated by cellular DNA polymerase into the host cell DNA, resulting in cell death (68). A theoretical possibility is that the major antiviral effect of these drugs is mediated through nonspecific cellular toxicity rather than inhibition of viral replication per se. To further examine this, 293T EBV-negative cells were transfected with BZLF1, BRLF1, and gp110 and treated with different doses of GCV or ACV, and the viability of cells was determined by trypan blue staining 2 days later. Compared to the solvent control, neither GCV nor ACV treatment had an obvious effect on 293T cell viability at the doses used to inhibit viral replication in these short-term assays (Fig. 6A and B).
FIG. 6.
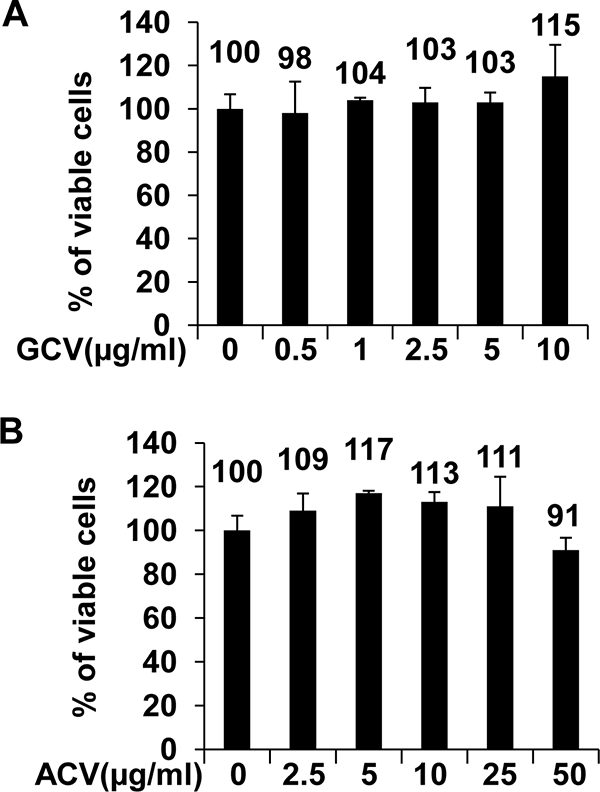
GCV and ACV are not toxic to 293T cells at the doses used to inhibit EBV replication. 293T EBV-negative cells were transfected with BZLF1/BRLF1/gp110 expression vectors and then treated with no drug or different doses of GCV (A) or ACV (B). Forty-eight hours later, cells were stained with trypan blue. The percentage of viable cells without drug treatment is set as 100%.
DISCUSSION
Although GCV and ACV both inhibit the lytic form of EBV replication in vitro and have been used to treat EBV infection in patients, it has remained unclear exactly how these drugs are converted to their active forms in lytically replicating EBV-infected cells. The conversion of these two nucleoside analogues to their monophosphorylated forms, which is the rate-limiting first step in their activation, is performed by the viral thymidine kinase in HSV-infected cells (20) but mediated by the viral protein kinase (UL97) in HCMV-infected cells (53, 75). Whereas HSV-1 TK is a polynucleoside kinase, UL97 is not known to be a nucleoside kinase at all, although it phosphorylates the acyclic purine analogs ACV and GCV. ACV and GCV are then subsequently further phosphorylated by cellular enzymes to become nucleotide triphosphates, which inhibit viral DNA polymerase-mediated viral DNA replication both as competitive alternative substrates for GTP and (in the case of ACV) as a DNA chain terminator (17, 29). Since EBV encodes both a viral thymidine kinase and a viral protein kinase (homologous to UL97), one or both of these viral proteins might potentially be required for ACV and/or GCV activity against EBV. In this paper, we used a genetic approach to inhibit expression of either EBV-TK or EBV-PK in cells with lytic EBV infection and determined whether either protein is required for ACV and/or GCV antiviral activity. Our results show that EBV-PK but not the EBV-TK is required for the antiviral effects of both ACV and GCV against EBV.
