

Abstract
Leukocyte access into the central nervous system (CNS) parenchyma is tightly regulated by the blood-brain barrier (BBB). Leukocyte migration through the endothelial cell wall into the perivascular space is well characterized; however, mechanisms regulating their penetration through the glia limitans into the parenchyma are less well studied, and the role of monocytes relative to neutrophils is poorly defined. Acute viral encephalitis was thus induced in CCL2-deficient (CCL2−/−) mice to specifically abrogate monocyte recruitment. Impaired monocyte recruitment prolonged T cell retention in the perivascular space, although no difference in overall CNS accumulation of CD4 or CD8 T cells was detected by flow cytometry. Delayed penetration to the CNS parenchyma was not associated with reduced or altered expression of either matrix metalloproteinases (MMP) or the T cell chemoattractants CXCL10 and CCL5. Nevertheless, decreased parenchymal leukocyte infiltration delayed T cell-mediated control of virus replication as well as clinical disease. These data are the first to demonstrate that the rapid monocyte recruitment into the CNS during viral encephalitis is dispensable for T cell migration across the blood vessel endothelium. However, monocytes facilitate penetration through the glia limitans. Thus, the rapid monocyte response to viral encephalitis constitutes an indirect antiviral pathway by aiding access of effector T cells to the site of viral infection.
The blood-brain barrier (BBB) is a key feature contributing to the immune-specialized environment of the central nervous system (CNS); others are the paucity of dendritic cells, low major histocompatibility complex (MHC) expression, and relative lack of lymphatic drainage (10, 15). The complex composition of the BBB tightly regulates CNS leukocyte entry under physiological conditions (4). However, disruption of the BBB induced by infection, trauma, or autoimmunity is critical in initiating parenchymal inflammation. While leukocyte entry into the CNS parenchyma is beneficial in controlling microbial infections, dysregulated recruitment is associated with chronic neuroinflammatory diseases such as HIV-associated neurological disorders and multiple sclerosis (MS) (13, 51). Several distinct physical barriers must be breached during leukocyte migration into the parenchyma. At postcapillary venules of the BBB, where leukocytes extravasate from blood into the CNS (4, 10, 34), cell migration is regulated at two stages (34). First, activated leukocytes enter the perivascular space by migrating across the vessel wall composed of endothelial cells connected by tight junctions and associated with a basement membrane (29). This process involves tethering/rolling, activation, adhesion, and diapedesis and is regulated by adhesion molecules, chemokines, and chemokine receptors (10, 29). Once in the perivascular space, inflammatory cells must further penetrate the glia limitans to enter the CNS parenchyma. This barrier is composed of astrocyte foot processes associated with a distinct basement membrane (24, 41). In contrast to the well-defined mechanisms regulating migration across the endothelial cell layer, factors governing migration through the glia limitans are less well described. As leukocyte access to the CNS parenchyma is associated with clinical symptoms during inflammatory disorders (46, 48) but is also necessary for antimicrobial control, understanding the components regulating parenchymal leukocyte entry may lead to more refined therapeutic strategies controlling this process.
A role for monocytes in facilitating transmigration across the glia limitans was noted by prevention of clinical disease in the absence of monocytes in the experimental autoimmune encephalitis (EAE) model of MS due to leukocyte accumulation in the perivascular space (48). In contrast, monocyte depletion does not alter parenchymal T cell infiltration after trauma-induced brain inflammation (14). These opposing data suggest that monocyte-dependent migration into the CNS parenchyma depends upon the nature of the CNS insult. Monocytes are a component of viral encephalitis in humans and animal models, including HIV, simian immunodeficiency virus (9, 11, 27), and West Nile virus encephalitis (17). In vitro data further suggest that the chemokine CCL2 (monocyte chemoattractant protein 1 [MCP-1]), which is essential for monocyte recruitment (26), enhances the ability of peripheral lymphocytes from HIV-infected patients to cross the BBB (9, 11, 27). However, a specific role of monocytes in glia limitans disruption during viral encephalitis has not been addressed.
A well-characterized model of viral encephalitis was chosen to better define the role of monocytes in facilitating lymphocyte access to the CNS parenchyma. Mice infected with the nonfatal neurotropic JHM strain of mouse hepatitis virus (JHMV) develop an acute encephalitis associated with immune-mediated primary demyelination (6). Neutrophils and monocytes are the first cells to infiltrate the CNS (6), consistent with early upregulation of CCL2 and the neutrophil chemoattractants CXCL1 (KC) and CXCL2 (macrophage-inflammatory protein 2 [MIP-2α]) (25). Depletion of neutrophils and inflammatory monocytes during acute JHMV infection decreased BBB permeability and CNS leukocyte infiltration (54). However, the relative contribution of neutrophils and monocytes in CNS access remains unresolved. A specific role for monocytes in BBB disruption was analyzed following JHMV infection of CCL2 deficient (CCL2−/−) mice. The absence of CCL2 specifically disrupts recruitment of blood-derived monocytes into parenchymal tissues (20, 26), while neutrophil recruitment remains intact. Importantly, as circulating monocytes and lymphoid tissue macrophages are not affected, priming of antiviral T cell responses in cervical lymph nodes is not expected to be impaired. Altered T cell access to the CNS parenchyma can thus be directly attributed to monocyte-dependent functions.
Impaired parenchymal T cell access in infected CCL2−/− mice correlated with enhanced confinement within the perivascular space and a delay in both onset of clinical symptoms and control of virus replication. Furthermore, monocyte-mediated disruption of the glia limitans could not be attributed to enhanced or altered activity of matrix metalloproteinases (MMPs). These data are the first to demonstrate a critical contribution of monocytes in aiding lymphocyte access across the glia limitans into the CNS parenchyma during acute viral encephalitis. Furthermore, while monocyte-facilitated migration of activated lymphocytes through the glia limitans is beneficial in early viral intervention, this process contributes to clinical disease. Overall the results support a common role of monocytes in disrupting the BBB via alterations in the glia limitans independent of the distinct stimuli initiating and propagating leukocyte CNS recruitment during autoimmune-mediated and virus-induced inflammation.
MATERIALS AND METHODS
Mice.
C57BL/6 mice were obtained from the National Cancer Institute (Frederick, MD). Homozygous CCL2-deficient (CCL2−/−) mice were originally obtained from B. J. Rollins (Dana-Farber Cancer Institute, Boston MA) (26) and were backcrossed for eight generations to C57BL/6 mice (22). All mice were used at 6 to 7 weeks of age. All procedures were performed in compliance with the Cleveland Clinic Institutional Animal Care and Use Committee approved protocols.
Virus.
The glia-tropic JHMV-neutralizing monoclonal antibody (MAb)-derived 2.2v-1 variant was used for all infections (12). Virus was propagated in the presence of MAb J2.2 to prevent reversion to the highly lethal parental virus and plaque assayed on monolayers of DBT cells (43). Mice were infected in the left hemisphere with 250 PFU of JHMV diluted in endotoxin-free Dulbecco's phosphate-buffered saline (PBS) in a final volume of 30 μl. Clinical disease severity was graded daily as previously described (12): 0, healthy; 1, hunched back; 2, partial hind limb paralysis or inability to maintain the upright position; 3, complete hind limb paralysis; 4, moribund or dead. Virus titers in the CNS were determined as described previously (43). Briefly, brains were homogenized individually in Dulbecco's PBS using Ten Broeck tissue homogenizers. Homogenates were clarified by centrifugation at 400 × g for 7 min at 4°C. Supernatants were stored at −70°C until assayed for infectious virus by plaque assays.
