

Abstract
Measles virus (MV) causes transient severe immunosuppression in patients, which may lead to secondary viral and bacterial infections, largely accounting for measles-related morbidity and mortality. MV is known to infect immune cells by using the human signaling lymphocyte activation molecule (SLAM; also called CD150) as a cellular receptor, but the mechanism by which MV causes immunosuppression is not well understood. We show that MV infection of SLAM knock-in mice, in which the V domain of mouse SLAM was replaced by the V domain of human SLAM, crossed with alpha/beta-interferon receptor knockout mice, reproduced many immunological alterations observed in human patients. These included lymphopenia, inhibition of T-cell proliferation and antibody production, increased production of the Th2 cytokine interleukin-4 (IL-4) and the immunosuppressive cytokine IL-10, and suppression of contact hypersensitivity. Gross redistribution of lymphocytes among lymphoid tissues was not apparent in infected mice, nor was an increase of regulatory T cells. The numbers of lymphocytes in lymph nodes remained almost unchanged after MV infection, despite enhanced apoptosis, suggesting that lymph nodes were replenished with lymphocytes from the peripheral blood, which may have contributed to the observed lymphopenia in the spleen. Blocking of IL-10 by use of an anti-IL-10 receptor antibody ameliorated suppression of contact hypersensitivity in infected mice. These results indicate that SLAM knock-in mice lacking the expression of the alpha/beta-interferon receptor serve as a useful small animal model with which to elucidate MV-induced immunosuppression.
Measles virus (MV), a member of the Morbillivirus genus in the Paramyxoviridae family, causes an acute febrile disease with a generalized skin rash accompanied by transient immunosuppression, which may lead to secondary viral and bacterial infections, largely accounting for measles-related morbidity and mortality (8). von Pirquet first made a scientific observation of virus-induced immunosuppression when he showed that children who had been positive for the tuberculin skin test failed to mount a response to tuberculin during the course of measles (35). Additionally, children with measles were found to show a significantly reduced antibody response to immunization with the H and O antigens of Salmonella enterica serovar Typhi (37). MV infection is also characterized by lymphopenia, suppression of mitogen-induced and antigen-specific lymphocyte proliferation ex vivo, and cytokine imbalance skewed toward a Th2 response (8, 13, 26). The exact mechanism underlying these immunological alterations and the extent to which they are related to immunosuppression in vivo are not well understood.
MV predominantly infects immune cells such as lymphocytes, dendritic cells (DCs), and macrophages by using the human signaling lymphocyte activation molecule (SLAM; also called CD150) as a receptor (4, 33, 38). Several groups have produced SLAM transgenic mice, using various promoters, as small animal models with which to study MV pathogenesis (11, 12, 27, 31, 36). To facilitate MV replication, these transgenic mice are made defective in alpha/beta-interferon (IFN-α/β) signaling (12, 31, 36) or used when they are newborn (11, 27). Recently, we reported that SLAM knock-in mice, in which the V domain of mouse SLAM was replaced by the V domain of human SLAM, efficiently supported MV replication in lymphoid tissues when crossed with mice lacking the IFN-α/β receptor (IFNAR) (22). Splenocytes from these mice infected with MV demonstrated suppression of proliferative responses to concanavalin A (22).
In this study, we further examined MV-induced immunological alterations in IFNAR−/− SLAM knock-in mice (KI mice). We found that enhanced apoptosis of lymphocytes and increased production of the immunosuppressive cytokine interleukin-10 (IL-10) may be responsible for some of the alterations.
MATERIALS AND METHODS
Mice.
Generation of KI mice (SLAM knock-in mice crossed with IFNAR−/− mice) has been described previously (22). All mice used were 8 to 10 weeks of age, and animal experiments were reviewed by the Institutional Committee of Ethics on Animal Experiments and carried out according to the Guidelines for Animal Experiments of the Faculty of Medicine, Kyushu University, Japan.
Viruses and cells.