The conflicting reports about the substrate specificity of EBV-TK with regard to GCV and ACV (33, 52, 59, 78) have led to confusion in the field about whether EBV-TK and/or EBV-PK mediates ACV and/or GCV sensitivity in EBV-infected cells. We have demonstrated here that loss of EBV-TK activity in the context of the intact EBV genome does not in any way impair the ability of the virus to respond to either ACV or GCV (in fact, the IC50s of both ACV and GCV are slightly lower in cells infected with the TKmut virus than in cells infected with the WT virus). Thus, EBV-TK is certainly not required for either ACV or GCV activation in cells with lytic EBV infection. One potential explanation for this result is that both EBV-TK and EBV-PK independently induce enough ACV and GCV phosphorylation in EBV-infected cells that loss of either viral protein alone does not affect ACV/GCV susceptibility. However, this possibility is made unlikely by the finding that cells infected with EBV PKmut (which can still express EBV-TK at a normal level) are highly resistant to ACV and GCV. Nevertheless, since EBV-TK and EBV-PK have been reported to directly interact in yeast two-hybrid assays (8), consistent with the observation that EBV-TK is a substrate of EBV-PK (87), it remains theoretically possible that only a PK-phosphorylated form of EBV-TK can enhance ACV/GCV phosphorylation in cells, since EBV TK phosphorylated by cellular enzymes is without activity (33; J. D. Fingeroth, unpublished data).
Although TKmut did not show enhanced resistance to ACV/GCV (consistent with a previous report that GCV and ACV are not substrates for EBV-TK [33]), it did confer some susceptibility to the antiviral effect of the thymidine analogue, BrdU, a known substrate of EBV-TK. These results indicate that as previously suggested, thymidine nucleoside analogs (rather than guanine nucleoside analogues, such as ACV/GCV) may prove useful as anti-EBV drugs that take advantage of the EBV-TK activity (33). If so, EBV isolates that become resistant to ACV/GCV may not necessarily be resistant to thymidine nucleoside analogues, and the antiviral effects of the guanine nucleoside analogues and the thymidine nucleoside analogues together might prove synergistic for EBV and decrease the likelihood that resistance develops.
Our results here also suggest that the EBV-encoded serine/threonine protein kinase, like the HCMV UL97 kinase, induces enough GCV phosphorylation when expressed at a normal level in lytically infected cells to be clinically useful. Although the HCMV UL97 kinase is generally not thought to phosphorylate ACV efficiently enough for this drug to be used for treatment of HCMV infection in patients, ACV is currently considered by some to be the treatment of choice for lytic EBV infection in humans (37). Notably, we found that it is EBV-PK, and not EBV-TK, which mediates the susceptibility of EBV to ACV. This result suggests that either EBV-PK phosphorylates ACV more efficiently than the HCMV UL97 kinase or EBV lytic replication is better inhibited than HCMV replication by very low levels of triphosphorylated ACV. A head-to-head comparison will clarify this issue.
Although EBV-PK has a relatively low sequence homology with UL97, EBV-PK can partially complement a UL97 defect in the HCMV genome (67), and EBV-PK and HCMV UL97 may share many similar functions during lytic viral replication. The CMV UL97 kinase and EBV-PK are both required for nuclear egress of the viruses in most cell lines (35, 62), and shared cellular substrates of these two kinases include nuclear lamin A/C (35, 49), cellular elongation factor 1δ (42-44), and pRB (39; C. V. Kuny, K. Chinchilla, M. Culbertson, and R. F. Kalejta, submitted for publication). The unusual ability of the EBV PKmut virus to efficiently produce infectious viral particles in 293T cells allowed us to study the role of EBV-PK in mediating ACV/GCV susceptibility. As reported in another article, we found that both the large and small forms of the SV40 T antigens contribute to the rescue of the PKmut virus in 293T cells (57). Similarly, another group recently reported that an HCMV mutant unable to express the UL97 kinase can be partially rescued by the human papillomavirus E7 viral protein (41). Interestingly, this paper also showed that the antiviral effect of maribavir (a drug which inhibits UL97 kinase activity) (7) is highly attenuated in the presence of E7 (41). Since maribavir has also been reported to inhibit lytic EBV replication in some cell types (26, 30), it will obviously be of interest to determine if the PKmut virus is resistant to this drug in cells (such as 293T cells) where it is able to replicate.
Our results, as well as those of previous studies (45), also raise the question of whether GCV might be a more effective drug than ACV for treating lytic EBV infection in patients. We found that the IC50 of ACV for WT EBV was 4.1 μM, while the IC50 of GCV was 1.5 μM. Previous studies using Southern blot methods to measure the amount of lytic EBV viral replication also reported that the IC50 of ACV for WT EBV is considerably higher (7 to 10 μM) (71, 86) than the IC50 of GCV (found to be 1.0 μM) (11, 56). The definition of in vitro resistance of HSV isolates to ACV has varied in the literature from 4.4 to 13.2 μM, according to the method selected and various other factors (14, 22, 23). The IC50 for the EBV-PK mutant virus of acyclovir (36.4 μM) would clearly categorize it as resistant to ACV. Nevertheless, the relatively high IC50 of ACV for WT EBV, although considerably less than that for HCMV (77 ± 2.1 μM), is much higher than that for HSV (IC50 = 0.45 to 1.47 μM), raising the possibility that oral ACV might be only marginally active against lytic EBV infection in patients, particularly since the reported peak ACV serum level in patients receiving an 800-mg dose of oral acyclovir is only 7 μM (37).