Neutrophil depletion.
Neutrophils were depleted by intraperitoneal administration of 250 μg of anti-Ly6G (clone 1A8) MAb at day −1, at the time of infection, and every other day postinfection (p.i.). Control animals received the same amount of a rat IgG2a isotype control MAb.
Isolation of CNS mononuclear cells.
After perfusion with PBS, brains were homogenized as described above. Cell pellets were resuspended in RPMI containing 25 mM HEPES (pH 7.2) and adjusted to 30% Percoll (Pharmacia, Uppsala, Sweden). A 1-ml underlay of 70% Percoll was added before centrifugation at 800 × g for 30 min at 4°C. Cells were recovered from the 30%-70% interface (5) and washed with RPMI before analysis.
Flow cytometry.
CNS cell suspensions were blocked with anti-mouse CD16/CD32 (clone 2.4G2; BD Pharmingen) MAb on ice for 15 min before staining. Cells were stained with fluorescein isothiocyanate (FITC)-, phycoerythrin (PE)-, peridinin chlorophyll protein (PerCP)-, or allophycocyanin (APC)-conjugated MAb for 30 min on ice in PBS containing 0.1% bovine serum albumin. Expression of surface markers was characterized with MAbs (all from BD Pharmingen except when indicated) specific for CD45 (clone Ly-5), CD4 (clone GK1.5), CD8 (clone 53-6.7), CD11b (clone M1/70), F4/80 (Serotec, Raleigh, NC), Ly-6G (clone 1A8), NK1.1 (clone PK136), and I-A/I-E (clone 2G9). Virus-specific CD8 T cells were identified using H-2Db/S510 MHC class I tetramers as described previously (5). Samples were analyzed using a FACSCalibur flow cytometer and CellQuest Software (BD Biosciences, Mountain View, CA).
Cell sorting.
CNS cells isolated from infected mice at day 3 p.i. were stained as described above with FITC-Ly6G, PE-F4/80, PerCP-CD11b, and APC-CD45. Neutrophils (Ly-6G+ F4/80− CD11b+) and macrophages (Ly-6G− F4/80+ CD11b+) were purified using a FACSAria (BD Biosciences). Totals of 88,000 neutrophils and 424,000 macrophages were isolated from six mice, with 99.6% and 98.7% purity, respectively.
Histopathological analysis.
After PBS perfusion, brains and spinal cords were fixed in 10% neutral buffered formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin as described previously (35). For analysis of CD45 and CD4 distribution, mice were perfused with ice-cold PBS followed by 4% paraformaldehyde (PFA). Brains and spinal cords were dissected, fixed for 1 h in 4% PFA at 4°C, and then incubated with sucrose gradients as follows: 30 min with 15% sucrose at room temperature, 30 min with 20% sucrose at 4°C, and overnight with 30% sucrose at 4°C. Tissues were then stored in cryoprotection solution until preparation of 30-μm microtome sections. For analysis of CD8 distribution, mice were perfused with ice-cold PBS. Brains and spinal cords were then quickly frozen in liquid nitrogen in OCT and kept at −80°C until 10-μm sections were prepared. Sections were fixed with methanol for 5 min for CD8 staining, treated with 1% Triton X-100 for 30 min and with blocking solution for 30 min, and then stained with rabbit anti-mouse laminin (Cedarlane Laboratories, Ontario, Canada) or rat anti-mouse CD45 (Serotec), CD4 (BD Pharmingen), or CD8 MAb overnight at 4°C. Alexa Fluor 594 goat anti-rabbit (Invitrogen, Carlsbad, CA) and biotinylated rat anti-mouse (Vector Laboratories) immunoglobulins were added and left for 1 h, followed by streptavidin-Alexa 488 (BD Pharmingen). Sections were mounted with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories) and analyzed using a Leica DM4000B fluorescence microscope. For quantification, 10 pictures per animal were analyzed in areas of inflammation at each time point.
Zymography.
Zymography was performed as described previously (54). Briefly, cells purified from the CNS were resuspended in lysis buffer (1% Triton X-100, 300 mM NaCl, 50 mM Tris [pH 7.4]), and lysates from 2.5 × 105 cells were separated on 10% acrylamide gels containing 1% gelatin (Bio-Rad, Hercules, CA). Following electrophoresis, gels were consecutively placed in 1× renaturing buffer (Bio-Rad) for 30 min at room temperature, 1× developing buffer (Bio-Rad) for 20 min at room temperature, and then overnight at 37°C. Gels were then stained in 0.25% Coomassie brilliant blue R-250 (Bio-Rad) and destained with the destain solution (Bio-Rad).
Gene expression analysis.
RNA was prepared from individual brains of three mice per group by extraction with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. First-strand cDNA synthesis used SuperScript II reverse transcriptase (Invitrogen) with oligo(dT)12-18 primers (Invitrogen). Semiquantitative RNA expression was assessed using a LightCycler and a SYBR green kit (Roche, Basel, Switzerland) and the following primer sets: CXCL10, 5′-GACGGTCCGCTGCAACTG-3′ (forward) and 5′-GCTTCCCTATGGCCCTCATT-3′ (reverse); CCL5, 5′-GCAAGTGCTCCAATCTTGCA-3′ (forward) and 5′-CTTCTCTGGGTTGGCACACA-3′ (reverse); CCL3, 5′-CCAAGTCTTCTCAGCGCCAT-3′ (forward) and 5′-GAATCTTCCGGCTGTAGGAGAAG-3′ (reverse); CXCL12, 5′-CCAGAGCCAACGTCAAGCAT-3′ (forward) and 5′-CAGCCGTGCAACAATCTGAA-3′ (reverse); MMP2, 5′-TTCCCTAAGCTCATCGCAGACT-3′ (forward) and 5′-CACGCTCTTGAGACTTTGGTTCT-3′ (reverse); MMP3, 5′-TTTAAAGGAAATCAGTTCTGGGCTATA-3′ (forward) and 5′-CGATCTTCTTCACGGTTGCA-3′ (reverse); MMP7, 5′-TGGCTTCGAAGGAGAGATC-3′ (forward) and 5′-CGAAGGCATGACCTAGAGTGTTC-3′ (reverse); MMP12, 5′-GGAGCTCACGGAGACTTCAACT-3′ (forward) and 5′-CCTTGAATACCAGGTCCAGGATA-3′ (reverse); MMP14, 5′-TAAGCACTGGGTGTTTGACGAA-3′ (forward) and 5′-CCCTCGGCCAAGCTCCT-3′ (reverse); tissue inhibitor of MMP 1 (TIMP1), 5′-CCAGAGCCGTCACTTTGCTT-3′ (forward) and 5′-AGGAAAAGTAGACAGTGTTCAGGCTT-3′ (reverse); TIMP2, 5′-ACGCTTAGCATCACCCAGAAG-3′ (forward) and 5′-TGGGACAGCGAGTGATCTTG-3′ (reverse); and TIMP3, 5′-ATCCCCAGGATGCCTTCTG-3′ (forward) and 5′-CCCTCCTTCACCAGCTTCTTT-3′ (reverse). The linearity of each primer pair was confirmed to have a correlation coefficient of >0.98 by measuring 5-fold dilutions of cDNA samples. Levels of mRNA expression were normalized to ubiquitin mRNA and converted to a linearized value using the formula [1.8e(CTubiquitin − CTgene x)] × 105 as previously described (53), where CT is the threshold cycle value.