The MV used in this study was a recombinant virus (IC323) based on the wild-type IC-B strain (32), which has been shown to be virulent in monkeys (4, 32). The virus was grown on Vero/hSLAM cells (23), and virus titers were determined by plaque assay on Vero/hSLAM cells. For some experiments, IC323 expressing enhanced green fluorescent protein (EGFP) was used to monitor infection levels. Splenocytes and lymph node cells isolated from mice were cultured in RPMI 1640 medium (MP Biomedicals LLC) containing 10% fetal bovine serum, 100 μM 2-mercaptoethanol (Sigma), 100 U/ml penicillin, and 100 μg/ml streptomycin. DCs were derived from bone marrow (BM) cells. Briefly, BM cells were prepared from mice, and red blood cells were removed by treatment with 1.66% NH4Cl. The BM cells were then suspended with RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 μM 2-mercaptoethanol, 10 ng/ml recombinant mouse granulocyte-macrophage colony-stimulating factor (Peprotech), 100 U/ml penicillin, and 100 μg/ml streptomycin. Approximately 1 × 106 BM cells were seeded into each well of a 24-well plate. Nonadherent cells were discarded, and the remaining cells were fed with fresh medium. Following 6 days of incubation, nonadherent and loosely adherent cells were harvested and used as immature DCs. Mononuclear cells were prepared from the liver by Percoll density gradient centrifugation (GE Healthcare), and those from the lung were prepared by collagenase treatment of the tissue.
MV infection.
For in vivo infection, 500 μl of solution containing 1 × 106 PFU of MV was injected into the peritoneal cavity of each mouse. For in vitro experiments, DCs (stimulated with 100 ng/ml lipopolysaccharide for 16 h) were infected with MV at a multiplicity of infection (MOI) of 1. After 1 h of incubation, the cells were washed twice with phosphate-buffered saline (PBS) and cultured in medium for the indicated times.
Flow cytometry.
Single-cell suspensions of splenocytes and lymph node cells were prepared from mice and incubated with anti-mouse CD16/32 monoclonal antibody (MAb) (2.4G2; Pharmingen) to block nonspecific IgG binding to Fc receptors. The following MAbs were used to detect the respective cell surface molecules: fluorescein isothiocyanate (FITC)-conjugated anti-Thy1.2 (53.2.1; eBioscience), FITC-conjugated anti-CD4 (GK1.5; eBioscience), FITC-conjugated anti-B220 (RA3-6B2; eBioscience), and anti-FoxP3 (3G3; Abcam). Phycoerythrin-conjugated secondary antibody was used following staining with anti-FoxP3 antibody. For the detection of apoptosis, lymph node cells were stained with FITC-conjugated annexin V and propidium iodide (AbD Serotec). Flow cytometric analysis was performed on a FACSCalibur instrument (BD Biosciences).
Cell proliferation assay.
Splenocytes and lymph node cells were isolated from infected mice. CD4+ T cells were prepared using a negative-selection column (Miltenyi Biotec), stimulated in plates coated with 3 μg/ml of anti-CD3 MAb (145.2C11; BD Pharmingen) for 48 h, and pulsed with 1 μCi of [3H]thymidine for the last 6 h of incubation. The cells were then harvested on glass fiber filters, and the incorporated [3H]thymidine was measured using a liquid scintillation counter.
Measurement of antibody production.
Mice were infected with MV or left uninfected, and at 5 days postinfection (p.i.), individual mice were immunized intraperitoneally (i.p.) with 100 μg ovalbumin (OVA; Sigma) emulsified in Freund's complete adjuvant (Sigma). To determine the levels of anti-OVA antibodies in sera collected 2 weeks after immunization, 96-well plates were coated with 10 μg/ml OVA (200 μl/well) and incubated overnight at 4°C. After being washed with PBS, the plates were blocked with 5% nonfat milk at room temperature for 2 h. Serially diluted sera were added to the wells of plates (100 μl/well) and incubated at room temperature for 1 h. After washing of the plates, horseradish peroxidase-labeled anti-mouse IgG (Bio-Rad) was added and incubated at room temperature for 1 h, and peroxidase substrate (2,2′-azino-bis-3-ethylbenthiazoline-6-sulfonic acid tablets; Sigma) was added to each well. After a 30-min incubation, absorbance at a wavelength of 405 nm was determined with a microplate reader (Bio-Rad).
Measurement of cytokine production.
CD4+ T cells were isolated from the spleens of infected mice by use of a negative-selection column and were stimulated in plates coated with 3 μg/ml of anti-CD3 MAb for 48 h. The culture supernatants were collected and analyzed for the presence of IL-4, IFN-γ, IL-10, and transforming growth factor beta (TGF-β), using enzyme-linked immunosorbent assay (ELISA) kits (eBioscience) according to the manufacturer's instructions. The culture supernatants of infected and uninfected DCs were collected at 24 and 48 h p.i. and were analyzed for IL-10 and IL-12 by use of ELISA kits (eBioscience) according to the manufacturer's instructions.