In the case of GCV, the reported peak serum level of drug following a dose of oral valganciclovir (900 mg) is 23 to 26 μM (37). HCMV isolates are considered to be GCV sensitive at IC50s of <6 μM (12). Although the IC50 of GCV for WT EBV is clearly within the range predicted for GCV susceptibility in patients, the high IC50 of the PKmut EBV virus (19.6 μM) suggests that this mutant would be resistant to GCV therapy in patients.
A study directly comparing the ability of ACV versus that of GCV to treat lytic EBV infection in humans has not been performed. Interestingly, in most studies, ACV treatment has not been found to benefit patients with infectious mononucleosis (IM) (77). In a number of these studies (2, 79, 80), the peak level of ACV achieved in the serum was likely less than the IC50 for EBV (since patients received doses of 600 to 800 mg orally five times per day), which could possibly explain the lack of clinical benefit in these studies. Nevertheless, the lack of clinical benefit from oral ACV treatment in patients with IM may reflect not only the relatively low susceptibility of EBV to this drug, as well as the low oral bioavailability of ACV, but the fact that many of the symptoms of IM are mediated by the host immune response to virally infected cells with the latent form of infection. We predict that either oral valacyclovir therapy, which results in much higher serum levels of acyclovir (up to 22 μM after a dose of 1,000 mg), or oral valganciclovir therapy would be considerably more effective than oral acyclovir for treating lytic EBV infection in patients in circumstances where it is indicated. A small study using valacyclovir to treat patients with IM suggested that it may improve clinical symptoms (4). Valacyclovir was reported to effectively treat most cases of oral hairy leukoplakia (a lytic EBV infection on the tongue that occurs in immunosuppressed patients), although a few cases were resistant to this drug (82). Our results suggest that a clinical study comparing the efficacy of valganciclovir with that of valacyclovir for treating patients who are severely ill with uncontrolled IM or who have unusual complications of primary EBV infection, such as viral meningoencephalitis, may be warranted. Nevertheless, in patients with less-severe forms of IM, antiviral therapy is probably not indicated.
Acknowledgments
We thank our collaborators, Xianming Yu and Janet Mertz, at the University of Wisconsin—Madison for help in constructing the EBV-PK and -TK mutants. Thanks to Henri-Jacques Delecluse for providing us with valuable reagents.
This work was supported by grants R01-CA58853 (to S.C.K.), R01-H6064851 (to J.S.P. and S.C.K.), and P01-CA019014 (to S.C.K.), training grant T32-AI078985 (to S.R.H.), and grants R01-DE12186 (to J.D.F.), K24 CA 80935 (to J.D.F.), and R21-AI072221 (to E.G.).
Footnotes
Published ahead of print on 24 February 2010.