RESULTS
Inflammatory monocytes dominate CNS inflammatory cells early in infection.
Neutrophils and monocytes are the first cells to infiltrate the CNS following infection with the sublethal glia-tropic JHMV (6). Whereas neutrophils, characterized as CD45hi Ly6Clow F4/80− Ly6Ghi (6, 45), represented a small percentage of CNS infiltrating leukocytes following infection (Fig. 1A) (∼6% at day 3 p.i.), monocytes exhibiting a CD45hi Ly6Chi phenotype comprised ∼65% of the total inflammatory population at days 3 and 5 p.i. (Fig. 1A to C). This high frequency subsequently declined to ∼20% by day 10 p.i. as T cells were recruited into the infected CNS. A minor proportion of Ly6Chi F4/80+ monocytes expressed the MHC class II at day 3 p.i. (Fig. 1B and C), which nevertheless increased during the course of infection (Fig. 1C). These data confirmed that Ly6Chi monocytes, defined as inflammatory cells (44, 45), migrate into the CNS, where they represent the major population at early times p.i.
FIG. 1.

Monocytes represent the major population of CNS-infiltrating leukocytes early after infection. (A) Representative density plot of CNS-derived cells at 3 day p.i. stained with anti-CD45 and Ly-6C MAbs. CNS-infiltrating leukocytes (CD45hi) contain a prominent population of Ly6Chi monocytes (R8; 68.13% of CD45hi) and a minor proportion of Ly6Cint neutrophils (R7; 5.25% of CD45hi). (B) F4/80 and MHC class II expression within the CD45hi Ly6Cint and the CD45hi Ly6Chi myeloid populations at days 3 and 5 p.i. (C) Kinetics of MHC class II expression by CD45hi Ly6Chi F4/80+ monocytes during the course of infection. Data represent means for three mice at each time point.
CCL2 is specifically required for CNS monocyte recruitment.
The contribution of monocytes in regulating leukocyte accumulation in the CNS following virus infection was assessed by flow cytometry. Recruitment of CD45hi bone marrow-derived leukocytes into the CNS of CCL2−/− infected mice was significantly reduced relative to that in wild-type (WT) mice at days 3 and 5 p.i. (Fig. 2). However, CNS infiltration was similar in both groups by day 7 p.i. and subsequently declined with similar kinetics. As monocytes comprised the major population of infiltrating cells early in WT mice, decreased CNS leukocyte recruitment in the absence of CCL2 was most likely attributable to a deficit in monocytes. However, in addition to monocytes, CCL2 also attracts natural killer (NK) cells and T lymphocytes (18) by binding to CCR2. To verify that CCL2 deficiency affects predominantly monocytes, CNS accumulations of monocytes (F4/80+), neutrophils (Ly6G+ F4/80−), NK cells, and CD4 and CD8 T cells in infected CCL2−/− and control mice were compared. In WT mice, monocytes were rapidly recruited, remained stable until day 7 p.i., and decreased to ∼20% of the infiltrating cells by day 10 p.i. (Fig. 2). In contrast, monocyte infiltration into the CNS of infected CCL2−/− mice was reduced by ∼80% at all time points p.i.
FIG. 2.

CCL2 is required for CNS monocyte recruitment. CNS inflammation in infected CCL2−/− and WT was mice analyzed by flow cytometry at the indicated times p.i. Numbers of total inflammatory leukocytes (CD45hi), macrophages (F4/80+), neutrophils (Ly-6G+), CD4+ T cells, CD8+ T cells, and Db/S510 tetramer-positive virus-specific CD8 T cells per brain are shown. Data represent means (± standard errors of the means) from three separate experiments with three pooled mice per time point per experiment (n = 9 per group). *, P < 0.05.
Although Ly-6G+ neutrophils were ∼10-fold fewer than monocytes within the CNS of WT mice, comparable numbers were present at day 3 p.i. in both WT and CCL2−/− mice (Fig. 2). However, while neutrophils declined to nearly the limit of detection by day 7 p.i. in WT mice, they persisted in the CNS of CCL2−/− mice as evidenced by a 2.5-fold increase, potentially compensating for the absence of monocytes (Fig. 2). Similar NK cell numbers in both groups supported a redundant role of CCL2 in NK cell trafficking (data not shown). CD4 and CD8 T cells began to accumulate within the CNS at day 5 p.i. and peaked between days 7 and 10 p.i. in both WT and CCL2−/− mice (Fig. 2). Similar CD4 and CD8 T cell infiltration in both groups, including CD8 T cells specific for the viral spike epitope (S510), demonstrated that T cell recruitment into the CNS is not affected by the absence of CCL2 or other chemokines potentially secreted by monocytes. There was also no indication of reduced Db/S510 tetramer reactivity on CD8 T cells in cervical lymph nodes or spleen in CCL2−/− mice (data not shown), supporting unimpaired peripheral virus-specific CD8 T cell activation and expansion. CCL2 thus regulates early CNS infiltration of bone marrow-derived cells during viral encephalitis mainly by influencing monocyte recruitment. The resulting decreased overall infiltration is nevertheless overcome as T cells accumulate to high frequencies.
Impaired monocyte recruitment delays disease onset and virus clearance.
Monocytes have been implicated in exacerbating encephalitis by numerous functions, including release of MMPs, tumor necrosis factor, and inducible nitric oxide synthase (28, 50). The onset of encephalitic symptoms, characterized by hunched posture and ruffled fur, was clearly different between the two groups (Fig. 3A). Whereas WT mice began to exhibit encephalitis at day 6 p.i. (Fig. 3A), CCL2−/− mice did not develop clinical symptoms until day 7 p.i., indicating a short yet consistent delay in disease onset (Fig. 3A). Nevertheless, both groups displayed identical signs of paralysis at day 9 and subsequent times p.i. (Fig. 3A). Mortality rates (∼10%) did not differ between the two groups (data not shown).
FIG. 3.