Quantitative real-time reverse transcription-PCR (RT-PCR).
Total RNA was isolated from cells by use of Trizol (Invitrogen), and 2 μg of the RNA was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Promega) and random primers (Takara) after treatment with DNase I (Promega). Quantitative real-time PCR was performed using SYBR Premix Ex Taq II (Takara) and a Light Cycler 1.5 (Roche). The following primers were used: 5′-ATGCTCCTAGAGCTGCGGACT-3′ (forward) and 5′-CCTGCATTAAGGAGTCGGTTAG-3′ (reverse) for IL-10 and 5′-CAGAAGCTAACCATCTCCTGGTTTG-3′ (forward) and 5′-CCGGAGTAATTTGGTGCTCCACAC-3′ (reverse) for IL-12 p40. All data were normalized with the expression level of β-actin.
Generation of anti-mouse IL-10R MAb.
To generate an anti-mouse IL-10 receptor (IL-10R) MAb, rats were immunized with 293T cells transfected with cDNA encoding mouse IL-10R. Hybridoma cells producing anti-mouse IL-10R antibodies were screened for the ability to bind to 293T cells expressing mouse IL-10R. Antibodies were further selected for the ability to block IL-10-dependent proliferation of mouse MC/9 cells (34). Anti-IL-10R MAb was purified by ammonium sulfate fractionation and protein G affinity chromatography.
Assay for contact hypersensitivity.
Contact hypersensitivity to 2,4-dinitro-1-fluorobenzene (DNFB; Sigma) was determined as described previously (20, 28), with modifications. Briefly, 50 μl of 0.5% DNFB in ethanol was applied to the shaved ventral skin of mice (day 0). On day 1, sensitized mice were infected with MV or left uninfected. On day 6, 10 μl of 1% DNFB in olive oil was applied to both sides of the right ear, and olive oil alone was applied to the left ear. For some experiments, 500 μg of rat anti-mouse IL-10R MAb or control rat IgG was injected i.p. into individual mice at day 4. Either clone YL03.1B1.3 (kindly provided by Schering-Plough Biopharma) or our own preparation (described above) was used as an anti-mouse IL-10R MAb. The ear thickness of mice was measured by an examiner before and after challenge, in a blinded fashion. Ear swelling was calculated as follows: (E − E0)right ear − (E − E0)left ear, where E and E0 represent the ear thicknesses after and before challenge, respectively. Ears treated with DFNB were also examined histopathologically.
RESULTS
MV-induced immunosuppression in KI mice.
We first examined whether MV infection affects lymphocyte counts in KI mice, as is the case in humans (8). Spleens were obtained from KI mice at various time points after MV infection, and the total numbers of splenocytes (lymphocytes and monocytes/macrophages) were determined. We found that the numbers of splenocytes did not change significantly until 14 days p.i. However, the proportions of different cell types varied, as shown by flow cytometry analysis. At 5 days p.i., the proportion of T cells (Thy1.2+ cells) in KI mice decreased to less than one-third of that in uninfected KI mice, whereas it was hardly altered in unsusceptible IFNAR−/− mice (wild-type [WT] mice) (Fig. 1A). In KI mice, the absolute numbers of T cells did not change significantly at 2 days p.i. but were decreased at 5 days p.i. and returned to their original levels at 14 days p.i. (Fig. 1B). The results indicate that there was a transient decrease in the proportion as well as the absolute number of T cells in the spleens of MV-infected KI mice. On the other hand, the absolute numbers of B cells (B220+ cells) remained more or less constant in those mice (Fig. 1B).
FIG. 1.