REFERENCES
- 1.Ambinder, R. F. 2007. Epstein-Barr virus and Hodgkin lymphoma. Hematol. Am. Soc. Hematol. Educ. Prog. 2007:204-209. [DOI] [PubMed] [Google Scholar]
- 2.Andersson, J., B. Skoldenberg, W. Henle, J. Giesecke, A. Ortqvist, I. Julander, E. Gustavsson, B. Akerlund, S. Britton, and I. Ernberg. 1987. Acyclovir treatment in infectious mononucleosis: a clinical and virological study. Infection 15(Suppl. 1):S14-S20. [DOI] [PubMed] [Google Scholar]
- 3.Andersson, J. P. 1991. Clinical aspects on Epstein-Barr virus infection. Scand. J. Infect. Dis. Suppl. 80:94-104. [PubMed] [Google Scholar]
- 4.Balfour, H. H., Jr., K. M. Hokanson, R. M. Schacherer, C. M. Fietzer, D. O. Schmeling, C. J. Holman, H. E. Vezina, and R. C. Brundage. 2007. A virologic pilot study of valacyclovir in infectious mononucleosis. J. Clin. Virol. 39:16-21. [DOI] [PubMed] [Google Scholar]
- 5.Bestman-Smith, J., I. Schmit, B. Papadopoulou, and G. Boivin. 2001. Highly reliable heterologous system for evaluating resistance of clinical herpes simplex virus isolates to nucleoside analogues. J. Virol. 75:3105-3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biron, K. K., J. A. Fyfe, S. C. Stanat, L. K. Leslie, J. B. Sorrell, C. U. Lambe, and D. M. Coen. 1986. A human cytomegalovirus mutant resistant to the nucleoside analog 9-([2-hydroxy-1-(hydroxymethyl)ethoxy]methyl)guanine (BW B759U) induces reduced levels of BW B759U triphosphate. Proc. Natl. Acad. Sci. U. S. A. 83:8769-8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biron, K. K., R. J. Harvey, S. C. Chamberlain, S. S. Good, A. A. Smith III, M. G. Davis, C. L. Talarico, W. H. Miller, R. Ferris, R. E. Dornsife, S. C. Stanat, J. C. Drach, L. B. Townsend, and G. W. Koszalka. 2002. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action. Antimicrob. Agents Chemother. 46:2365-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calderwood, M. A., K. Venkatesan, L. Xing, M. R. Chase, A. Vazquez, A. M. Holthaus, A. E. Ewence, N. Li, T. Hirozane-Kishikawa, D. E. Hill, M. Vidal, E. Kieff, and E. Johannsen. 2007. Epstein-Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. U. S. A. 104:7606-7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, C. Y., Y. N. Chang, P. Ryan, M. Linscott, G. J. McGarrity, and Y. L. Chiang. 1995. Effect of herpes simplex virus thymidine kinase expression levels on ganciclovir-mediated cytotoxicity and the “bystander effect.” Hum. Gene Ther. 6:1467-1476. [DOI] [PubMed] [Google Scholar]
- 10.Chen, M. R., S. J. Chang, H. Huang, and J. Y. Chen. 2000. A protein kinase activity associated with Epstein-Barr virus BGLF4 phosphorylates the viral early antigen EA-D in vitro. J. Virol. 74:3093-3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng, Y. C., E. S. Huang, J. C. Lin, E. C. Mar, J. S. Pagano, G. E. Dutschman, and S. P. Grill. 1983. Unique spectrum of activity of 9-[(1,3-dihydroxy-2-propoxy)methyl]-guanine against herpesviruses in vitro and its mode of action against herpes simplex virus type 1. Proc. Natl. Acad. Sci. U. S. A. 80:2767-2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chou, S., R. H. Waldemer, A. E. Senters, K. S. Michels, G. W. Kemble, R. C. Miner, and W. L. Drew. 2002. Cytomegalovirus UL97 phosphotransferase mutations that affect susceptibility to ganciclovir. J. Infect. Dis. 185:162-169. [DOI] [PubMed] [Google Scholar]
- 13.Cohen, J. I. 2000. Epstein-Barr virus infection. N. Engl. J. Med. 343:481-492. [DOI] [PubMed] [Google Scholar]
- 14.Collins, P., and M. N. Ellis. 1993. Sensitivity monitoring of clinical isolates of herpes simplex virus to acyclovir. J. Med. Virol. Suppl. 1:58-66. [DOI] [PubMed] [Google Scholar]
- 15.Darby, G., B. A. Larder, K. F. Bastow, and H. J. Field. 1980. Sensitivity of viruses to phosphorylated 9-(2-hydroxyethoxymethyl)guanine revealed in TK-transformed cells. J. Gen. Virol. 48:451-454. [DOI] [PubMed] [Google Scholar]