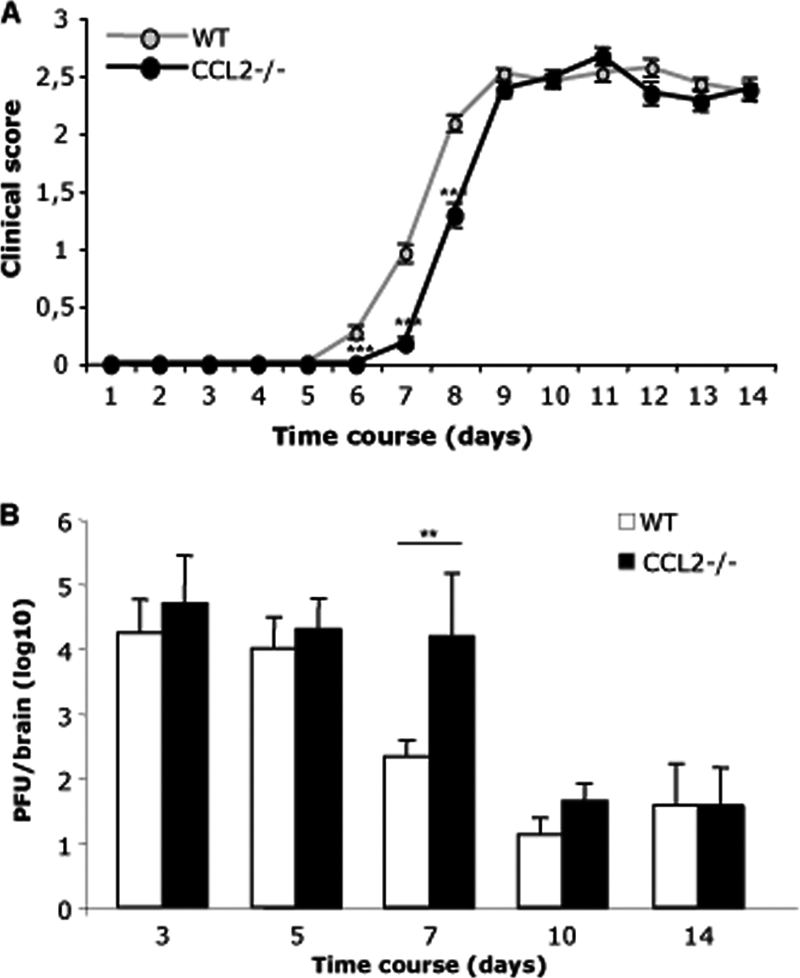
Impaired monocyte recruitment delays disease onset and virus clearance. (A) Clinical symptoms were monitored daily according to the following grades: 0, healthy; 1, hunched back; 2, partial hind limb paralysis or inability to maintain the upright position; 3, complete hind limb paralysis; 4, moribund or dead. Data represent means (± standard errors of the means) for 60 WT mice and 54 CCL2−/− mice from three separate experiments. ***, P < 0.001. (B) Virus replication in brains of WT and CCL2−/− mice analyzed by plaque assay. Data represent the averages (± standard deviations) for three mice per time point per experiment from two separate experiments. (n = 6 per group) **, P < 0.01.
Clinical signs of encephalitis correlate with CNS T cell infiltration and T cell-mediated antiviral function in infected WT mice (6). Despite initial containment by alpha/beta interferon (IFN-α/β)-mediated mechanisms (23), CD8 T cells play a major role in reducing viral replication via perforin- and IFN-γ-mediated mechanisms (6). Despite overall similar T cell accumulation in WT and CCL2−/− mice, the delayed disease onset suggested an altered antiviral function. CCL2−/− mice harbored CNS virus loads similar to those in WT mice at days 3 and 5 p.i., confirming no differences in initial viral replication and innate immune control (Fig. 3B). However, as infectious virus in the CNS of WT mice began to decrease at day 7 p.i., consistent with exertion of T cell effector function, replication remained uncontrolled in CCL2−/− mice. Nevertheless, this impaired viral control was subsequently overcome (Fig. 3B). The delay in virus control supported lagging T cell effector function within the CNS in the absence of CCL2, despite comparable virus-specific T cell activation and CNS recruitment. Furthermore, the delay in onset of clinical disease was also transient and was overcome 3 days later, when both groups displayed similar viral loads and disease symptoms (Fig. 3).
Leukocytes are retained in the perivascular space of CCL2−/− mice.
Delayed disease onset and virus clearance in the absence of monocytes suggested that effector T cells are impaired in accessing the parenchyma through the glia limitans. Potential T cell retention in the perivascular space was supported by the absence of clinical symptoms when monocyte recruitment is abrogated during EAE (46, 48). Indeed, perivascular leukocyte accumulation was profound in the CNS of CCL2−/− compared to WT mice at day 5 p.i. (Fig. 4A). To quantify relative leukocyte distributions, laminin within the basal membranes was visualized to define the borders imposed by endothelial cells on one side and the glia limitans on the other (Fig. 4B). Lymphocytes were too sparse at day 3 p.i. for consistent quantitative assessment. However, at day 5 p.i. a higher proportion of leukocytes accumulated within the perivascular space in CCL2−/− than in WT mice, correlating with a significant relative leukocyte reduction in the parenchyma (Fig. 4C). However, these differences resolved after 5 days p.i., (Fig. 4C), as evidenced by equalized leukocyte distribution in both groups at day 7 and enhanced accumulation within the parenchyma by 10 p.i., concomitant with decreased cellularity within the perivascular space (Fig. 4C). These data imply that monocytes do not regulate leukocyte access at the endothelial barrier but rather play a vital role in promoting migration from the perivascular space through the glia limitans into parenchymal sites. The sparse monocyte CNS infiltration in CCL2−/− mice predicted that T cells were the major population accumulating in the perivascular space at day 5 p.i. A correlation between impaired parenchymal T cell access and delayed disease onset and virus control was thus assessed by directly studying the distribution of T cells. Despite their sparse numbers at day 5 p.i., retention of CD8 T cells in the perivascular space was increased in CCL2−/− mice compared to controls (Fig. 4D). No difference was observed at later time points p.i., consistent with the lack of retention by the entire inflammatory infiltrate. In both WT and CCL2−/− mice, the majority of CD8 T cells reached the CNS parenchyma by day 10 p.i. (Fig. 4D). CD4 T cells also exhibited impaired parenchymal infiltration in CCL2−/− mice. However, in contrast to CD8 T cells, CD4 T cells preferentially accumulated in the perivascular space until day 7 p.i. in CCL2−/− mice (Fig. 4E), possibly due to the expression of the tissue inhibitor of MMPs, TIMP1 (53). Although CD4 T cells eventually migrated to the parenchyma by day 10 p.i. in CCL2−/− mice, the percentage of parenchymal infiltration remained lower than that in WT mice, in which ∼80% of CD4 T cells were localized in the parenchyma at this time. These data supported delayed CD4 T cell trafficking to parenchyma compared to CD8 T cells (42). Retention of CD8 and CD4 T cells in the perivascular space of CCL2−/− mice thus directly correlated with the impaired ability of monocytes to promote T cell migration across the glia limitans.
FIG. 4.

Leukocyte retention in the perivascular space of CCL2−/− mice. (A) Brain sections of WT and CCL2−/− mice at day 5 p.i. were stained with hematoxylin and eosin (H&E). Perivascular inflammation was prominent in CCL2−/− compared to WT mice. Pictures are representative of three mice from each group. Scale bar, 300 μm. (B) Leukocyte localization was analyzed using anti-CD45 (green) and anti-laminin (red) antibodies. Scale bar, 25 μm. (C to E) CD45+ cells (C) and CD8 (D) and CD4 (E) T cells in perivascular space versus parenchyma quantified in CCL2−/− and WT mice at days 5, 7, and 10 p.i. For quantification, 10 pictures per animal were analyzed in areas of inflammation at each time point. Data are representative of three mice from each group (means ± standard errors of the means). *, P < 0.05.
Monocyte-mediated disruption of the glia limitans is MMP independent.
Retention of T cells in the perivascular space of CCL2−/− mice implicated monocytes in promoting migration through the glia limitans via chemokine secretion and/or proteolytic enzyme activity. However, transcription of neither CXCL10 (IP-10) or CCL5 (RANTES) mRNA, encoding two major T cell chemoattractants early during JHMV infection (25), was reduced in infected CCL2−/− compared to WT mice (Fig. 5). CCL3 (MIP-1α) mRNA, encoding a chemoattractant for both T cells and macrophages, was also similarly expressed early after infection in both groups (Fig. 5). Furthermore, CXCL12 (SDF-1), which is implicated in retaining inflammatory cells within the perivascular space during EAE and West Nile virus encephalitis (30, 31), decreased with similar kinetics in both WT and CCL2−/− mice during the course of infection (Fig. 5). Thus, impaired chemokine production did not account for delayed leukocyte infiltration into the CNS parenchyma in the absence of monocytes, suggesting a direct effect of monocytes on glia limitans integrity (33, 48).