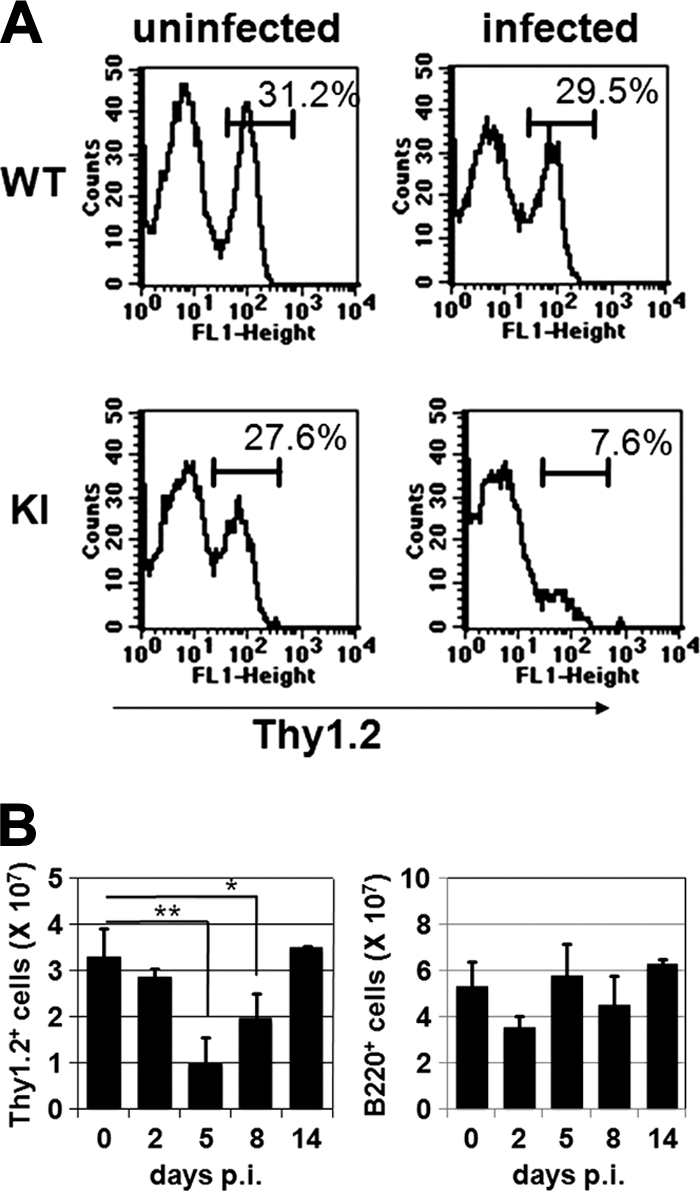
Lymphocyte counts in spleens of MV-infected mice. (A) Single-cell suspensions of splenocytes were prepared from uninfected WT and KI mice or from WT and KI mice 5 days after MV infection, stained with FITC-conjugated anti-Thy1.2 antibody, and analyzed by flow cytometry. The data shown are representative of >10 mice for each group. (B) Splenocytes were prepared from KI mice at the indicated time points after MV infection and were counted. Cells were stained with FITC-conjugated anti-Thy1.2 or anti-B220 antibody. The absolute numbers of T cells (left) and B cells (right) were calculated from total cell numbers and proportions of Thy1.2+ and B220+ cells, respectively. The value at each time point represents the mean ± standard deviation for four mice, and the data shown are representative of three different experiments. **, P < 0.01; *, P < 0.05 (significant differences based on Student's t test).
Next, we examined whether MV infection affected T-cell proliferation in these mice. KI and WT mice were infected with MV or left uninfected. CD4+ T cells were isolated from the spleens of these mice at 5 days p.i. and stimulated with anti-CD3 antibody. Proliferation of CD4+ T cells was significantly suppressed in infected KI mice but not in infected WT mice (Fig. 2A), consistent with our previous observations at 4 days p.i., obtained using a different assay (22). Similar to the decrease in splenic T cells, suppression of CD4+ T-cell proliferation was observed only at 5 days p.i. (Fig. 2B). Although splenic B-cell counts were not significantly altered (Fig. 1B), production of anti-OVA antibody was suppressed in MV-infected KI mice (Fig. 2C).
FIG. 2.