- 16.Datta, A. K., B. M. Colby, J. E. Shaw, and J. S. Pagano. 1980. Acyclovir inhibition of Epstein-Barr virus replication. Proc. Natl. Acad. Sci. U. S. A. 77:5163-5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Clercq, E. 2004. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2:704-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Turenne-Tessier, M., T. Ooka, G. de The, and J. Daillie. 1986. Characterization of an Epstein-Barr virus-induced thymidine kinase. J. Virol. 57:1105-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delecluse, H. J., T. Hilsendegen, D. Pich, R. Zeidler, and W. Hammerschmidt. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245-8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elion, G. B. 1983. The biochemistry and mechanism of action of acyclovir. J. Antimicrob. Chemother. 12(Suppl B):9-17. [DOI] [PubMed] [Google Scholar]
- 21.Elion, G. B., P. A. Furman, J. A. Fyfe, P. de Miranda, L. Beauchamp, and H. J. Schaeffer. 1977. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc. Natl. Acad. Sci. U. S. A. 74:5716-5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Englund, J. A., M. E. Zimmerman, E. M. Swierkosz, J. L. Goodman, D. R. Scholl, and H. H. Balfour, Jr. 1990. Herpes simplex virus resistant to acyclovir. A study in a tertiary care center. Ann. Intern. Med. 112:416-422. [DOI] [PubMed] [Google Scholar]
- 23.Erlich, K. S., J. Mills, P. Chatis, G. J. Mertz, D. F. Busch, S. E. Follansbee, R. M. Grant, and C. S. Crumpacker. 1989. Acyclovir-resistant herpes simplex virus infections in patients with the acquired immunodeficiency syndrome. N. Engl. J. Med. 320:293-296. [DOI] [PubMed] [Google Scholar]
- 24.Fixman, E. D., G. S. Hayward, and S. D. Hayward. 1992. trans-acting requirements for replication of Epstein-Barr virus ori-Lyt. J. Virol. 66:5030-5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fyfe, J. A., P. M. Keller, P. A. Furman, R. L. Miller, and G. B. Elion. 1978. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine. J. Biol. Chem. 253:8721-8727. [PubMed] [Google Scholar]
- 26.Gershburg, E., K. Hong, and J. S. Pagano. 2004. Effects of maribavir and selected indolocarbazoles on Epstein-Barr virus protein kinase BGLF4 and on viral lytic replication. Antimicrob. Agents Chemother. 48:1900-1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gershburg, E., M. Marschall, K. Hong, and J. S. Pagano. 2004. Expression and localization of the Epstein-Barr virus-encoded protein kinase. J. Virol. 78:12140-12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gershburg, E., and J. S. Pagano. 2008. Conserved herpesvirus protein kinases. Biochim. Biophys. Acta 1784:203-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gershburg, E., and J. S. Pagano. 2005. Epstein-Barr virus infections: prospects for treatment. J. Antimicrob. Chemother. 56:277-281. [DOI] [PubMed] [Google Scholar]
- 30.Gershburg, E., and J. S. Pagano. 2002. Phosphorylation of the Epstein-Barr virus (EBV) DNA polymerase processivity factor EA-D by the EBV-encoded protein kinase and effects of the L-riboside benzimidazole 1263W94. J. Virol. 76:998-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gershburg, E., S. Raffa, M. R. Torrisi, and J. S. Pagano. 2007. Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J. Virol. 81:5407-5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill, M. B., J. L. Kutok, and J. D. Fingeroth. 2007. Epstein-Barr virus thymidine kinase is a centrosomal resident precisely localized to the periphery of centrioles. J. Virol. 81:6523-6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gustafson, E. A., A. C. Chillemi, D. R. Sage, and J. D. Fingeroth. 1998. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 42:2923-2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gutierrez, M. I., J. G. Judde, I. T. Magrath, and K. G. Bhatia. 1996. Switching viral latency to viral lysis: a novel therapeutic approach for Epstein-Barr virus-associated neoplasia. Cancer Res. 56:969-972. [PubMed] [Google Scholar]
- 35.Hamirally, S., J. P. Kamil, Y. M. Ndassa-Colday, A. J. Lin, W. J. Jahng, M. C. Baek, S. Noton, L. A. Silva, M. Simpson-Holley, D. M. Knipe, D. E. Golan, J. A. Marto, and D. M. Coen. 2009. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 5:e1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammerschmidt, W., and B. Sugden. 1988. Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 55:427-433. [DOI] [PubMed] [Google Scholar]
- 37.Hayden, F. G. 2005. Antiviral drugs (other than antiretrovirials), p. 514-550. In G. L. Mandell, J. E. Bennett, and R. Dolin (ed.), Principles and practice of infectious diseases. Elsevier Inc., Philadelphia, PA.