FIG. 5.

Monocytes do not affect mRNA CNS chemokine expression. Expression of CXCL10, CCL5, CCL3, and CXCL12 chemokines relative to ubiquitin mRNA in brains of naive (n = 4) and WT and CCL2−/− mice at days 3, 5, 7, and 10 p.i. (n = 3 per time point) was measured by real-time PCR. No differences in CXCL10, CCL5, or CCL3 upregulation, or in CXCL12 downregulation, were observed between WT and CCL2−/− mice during the course of infection.
Monocytes/macrophages are a source of MMPs during HIV CNS infections (50) and autoimmune-mediated inflammation (2, 47, 49). Several reports support a direct role of MMPs in mediating glia limitans permeability (1, 46). A correlation between leukocyte accumulation in the perivascular space and reduction of MMP expression and/or activity in CCL2−/− mice was thus tested. Distinct from other neuroinflammatory disorders (19, 40), JHMV infection is associated with a more limited pattern of MMP expression, restricted to MMP9, -3, and -12 (53). Neutrophil/monocyte-mediated loss of BBB integrity implicated neutrophils as the specific source of MMP9 (54). To test whether monocytes constitute an alternate source of MMP9, neutrophils and macrophages were purified from the CNS of infected WT mice. MMP9 activity was associated only with neutrophils and not with monocytes (Fig. 6A). Furthermore, MMP9 activity was not reduced in the CNS of CCL2−/− mice compared to controls at 3 and 5 days p.i. (data not shown), supporting the finding that monocytes recruited into the CNS during viral encephalitis do not express this enzyme at significant levels. Dysregulation of MMP3, expressed primarily by astrocytes, and MMP12, expressed by CNS resident cells as well as infiltrating leukocytes following JHMV infection (53), was also tested. However, the absence of monocytes did not alter the kinetics or levels of MMP12. MMP3 mRNA increased in CCL2−/− mice at days 3 and 5 p.i. (Fig. 6B), suggesting a compensatory effect. To account for potential upregulation of other compensatory MMPs in the absence of monocytes, MMP2, MMP7, and MMP14 were analyzed based on their upregulation during other CNS inflammatory disorders (19, 40). However, MMP2, constitutively expressed in the CNS, was not increased (Fig. 6B), nor was expression of MMP7 and MMP14 mRNAs induced in CCL2−/− mice at any time point (data not shown), consistent with previous data for WT mice (53). Expression of the MMP inhibitors TIMP1, -2, and -3 was also examined. TIMP1, expressed mainly by CD4 T cells infiltrating the JHMV-infected CNS (53), peaked at day 7 p.i. in both groups, and its expression did not significantly differ between the two groups at any time p.i. (Fig. 6B). TIMP2 and -3 mRNA expression was also not upregulated during JHMV infection in either WT or CCL2−/− mice, consistent with previous results (53). These data indicate that monocytes are not a major source of MMPs or TIMPs during JMHV infection and that their role in glia limitans disruption is MMP independent.
FIG. 6.

MMP-independent disruption of glia limitans. (A) MMP9 activity was analyzed by zymography from purified populations of monocytes and neutrophils isolated from WT mice at 3 days p.i. (B) Relative mRNA expression of MMP12, MMP3, MMP2, and TIMP1 was analyzed by quantitative real-time PCR. Total RNA was extracted from brains of naive mice (n = 4) and WT and CCL2−/− mice at days 3, 5, 7, and 10 p.i. (n = 3 for each time point).
Neutrophils do not compensate for the absence of CNS infiltrating monocytes.
Depletion of both neutrophils and monocytes prior to a lethal JHMV infection resulted in a drastic decrease of both BBB permeability and CNS inflammation (54). Retention of neutrophils at day 7 p.i. in the CNS of sublethally infected CCL2−/− mice thus suggested a potential compensatory mechanism facilitating parenchymal T cell access in the absence of monocytes. Neutrophils were therefore depleted from infected CCL2−/− mice to assess their contribution to CNS myeloid and T cell recruitment. Although flow cytometric analysis of CNS derived cells confirmed neutrophil depletion in CCL2−/− mice (Fig. 7A), their absence did not alter CNS cellular inflammation compared to that in neutrophil-sufficient CCL2−/− mice at days 3 and 5 p.i.; an overall reduction of CNS inflammation was observed at later times p.i. (Fig. 7B). Similarly, neutrophil depletion in WT mice reduced CNS leukocyte recruitment mainly between days 7 and 14 p.i. (Fig. 7B). The absence of neutrophils thus affected CNS leukocyte migration at the peak of inflammation (day 7 to 10 p.i.) but not at early time points. Importantly, neutrophils did not compensate for the absence of monocytes in CCL2−/− mice at early time points. Moreover, specific neutrophil depletion from WT mice demonstrated no difference in disease severity. Similarly, neutrophil depletion from CCL2−/− mice did not alter the delayed disease onset observed in the neutrophil-sufficient control group (data not shown). However, no differences were observed at later time points, as both neutrophil-depleted and control CCL2−/− showed signs of hind limb paralysis similar to those in WT mice. Moreover, treated and untreated CCL2−/− mice both controlled virus replication to below the limit of detection by day 14 p.i. (data not shown).
FIG. 7.