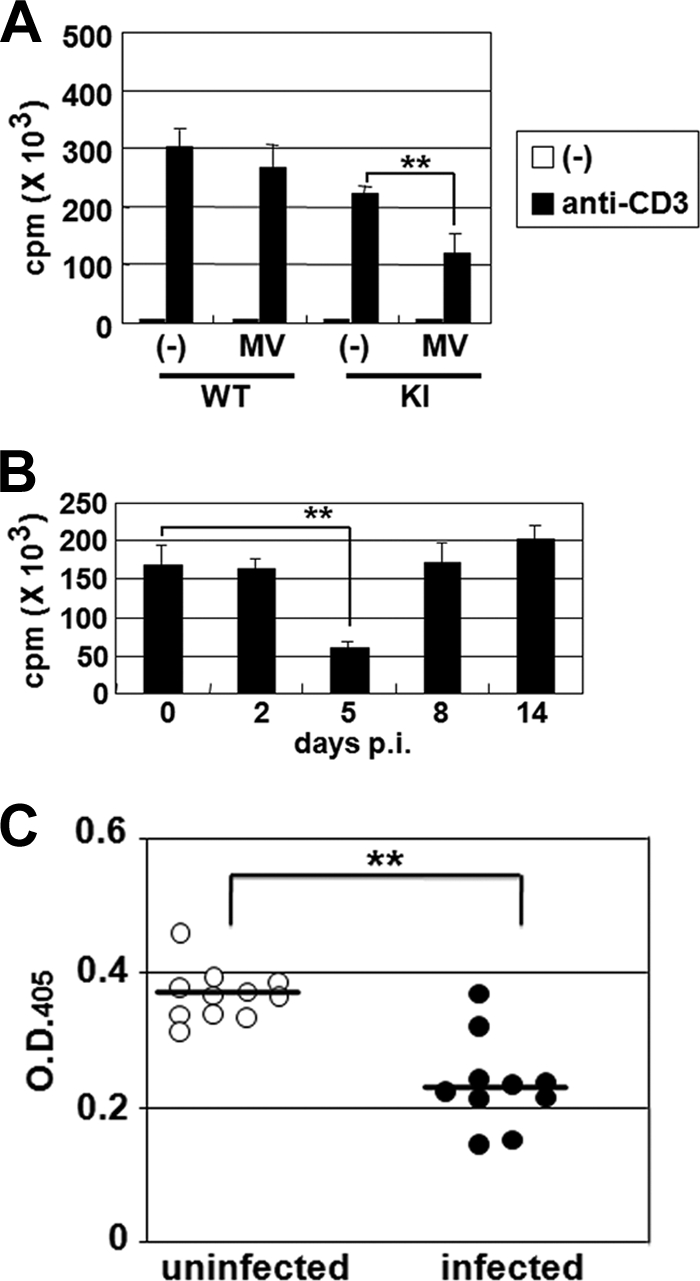
T-cell proliferation and antibody production in MV-infected mice. (A) CD4+ T cells were isolated from the spleens of uninfected WT and KI mice or from those of WT and KI mice 5 days after i.p. infection with MV. The cells (2 × 105 cells/well) were stimulated in plates coated with anti-CD3 antibody for 48 h and were pulsed with [3H]thymidine for the last 6 h of incubation. Thymidine uptake was measured in triplicate for each sample. The value for each group represents the mean ± standard deviation for four mice, and the data shown are representative of three different experiments. (B) CD4+ T cells were isolated from the spleens of KI mice at the indicated times after MV infection. The cells were stimulated in plates coated with anti-CD3 antibody for 48 h and were pulsed with [3H]thymidine for the last 6 h. Thymidine uptake was measured in triplicate for each sample. The value at each time point represents the mean ± standard deviation for four mice, and the data shown are representative of three different experiments. (C) KI mice were infected with MV or left uninfected and were immunized with OVA at 5 days p.i. The levels of anti-OVA antibodies were determined 2 weeks after immunization. Symbols indicate individual mice. The data shown are representative of two different experiments. **, P < 0.01 (significant difference based on Student's t test).
We also examined the effect of MV infection on the contact hypersensitivity response in mice. KI mice were sensitized with DNFB, infected with MV on the next day or left uninfected, and then challenged on their ears with DNFB at 5 days p.i. Ear thicknesses of MV-infected KI mice 48 h after the challenge were significantly suppressed compared with those of uninfected mice (Fig. 3A). Although infiltration of mononuclear cells varied even among the same group of mice, it tended to be diminished in the ears of infected mice (Fig. 3B).
FIG. 3.