- 38.Hong, G. K., H. J. Delecluse, H. Gruffat, T. E. Morrison, W. H. Feng, A. Sergeant, and S. C. Kenney. 2004. The BRRF1 early gene of Epstein-Barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. J. Virol. 78:4983-4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hume, A. J., J. S. Finkel, J. P. Kamil, D. M. Coen, M. R. Culbertson, and R. F. Kalejta. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320:797-799. [DOI] [PubMed] [Google Scholar]
- 40.Israel, B. F., and S. C. Kenney. 2003. Virally targeted therapies for EBV-associated malignancies. Oncogene 22:5122-5130. [DOI] [PubMed] [Google Scholar]
- 41.Kamil, J. P., A. J. Hume, I. Jurak, K. Munger, R. F. Kalejta, and D. M. Coen. 2009. Human papillomavirus 16 E7 inactivator of retinoblastoma family proteins complements human cytomegalovirus lacking UL97 protein kinase. Proc. Natl. Acad. Sci. U. S. A. 106:16823-16828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kato, K., Y. Kawaguchi, M. Tanaka, M. Igarashi, A. Yokoyama, G. Matsuda, M. Kanamori, K. Nakajima, Y. Nishimura, M. Shimojima, H. T. Phung, E. Takahashi, and K. Hirai. 2001. Epstein-Barr virus-encoded protein kinase BGLF4 mediates hyperphosphorylation of cellular elongation factor 1delta (EF-1delta): EF-1delta is universally modified by conserved protein kinases of herpesviruses in mammalian cells. J. Gen. Virol. 82:1457-1463. [DOI] [PubMed] [Google Scholar]
- 43.Kawaguchi, Y., K. Kato, M. Tanaka, M. Kanamori, Y. Nishiyama, and Y. Yamanashi. 2003. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J. Virol. 77:2359-2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawaguchi, Y., T. Matsumura, B. Roizman, and K. Hirai. 1999. Cellular elongation factor 1delta is modified in cells infected with representative alpha-, beta-, or gammaherpesviruses. J. Virol. 73:4456-4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keever-Taylor, C. A., B. Behn, S. Konings, R. Orentas, B. Davies, and D. Margolis. 2003. Suppression of EBV release from irradiated B lymphoblastoid cell-lines: superior activity of ganciclovir compared with acyclovir. Cytotherapy 5:323-335. [DOI] [PubMed] [Google Scholar]
- 46.Kit, S., M. Sheppard, H. Ichimura, S. Nusinoff-Lehrman, M. N. Ellis, J. A. Fyfe, and H. Otsuka. 1987. Nucleotide sequence changes in thymidine kinase gene of herpes simplex virus type 2 clones from an isolate of a patient treated with acyclovir. Antimicrob. Agents Chemother. 31:1483-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kutok, J. L., and F. Wang. 2006. Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. 1:375-404. [DOI] [PubMed] [Google Scholar]
- 48.Lebkowski, J. S., S. Clancy, and M. P. Calos. 1985. Simian virus 40 replication in adenovirus-transformed human cells antagonizes gene expression. Nature 317:169-171. [DOI] [PubMed] [Google Scholar]
- 49.Lee, C. P., Y. H. Huang, S. F. Lin, Y. Chang, Y. H. Chang, K. Takada, and M. R. Chen. 2008. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 82:11913-11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin, J. C., E. DeClercq, and J. S. Pagano. 1987. Novel acyclic adenosine analogs inhibit Epstein-Barr virus replication. Antimicrob. Agents Chemother. 31:1431-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin, J. C., N. D. Sista, F. Besencon, J. Kamine, and J. S. Pagano. 1991. Identification and functional characterization of Epstein-Barr virus DNA polymerase by in vitro transcription-translation of a cloned gene. J. Virol. 65:2728-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Littler, E., and J. R. Arrand. 1988. Characterization of the Epstein-Barr virus-encoded thymidine kinase expressed in heterologous eucaryotic and procaryotic systems. J. Virol. 62:3892-3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Littler, E., A. D. Stuart, and M. S. Chee. 1992. Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature 358:160-162. [DOI] [PubMed] [Google Scholar]
- 54.Littler, E., J. Zeuthen, A. A. McBride, E. Trost Sorensen, K. L. Powell, J. E. Walsh-Arrand, and J. R. Arrand. 1986. Identification of an Epstein-Barr virus-coded thymidine kinase. EMBO J. 5:1959-1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marschall, M., M. Stein-Gerlach, M. Freitag, R. Kupfer, M. van den Bogaard, and T. Stamminger. 2002. Direct targeting of human cytomegalovirus protein kinase pUL97 by kinase inhibitors is a novel principle for antiviral therapy. J. Gen. Virol. 83:1013-1023. [DOI] [PubMed] [Google Scholar]