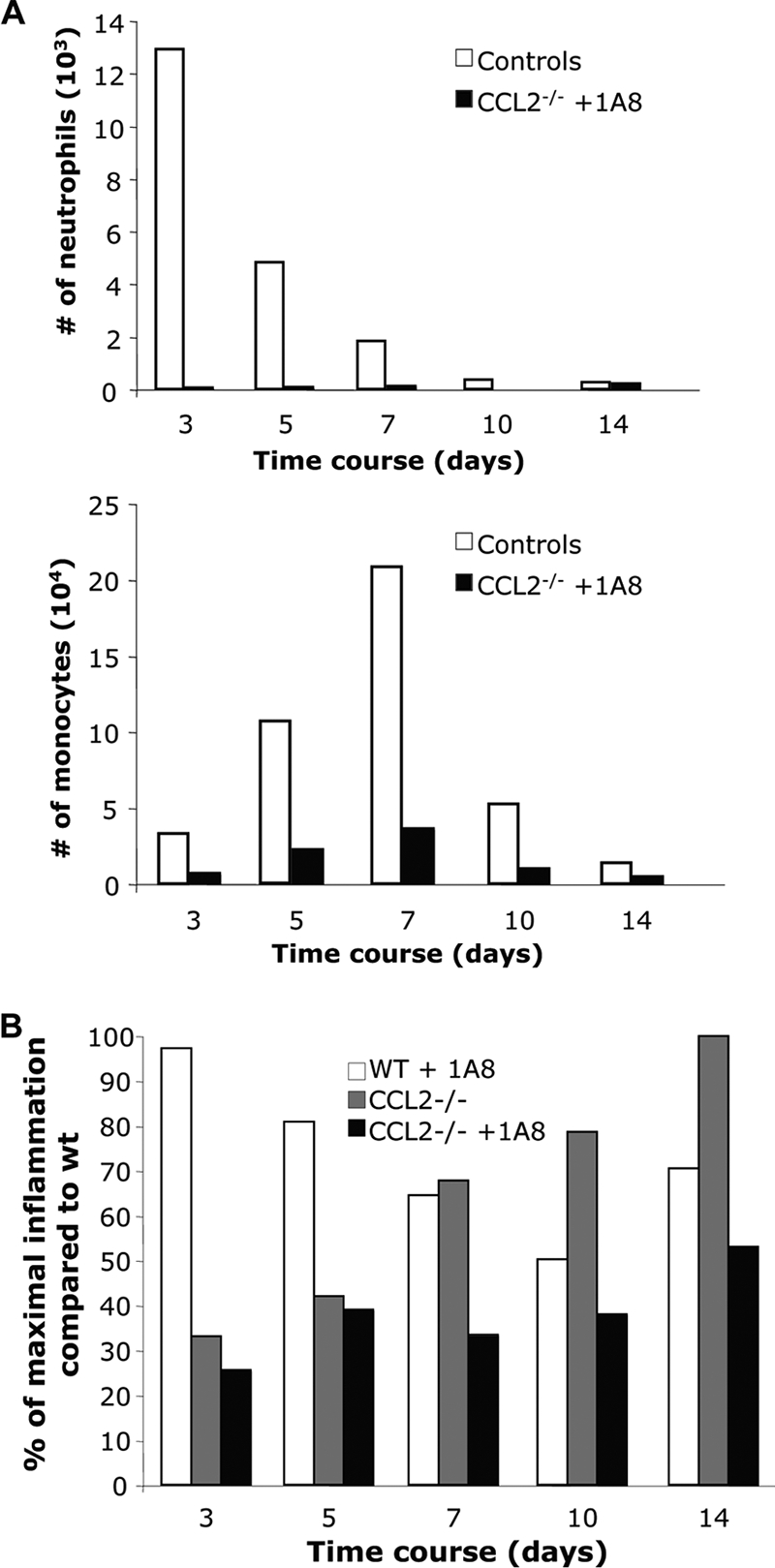
Neutrophils do not compensate for the absence of monocytes. (A) CNS inflammation of neutrophil-depleted CCL2−/− mice and controls (WT mice treated with isotype control antibody) analyzed by flow cytometry. Graphs represent the number of neutrophils (Ly-6G+) and macrophages (F4/80+) in the infiltrating CD45high population. Data represent means for three mice per group at each time point. (B) The percentage of maximal inflammation relative to that in WT mice was compared between CCL2−/− and CCL2−/− or WT neutrophil-depleted mice. Data represent the averages for three mice at each time point.
Taken together, these results implicate monocytes, not neutrophils, in facilitating early access of inflammatory cells into the parenchyma to control virus replication, albeit at the cost of enhancing clinical disease.
DISCUSSION
The BBB restrains parenchymal leukocyte entry and significantly alters the character of CNS inflammation (4). Leukocyte recruitment through the BBB is, however, crucial to protect the host during CNS infections. Nevertheless, uncontrolled CNS inflammation during neuroinflammatory disorders such as MS can mediate tissue damage. Thus, defining mechanisms that regulate migration into the CNS parenchyma is of particular interest to promote protective immune responses against CNS infection while restraining damaging immune responses leading to neuroinflammatory disorders.
Extensive studies of migration across the blood vessel wall into the perivascular space have identified potential therapeutic targets to modulate leukocyte trafficking into the CNS. For example, treatment of relapsing forms of MS with anti-alpha4-integrin reduced CNS leukocyte entry (1, 39). However, development of progressive multifocal leukoencephalopathy demonstrated how interference with CNS leukocyte trafficking can perturb the delicate immune balance controlling endogenous pathogens. While modulation of chemokine signaling represents another therapeutic target for neurological diseases (7, 32), the complexity of the chemokine system makes this a challenging approach. For example, although CCR2 is the only receptor for CCL2, several other chemokines signal to CCR2. Our data clearly support the notion that targeting the ligand has a different impact than targeting the chemokine receptor. Thus, CCL2 deficiency selectively impaired only monocyte recruitment into the CNS during virus-induced encephalitis. In contrast, CCR2−/− mice showed a drastic reduction of monocytes as well as T cells in the CNS during JHMV infection, resulting in uncontrolled virus replication and rapid mortality (8).
Few studies have focused on understanding the molecular and cellular mechanisms regulating leukocyte penetration through the glia limitans (34), which constitutes an alternative target for therapeutic strategies to ameliorate autoimmune-mediated disease (48). The present data are the first to demonstrate a critical role of monocytes in specifically breaching the glia limitans during acute viral encephalitis. During EAE, monocyte-facilitated parenchymal leukocyte access has been attributed to chemotactic signaling induced by monocyte-derived tumor necrosis factor (48). However, the absence of monocytes in JHMV-infected CCL2−/− mice did not correlate with decreased chemokine expression compared to that in controls, indicating a more direct role of monocytes in disrupting the glia limitans. In addition, no virus-infected cells, detected with the anti-JHMV MAb J3.3 specific for the carboxyl terminus of the virus nucleocapsid protein, could be found in the perivascular space of either controls or CCL2−/− mice (data not shown). These observations indicated that delayed T cell migration across the glia limitans of CCL2−/− mice was not due to a defect in T cell restimulation by antigen-presenting phagocytes in the perivascular space, as previously described to occur during EAE (3). Prime candidates in monocyte-mediated migration through the glia limitans are MMPs (46). However, no evidence for decreased or altered MMP mRNA expression in the absence of monocytes indicated that monocytes are not a primary source of MMPs during JHMV infection. Several hypotheses may thus explain leukocyte retention in the perivascular space of infected CCL2−/− mice. First, monocytes may secrete extracellular proteases other than MMPs, which may promote migration through the glia limitans. For example, the cysteine protease cathepsins (K, S, and L) can be mobilized extracellularly by macrophages (16, 37) and degrade collagen, a component of the glia limitans and extracellular matrix. Monocytes may also provide an indirect activation signal by inducing the release of tissue plasminogen activator. Thus, conversion of plasminogen into active plasmin by tissue plasminogen activator can lead to the degradation of other components of the glia limitans, such as laminin or fibronectin (16, 38). In addition, monocytes are a major source of reactive oxygen species, which can trigger MMP activation (36). Although our results indicated no difference in MMP mRNA expression in CCL2−/− mice compared to controls, we cannot rule out impaired cleavage of pro-MMP into activated MMP in the absence of monocytes. The identification of the molecule(s) released by monocytes to promote migration through the glia limitans is thus critical to develop more specifically targeted therapies for CNS disorders.
Neutrophil depletion prior to virus infection of the CCL2−/− mice did not alter viral pathogenesis compared to that in CCL2−/− control mice. Consistent with the case for neutrophil-depleted WT mice, depletion of neutrophils in CCL2−/− mice did not affect early CNS inflammation. Neutrophils thus did not compensate for the absence of monocytes and were not required to mediate sufficient leukocyte recruitment for antiviral control. These observations appear to contradict previous data showing a drastic reduction of CNS inflammation and BBB permeability, yet early mortality, after treatment with anti-Gr1 antibody (54). Nevertheless, the previous depletion studies were performed in mice infected with a more virulent JHMV variant causing lethal encephalitis, in which neutrophils are more abundant CNS inflammatory cells (∼20% of total CD45hi cells). In contrast, they represent only ∼5% of total infiltrating cells during the sublethal JHMV infection used in the present study. Lastly, the anti-Gr-1 antibody depletes not only neutrophils and monocytes but also activated CD8 T cells, preempting direct comparison of these studies. Nevertheless, a recent study using direct CXCR2 ablation to impair neutrophil recruitment also demonstrates enhanced mortality associated with impaired viral clearance in the sublethal infection model (21), supporting previous studies (54) and contradicting the present results. However, anti-CXCR2 treatment also affected monocyte recruitment, making it difficult to discern the respective effects of neutrophils versus monocytes on diminished T cell recruitment. Despite the apparent redundancy of neutrophils in enhancing parenchymal T cell access and viral clearance even in the absence of monocytes in our model, they clearly affected leukocyte accumulation at the peak of inflammation. In this context, it is of interest to note that MMP9 expression was specific for neutrophils and was not detected in monocytes. The precise role of MMP9 in enhancing leukocyte recruitment thus remains to be elucidated.