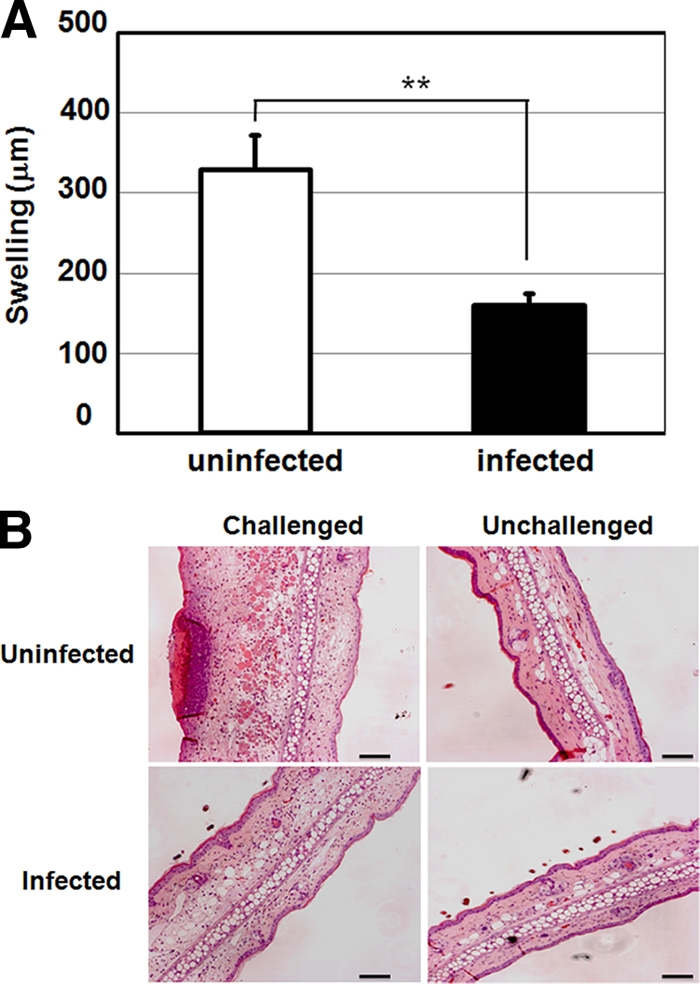
Contact hypersensitivity in MV-infected KI mice. (A) KI mice were sensitized with DNFB and infected with MV or left uninfected the next day. At 5 days p.i., DNFB was applied to the ears of mice. Ear thickness was measured 0 and 48 h after challenge. The value for each group represents the mean ± standard deviation for five mice, and the data shown are representative of three different experiments. **, P < 0.01 (significant difference based on Student's t test). (B) Tissue sections of the ears (challenged right and unchallenged left ears) were prepared from uninfected and infected KI mice 48 h after DNFB challenge and were stained with hematoxylin and eosin. Bar, 100 μm.
Profiles of cytokines produced by CD4+ T cells and DCs from MV-infected mice.
It has been reported that cytokine production is altered in measles patients (9, 19, 39). CD4+ T cells were isolated from the spleens of KI and WT mice at 5, 8, and 15 days p.i. and were stimulated with anti-CD3 antibody for 48 h. The supernatants of the stimulated CD4+ T cells were examined by ELISA for a Th1-type cytokine (IFN-γ), a Th2-type cytokine (IL-4), and the immunosuppressive cytokines IL-10 and TGF-β. CD4+ T cells from KI mice at 5 and 8 days p.i. produced similar levels of IFN-γ to those from similarly infected WT mice but produced much higher levels of IL-4 (Fig. 4A), indicating that MV infection causes a Th2 shift of the T-cell response in susceptible mice. Furthermore, production of IL-10, but not TGF-β, by CD4+ T cells was greatly increased in KI mice at 5 and 8 days p.i. compared with that in similarly infected WT mice (Fig. 4A). At 15 days p.i., differences in cytokine production levels were no longer found between splenic CD4+ T cells from WT and KI mice (Fig. 4A). Expression of IL-10 mRNA was also increased in lymph nodes obtained from infected KI mice at 5 and 8 days p.i. compared with that in lymph nodes from infected WT mice (Fig. 4B), indicating that production of IL-10 was also increased in vivo after MV infection. However, there was no difference in IL-10 mRNA levels between lymph nodes from two groups of mice at 14 days p.i. (Fig. 4B). BM-derived DCs were prepared from WT and KI mice and then infected with MV in vitro. Only ∼1% of DCs prepared from KI mice were infected, as determined by using MV expressing EGFP, while no DCs from WT mice were infected (data not shown). There were no apparent differences in IL-10 and IL-12 production at the protein level between DCs from WT and KI mice at 24 h p.i. (Fig. 4C) and 48 h p.i. (data not shown). However, DCs from KI mice produced increased levels of IL-10 mRNA but decreased levels of IL-12 mRNA compared with those from WT mice (Fig. 4C). All of these results indicate that MV infection of KI mice reproduces many immunological abnormalities observed in human patients.