- 56.Matthews, T., and R. Boehme. 1988. Antiviral activity and mechanism of action of ganciclovir. Rev. Infect. Dis. 10(Suppl 3):S490-S494. [DOI] [PubMed] [Google Scholar]
- 57.Meng, Q., S. R. Hagemeier, C. V. Kuny, R. F. Kalejta, and S. C. Kenney. 2010. Simian virus 40 T/t antigens and lamin A/C small interfering RNA rescue the phenotype of an Epstein-Barr virus protein kinase (BGLF4) mutant. J. Virol. 84:4524-4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Molesworth, S. J., C. M. Lake, C. M. Borza, S. M. Turk, and L. M. Hutt-Fletcher. 2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 74:6324-6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore, S. M., J. S. Cannon, Y. C. Tanhehco, F. M. Hamzeh, and R. F. Ambinder. 2001. Induction of Epstein-Barr virus kinases to sensitize tumor cells to nucleoside analogues. Antimicrob. Agents Chemother. 45:2082-2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moorman, N. J., D. O. Willer, and S. H. Speck. 2003. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J. Virol. 77:10295-10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morfin, F., and D. Thouvenot. 2003. Herpes simplex virus resistance to antiviral drugs. J. Clin. Virol. 26:29-37. [DOI] [PubMed] [Google Scholar]
- 62.Murata, T., H. Isomura, Y. Yamashita, S. Toyama, Y. Sato, S. Nakayama, A. Kudoh, S. Iwahori, T. Kanda, and T. Tsurumi. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75-81. [DOI] [PubMed] [Google Scholar]
- 63.Neuhierl, B., R. Feederle, W. Hammerschmidt, and H. J. Delecluse. 2002. Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and efficiency of infection. Proc. Natl. Acad. Sci. U. S. A. 99:15036-15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nugier, F., J. N. Colin, M. Aymard, and M. Langlois. 1992. Occurrence and characterization of acyclovir-resistant herpes simplex virus isolates: report on a two-year sensitivity screening survey. J. Med. Virol. 36:1-12. [DOI] [PubMed] [Google Scholar]
- 65.Pavic, I., A. Hartmann, A. Zimmermann, D. Michel, W. Hampl, I. Schleyer, and T. Mertens. 1997. Flow cytometric analysis of herpes simplex virus type 1 susceptibility to acyclovir, ganciclovir, and foscarnet. Antimicrob. Agents Chemother. 41:2686-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Perrine, S. P., O. Hermine, T. Small, F. Suarez, R. O'Reilly, F. Boulad, J. Fingeroth, M. Askin, A. Levy, S. J. Mentzer, M. Di Nicola, A. M. Gianni, C. Klein, S. Horwitz, and D. V. Faller. 2007. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 109:2571-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Romaker, D., V. Schregel, K. Maurer, S. Auerochs, A. Marzi, H. Sticht, and M. Marschall. 2006. Analysis of the structure-activity relationship of four herpesviral UL97 subfamily protein kinases reveals partial but not full functional conservation. J. Med. Chem. 49:7044-7053. [DOI] [PubMed] [Google Scholar]
- 68.Rubsam, L. Z., B. L. Davidson, and D. S. Shewach. 1998. Superior cytotoxicity with ganciclovir compared with acyclovir and 1-beta-D-arabinofuranosylthymine in herpes simplex virus-thymidine kinase-expressing cells: a novel paradigm for cell killing. Cancer Res. 58:3873-3882. [PubMed] [Google Scholar]
- 69.Saijo, M., T. Suzutani, M. Niikura, S. Morikawa, and I. Kurane. 2002. Importance of C-terminus of herpes simplex virus type 1 thymidine kinase for maintaining thymidine kinase and acyclovir-phosphorylation activities. J. Med. Virol. 66:388-393. [DOI] [PubMed] [Google Scholar]
- 70.Sarisky, R. T., Z. Gao, P. M. Lieberman, E. D. Fixman, G. S. Hayward, and S. D. Hayward. 1996. A replication function associated with the activation domain of the Epstein-Barr virus Zta transactivator. J. Virol. 70:8340-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schnute, M. E., D. J. Anderson, R. J. Brideau, F. L. Ciske, S. A. Collier, M. M. Cudahy, M. Eggen, M. J. Genin, T. A. Hopkins, T. M. Judge, E. J. Kim, M. L. Knechtel, S. K. Nair, J. A. Nieman, N. L. Oien, A. Scott, S. P. Tanis, V. A. Vaillancourt, M. W. Wathen, and J. L. Wieber. 2007. 2-Aryl-2-hydroxyethylamine substituted 4-oxo-4,7-dihydrothieno[2,3-b]pyridines as broad-spectrum inhibitors of human herpesvirus polymerases. Bioorg. Med. Chem. Lett. 17:3349-3353. [DOI] [PubMed] [Google Scholar]