The transient nature of delayed disease onset and control of virus replication in infected CCL2−/− mice constitutes a further enigma; disease severity and virus clearance were similar to those in infected WT mice at later time points, indicating that impaired T cell access to the parenchyma is overcome fairly rapidly. MMP expression by T cells (52) suggests that a critical number of activated CD4 and/or CD8 T cells may directly promote migration through the glia limitans in the absence of either monocytes or neutrophils.
In summary, these data are the first to demonstrate an important direct role of monocytes in promoting T cell migration across the glia limitans into the parenchyma during acute viral encephalitis. In the absence of monocytes, T cells accumulated transiently in the perivascular space, leading to delayed disease onset but also delayed virus control. In contrast to the EAE model, no overt defects in chemotactic signaling during virus infection in CCL2−/− mice indicated a direct effect of monocytes on glia limitans disruption. While dysregulation of MMP expression in the absence of monocytes was ruled out at the transcriptional level, direct effects on MMP proteolytic activity or involvement of proteases other than MMPs remain to be determined. Identification of mechanisms by which monocytes promote glia limitans disruption will be valuable in the design of less vigorous, more targeted therapeutic approaches to enhance T cell access to the CNS parenchyma or impair entry of potentially infected monocytes themselves in cases of infection, in addition to limiting this access during neuroinflammatory disorders.
Acknowledgments
This work was supported by Public Health Service grant PO1 NS018146 from the National Institutes of Health and by National MS Society fellowship grant FG 1791-A-1 (C.S.).
We thank Anna Rietsch, Wen Wei, and Sherry Yu for technical assistance.
Footnotes
Published ahead of print on 3 March 2010.
REFERENCES
- 1.Agrawal, S., P. Anderson, M. Durbeej, N. van Rooijen, F. Ivars, G. Opdenakker, and L. M. Sorokin. 2006. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J. Exp. Med. 203:1007-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bar-Or, A., R. K. Nuttall, M. Duddy, A. Alter, H. J. Kim, I. Ifergan, C. J. Pennington, P. Bourgoin, D. R. Edwards, and V. W. Yong. 2003. Analyses of all matrix metalloproteinase members in leukocytes emphasize monocytes as major inflammatory mediators in multiple sclerosis. Brain 126:2738-2749. [DOI] [PubMed] [Google Scholar]
- 3.Bartholomaus, I., N. Kawakami, F. Odoardi, C. Schlager, D. Miljkovic, J. W. Ellwart, W. E. Klinkert, C. Flugel-Koch, T. B. Issekutz, H. Wekerle, and A. Flugel. 2009. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 462:94-98. [DOI] [PubMed] [Google Scholar]
- 4.Bechmann, I., I. Galea, and V. H. Perry. 2007. What is the blood-brain barrier (not)? Trends Immunol. 28:5-11. [DOI] [PubMed] [Google Scholar]
- 5.Bergmann, C. C., J. D. Altman, D. Hinton, and S. A. Stohlman. 1999. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the central nervous system. J. Immunol. 163:3379-3387. [PubMed] [Google Scholar]
- 6.Bergmann, C. C., T. E. Lane, and S. A. Stohlman. 2006. Coronavirus infection of the central nervous system: host-virus stand-off. Nat. Rev. Microbiol. 4:121-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charo, I. F., and R. M. Ransohoff. 2006. The many roles of chemokines and chemokine receptors in inflammation. N Engl. J. Med. 354:610-621. [DOI] [PubMed] [Google Scholar]
- 8.Chen, B. P., W. A. Kuziel, and T. E. Lane. 2001. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J. Immunol. 167:4585-4592. [DOI] [PubMed] [Google Scholar]
- 9.Dallasta, L. M., L. A. Pisarov, J. E. Esplen, J. V. Werley, A. V. Moses, J. A. Nelson, and C. L. Achim. 1999. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am. J. Pathol. 155:1915-1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelhardt, B., and R. M. Ransohoff. 2005. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 26:485-495. [DOI] [PubMed] [Google Scholar]
- 11.Eugenin, E. A., K. Osiecki, L. Lopez, H. Goldstein, T. M. Calderon, and J. W. Berman. 2006. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and neuroAIDS. J. Neurosci. 26:1098-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleming, J. O., M. D. Trousdale, F. A. el-Zaatari, S. A. Stohlman, and L. P. Weiner. 1986. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J. Virol. 58:869-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frohman, E. M., M. K. Racke, and C. S. Raine. 2006. Multiple sclerosis—the plaque and its pathogenesis. N. Engl. J. Med. 354:942-955. [DOI] [PubMed] [Google Scholar]
- 14.Fux, M., N. van Rooijen, and T. Owens. 2008. Macrophage-independent T cell infiltration to the site of injury-induced brain inflammation. J. Neuroimmunol. 203:64-72. [DOI] [PubMed] [Google Scholar]
- 15.Galea, I., I. Bechmann, and V. H. Perry. 2007. What is immune privilege (not)? Trends Immunol. 28:12-18. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Touchard, A., T. D. Henry, G. Sangiorgi, L. G. Spagnoli, A. Mauriello, C. Conover, and R. S. Schwartz. 2005. Extracellular proteases in atherosclerosis and restenosis. Arterioscler. Thromb. Vasc. Biol. 25:1119-1127. [DOI] [PubMed] [Google Scholar]
- 17.Getts, D. R., R. L. Terry, M. T. Getts, M. Muller, S. Rana, B. Shrestha, J. Radford, N. Van Rooijen, I. L. Campbell, and N. J. King. 2008. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J. Exp. Med. 205:2319-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu, L., B. Rutledge, J. Fiorillo, C. Ernst, I. Grewal, R. Flavell, R. Gladue, and B. Rollins. 1997. In vivo properties of monocyte chemoattractant protein-1. J. Leukoc. Biol. 62:577-580. [DOI] [PubMed] [Google Scholar]
- 19.Hartung, H. P., and B. C. Kieseier. 2000. The role of matrix metalloproteinases in autoimmune damage to the central and peripheral nervous system. J. Neuroimmunol. 107:140-147. [DOI] [PubMed] [Google Scholar]
- 20.Held, K. S., B. P. Chen, W. A. Kuziel, B. J. Rollins, and T. E. Lane. 2004. Differential roles of CCL2 and CCR2 in host defense to coronavirus infection. Virology 329:251-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosking, M. P., L. Liu, R. M. Ransohoff, and T. E. Lane. 2009. A protective role for ELR+ chemokines during acute viral encephalomyelitis. PLoS Pathog. 5:e1000648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang, D. R., J. Wang, P. Kivisakk, B. J. Rollins, and R. M. Ransohoff. 2001. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J. Exp. Med. 193:713-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ireland, D. D., S. A. Stohlman, D. R. Hinton, R. Atkinson, and C. C. Bergmann. 2008. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 82:300-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janzer, R. C., and M. C. Raff. 1987. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 325:253-257. [DOI] [PubMed] [Google Scholar]