FIG. 4.

Cytokine production in MV-infected mice. (A) CD4+ T cells were isolated from the spleens of KI and WT mice at 5, 8, and 15 days p.i. and were stimulated with anti-CD3 antibody for 48 h. The levels of IL-4, IFN-γ, IL-10, and TGF-β in the culture supernatants were measured by ELISA. The value for each group represents the mean ± standard deviation for three mice, and the data shown are representative of two different experiments. **, P < 0.01 (significant differences based on Student's t test). (B) Total RNAs were extracted from lymph nodes of MV-infected KI and WT mice at the indicated times and analyzed for the expression of IL-10 mRNA by quantitative real-time RT-PCR. Data were normalized to the corresponding level of β-actin mRNA expression. The value at each time point represents the mean for three mice, and the data shown are representative of three different experiments. (C) DCs prepared from WT and KI mice were left uninfected (−) or infected with MV at an MOI of 1. The cells were cultured for 24 h, and the levels of IL-12 and IL-10 in the culture supernatants were measured by ELISA. The infected cells were also cultured for the indicated times, and the expression of IL-12 p40 and IL-10 mRNAs was analyzed by quantitative real-time RT-PCR. The data shown are representative of three independent experiments.
Enhanced apoptosis, but no increase in regulatory T cells, in MV-infected mice.
Under the experimental conditions used, <1% of cells in the spleens and lymph nodes from MV-infected KI mice were infected with MV at any time point, as determined using a fluorescent-MV or infectious-center assay (22). Therefore, direct infection per se cannot account for the observed immunological abnormalities. To gain insight into the mechanism of the decrease in splenic T cells, the spleens, lungs, livers, and inguinal lymph nodes were recovered from KI and WT mice at 5 days p.i., and the total numbers of cells in those organs were counted. In the case of the liver and lungs, mononuclear cells were further isolated and counted. The proportions of T and B cells were determined by flow cytometry to calculate the absolute numbers of the respective cell types. The total numbers of mononuclear cells were not significantly different in any organs between KI and WT mice (Fig. 5A). The absolute numbers of T cells were decreased in the spleens and lungs, but not the livers and lymph nodes, of KI mice compared with those in the corresponding organs of WT mice (Fig. 5B). The results indicate that the gross redistribution of T cells within the body (from the spleen to other organs) alone cannot account for the decrease in splenic T cells in MV-infected KI mice. There was no significant difference in the absolute numbers of B cells in any organs between KI and WT mice (Fig. 5C).
FIG. 5.

Possible mechanisms of MV-induced immunological alterations in KI mice. (A to C) Numbers of total mononuclear cells (A), T cells (B), and B cells (C) in various organs from MV-infected mice. Single-cell suspensions were prepared from the spleens, lungs, livers, and inguinal lymph nodes (LN) of MV-infected WT (white bars) and KI (black bars) mice at 5 days p.i. Total mononuclear cell numbers were counted, and T- and B-cell numbers were calculated from the proportions of Thy1.2+ and B220+ cells, respectively, as examined by flow cytometry. The value for each organ represents the mean ± standard deviation for three WT or KI mice, and the data shown are representative of two different experiments. **, P < 0.01; *, P < 0.05 (significant differences based on Student's t test). (D) Lymph node cells were prepared from WT and KI mice at the indicated times after MV infection, stained with annexin V and propidium iodide, and analyzed by flow cytometry. Proportions of annexin V-positive, propidium iodide-negative cells are shown. The value at each time point represents the mean ± standard deviation for four mice, and the data shown are representative of three different experiments. **, P < 0.01 (significant difference based on Student's t test). (E) Cells were prepared from the spleens and inguinal lymph nodes of WT (white bar) and KI (black bar) mice at the indicated times after MV infection, and the proportions of CD4+ FoxP3+ cells among total splenocytes and lymph node cells were determined by flow cytometry. The value at each time point represents the mean ± standard deviation for three mice, and the data shown are representative of two different experiments.