- 72.Shimizu, N., H. Yoshiyama, and K. Takada. 1996. Clonal propagation of Epstein-Barr virus (EBV) recombinants in EBV-negative Akata cells. J. Virol. 70:7260-7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smee, D. F., R. Boehme, M. Chernow, B. P. Binko, and T. R. Matthews. 1985. Intracellular metabolism and enzymatic phosphorylation of 9-(1,3-dihydroxy-2-propoxymethyl)guanine and acyclovir in herpes simplex virus-infected and uninfected cells. Biochem. Pharmacol. 34:1049-1056. [DOI] [PubMed] [Google Scholar]
- 74.Smith, R. F., and T. F. Smith. 1989. Identification of new protein kinase-related genes in three herpesviruses, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. J. Virol. 63:450-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sullivan, V., C. L. Talarico, S. C. Stanat, M. Davis, D. M. Coen, and K. K. Biron. 1992. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 358:162-164. [DOI] [PubMed] [Google Scholar]
- 76.Talarico, C. L., T. C. Burnette, W. H. Miller, S. L. Smith, M. G. Davis, S. C. Stanat, T. I. Ng, Z. He, D. M. Coen, B. Roizman, and K. K. Biron. 1999. Acyclovir is phosphorylated by the human cytomegalovirus UL97 protein. Antimicrob. Agents Chemother. 43:1941-1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Torre, D., and R. Tambini. 1999. Acyclovir for treatment of infectious mononucleosis: a meta-analysis. Scand. J. Infect. Dis. 31:543-547. [DOI] [PubMed] [Google Scholar]
- 78.Tung, P. P., and W. C. Summers. 1994. Substrate specificity of Epstein-Barr virus thymidine kinase. Antimicrob. Agents Chemother. 38:2175-2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tynell, E., E. Aurelius, A. Brandell, I. Julander, M. Wood, Q. Y. Yao, A. Rickinson, B. Akerlund, and J. Andersson. 1996. Acyclovir and prednisolone treatment of acute infectious mononucleosis: a multicenter, double-blind, placebo-controlled study. J. Infect. Dis. 174:324-331. [DOI] [PubMed] [Google Scholar]
- 80.van der Horst, C., J. Joncas, G. Ahronheim, N. Gustafson, G. Stein, M. Gurwith, G. Fleisher, J. Sullivan, J. Sixbey, S. Roland, et al. 1991. Lack of effect of peroral acyclovir for the treatment of acute infectious mononucleosis. J. Infect. Dis. 164:788-792. [DOI] [PubMed] [Google Scholar]
- 81.Vetsika, E. K., and M. Callan. 2004. Infectious mononucleosis and Epstein-Barr virus. Expert Rev. Mol. Med. 6:1-16. [DOI] [PubMed] [Google Scholar]
- 82.Walling, D. M., C. M. Flaitz, and C. M. Nichols. 2003. Epstein-Barr virus replication in oral hairy leukoplakia: response, persistence, and resistance to treatment with valacyclovir. J. Infect. Dis. 188:883-890. [DOI] [PubMed] [Google Scholar]
- 83.Westphal, E. M., A. Mauser, J. Swenson, M. G. Davis, C. L. Talarico, and S. C. Kenney. 1999. Induction of lytic Epstein-Barr virus (EBV) infection in EBV-associated malignancies using adenovirus vectors in vitro and in vivo. Cancer Res. 59:1485-1491. [PubMed] [Google Scholar]
- 84.Yang, C. S., M. J. Vitto, S. A. Busby, B. A. Garcia, C. T. Kesler, D. Gioeli, J. Shabanowitz, D. F. Hunt, K. Rundell, D. L. Brautigan, and B. M. Paschal. 2005. Simian virus 40 small t antigen mediates conformation-dependent transfer of protein phosphatase 2A onto the androgen receptor. Mol. Cell. Biol. 25:1298-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu, X., Z. Wang, and J. E. Mertz. 2007. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog. 3:e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zacny, V. L., E. Gershburg, M. G. Davis, K. K. Biron, and J. S. Pagano. 1999. Inhibition of Epstein-Barr virus replication by a benzimidazole L-riboside: novel antiviral mechanism of 5,6-dichloro-2-(isopropylamino)-1-beta-L-ribofuranosyl-1H-benzimidazole. J. Virol. 73:7271-7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu, J., G. Liao, L. Shan, J. Zhang, M. R. Chen, G. S. Hayward, S. D. Hayward, P. Desai, and H. Zhu. 2009. Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J. Virol. 83:5219-5231. [DOI] [PMC free article] [PubMed] [Google Scholar]