- 25.Lane, T. E., J. L. Hardison, and K. B. Walsh. 2006. Functional diversity of chemokines and chemokine receptors in response to viral infection of the central nervous system. Curr. Top. Microbiol. Immunol. 303:1-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu, B., B. J. Rutledge, L. Gu, J. Fiorillo, N. W. Lukacs, S. L. Kunkel, R. North, C. Gerard, and B. J. Rollins. 1998. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 187:601-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luabeya, M. K., L. M. Dallasta, C. L. Achim, C. D. Pauza, and R. L. Hamilton. 2000. Blood-brain barrier disruption in simian immunodeficiency virus encephalitis. Neuropathol. Appl. Neurobiol. 26:454-462. [DOI] [PubMed] [Google Scholar]
- 28.MacMicking, J., Q. W. Xie, and C. Nathan. 1997. Nitric oxide and macrophage function. Annu. Rev. Immunol. 15:323-350. [DOI] [PubMed] [Google Scholar]
- 29.Man, S., E. E. Ubogu, and R. M. Ransohoff. 2007. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. 17:243-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCandless, E. E., Q. Wang, B. M. Woerner, J. M. Harper, and R. S. Klein. 2006. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J. Immunol. 177:8053-8064. [DOI] [PubMed] [Google Scholar]
- 31.McCandless, E. E., B. Zhang, M. S. Diamond, and R. S. Klein. 2008. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc. Natl. Acad. Sci. U. S. A. 105:11270-11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller, R. J., W. Rostene, E. Apartis, G. Banisadr, K. Biber, E. D. Milligan, F. A. White, and J. Zhang. 2008. Chemokine action in the nervous system. J. Neurosci. 28:11792-11795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy, C. A., R. M. Hoek, M. T. Wiekowski, S. A. Lira, and J. D. Sedgwick. 2002. Interactions between hemopoietically derived TNF and central nervous system-resident glial chemokines underlie initiation of autoimmune inflammation in the brain. J. Immunol. 169:7054-7062. [DOI] [PubMed] [Google Scholar]
- 34.Owens, T., I. Bechmann, and B. Engelhardt. 2008. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 67:1113-1121. [DOI] [PubMed] [Google Scholar]
- 35.Parra, B., D. R. Hinton, N. W. Marten, C. C. Bergmann, M. T. Lin, C. S. Yang, and S. A. Stohlman. 1999. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J. Immunol. 162:1641-1647. [PubMed] [Google Scholar]
- 36.Pun, P. B., J. Lu, and S. Moochhala. 2009. Involvement of ROS in BBB dysfunction. Free Radic. Res. 43:348-364. [DOI] [PubMed] [Google Scholar]
- 37.Reddy, V. Y., Q. Y. Zhang, and S. J. Weiss. 1995. Pericellular mobilization of the tissue-destructive cysteine proteinases, cathepsins B, L, and S, by human monocyte-derived macrophages. Proc. Natl. Acad. Sci. U. S. A. 92:3849-3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reijerkerk, A., G. Kooij, S. M. van der Pol, T. Leyen, B. van Het Hof, P. O. Couraud, D. Vivien, C. D. Dijkstra, and H. E. de Vries. 2008. Tissue-type plasminogen activator is a regulator of monocyte diapedesis through the brain endothelial barrier. J. Immunol. 181:3567-3574. [DOI] [PubMed] [Google Scholar]
- 39.Rice, G. P., H. P. Hartung, and P. A. Calabresi. 2005. Anti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology 64:1336-1342. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg, G. A. 2002. Matrix metalloproteinases in neuroinflammation. Glia 39:279-291. [DOI] [PubMed] [Google Scholar]
- 41.Sixt, M., B. Engelhardt, F. Pausch, R. Hallmann, O. Wendler, and L. M. Sorokin. 2001. Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J. Cell Biol. 153:933-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stohlman, S. A., C. C. Bergmann, M. T. Lin, D. J. Cua, and D. R. Hinton. 1998. CTL effector function within the central nervous system requires CD4+ T cells. J. Immunol. 160:2896-2904. [PubMed] [Google Scholar]
- 43.Stohlman, S. A., D. R. Hinton, B. Parra, R. Atkinson, and C. C. Bergmann. 2008. CD4 T cells contribute to virus control and pathology following central nervous system infection with neurotropic mouse hepatitis virus. J. Virol. 82:2130-2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sunderkotter, C., T. Nikolic, M. J. Dillon, N. Van Rooijen, M. Stehling, D. A. Drevets, and P. J. Leenen. 2004. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 172:4410-4417. [DOI] [PubMed] [Google Scholar]
- 45.Templeton, S. P., T. S. Kim, K. O'Malley, and S. Perlman. 2008. Maturation and localization of macrophages and microglia during infection with a neurotropic murine coronavirus. Brain Pathol. 18:40-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toft-Hansen, H., R. Buist, X. J. Sun, A. Schellenberg, J. Peeling, and T. Owens. 2006. Metalloproteinases control brain inflammation induced by pertussis toxin in mice overexpressing the chemokine CCL2 in the central nervous system. J. Immunol. 177:7242-7249. [DOI] [PubMed] [Google Scholar]
- 47.Toft-Hansen, H., R. K. Nuttall, D. R. Edwards, and T. Owens. 2004. Key metalloproteinases are expressed by specific cell types in experimental autoimmune encephalomyelitis. J. Immunol. 173:5209-5218. [DOI] [PubMed] [Google Scholar]
- 48.Tran, E. H., K. Hoekstra, N. van Rooijen, C. D. Dijkstra, and T. Owens. 1998. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J. Immunol. 161:3767-3775. [PubMed] [Google Scholar]
- 49.Vos, C. M., E. S. van Haastert, C. J. de Groot, P. van der Valk, and H. E. de Vries. 2003. Matrix metalloproteinase-12 is expressed in phagocytotic macrophages in active multiple sclerosis lesions. J. Neuroimmunol. 138:106-114. [DOI] [PubMed] [Google Scholar]
- 50.Webster, N. L., and S. M. Crowe. 2006. Matrix metalloproteinases, their production by monocytes and macrophages and their potential role in HIV-related diseases. J. Leukoc. Biol. 80:1052-1066. [DOI] [PubMed] [Google Scholar]
- 51.Williams, K. C., and W. F. Hickey. 2002. Central nervous system damage, monocytes and macrophages, and neurological disorders in AIDS. Annu. Rev. Neurosci. 25:537-562. [DOI] [PubMed] [Google Scholar]
- 52.Yong, V. W., C. A. Krekoski, P. A. Forsyth, R. Bell, and D. R. Edwards. 1998. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci. 21:75-80. [DOI] [PubMed] [Google Scholar]
- 53.Zhou, J., N. W. Marten, C. C. Bergmann, W. B. Macklin, D. R. Hinton, and S. A. Stohlman. 2005. Expression of matrix metalloproteinases and their tissue inhibitor during viral encephalitis. J. Virol. 79:4764-4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou, J., S. A. Stohlman, D. R. Hinton, and N. W. Marten. 2003. Neutrophils promote mononuclear cell infiltration during viral-induced encephalitis. J. Immunol. 170:3331-3336. [DOI] [PubMed] [Google Scholar]