It has been reported that MV infection causes apoptosis of infected as well as uninfected bystander cells (1, 6, 29). The proportion of cells undergoing apoptosis among the total cells was significantly increased at 5 and 8 days p.i. in lymph nodes from KI mice, but not in those from WT mice (Fig. 5D).
Regulatory T cells have been implicated in immunosuppression during MV infection (28, 39). We examined the proportion of CD4+ FoxP3+ regulatory T cells among splenocytes and inguinal lymph node cells, but no increase in CD4+ FoxP3+ cells was observed in KI mice after MV infection (Fig. 5E).
Amelioration of MV-induced inhibition of the contact hypersensitivity response with anti-IL-10R antibody.
As described above, cells from MV-infected KI mice produced larger amounts of the immunosuppressive cytokine IL-10 than did cells from control mice. To determine whether IL-10 is involved in MV-induced immunosuppression, the effect of anti-IL-10R antibody on the contact hypersensitivity response was examined. The administration of anti-IL-10R antibody at 3 days p.i. (2 days before the challenge with DNFB) was included in the experimental protocol for the measurement of the contact hypersensitivity response. Due to treatment with anti-IL-10R antibody, ear swelling in MV-infected KI mice was restored to the same levels as those in uninfected mice (Fig. 6A). In contrast, the administration of anti-IL-10R antibody did not affect the number and proliferation of T cells in MV-infected KI mice (Fig. 6B and C).
FIG. 6.
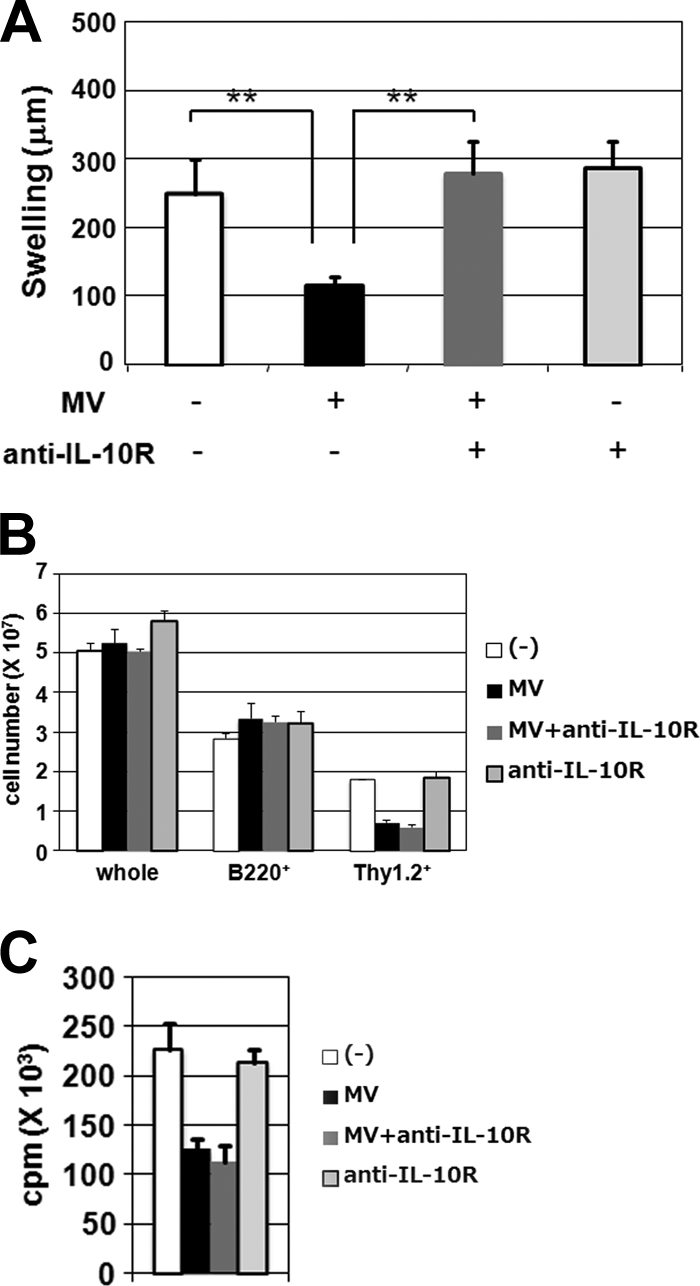
Effect of anti-IL-10R antibody on MV-induced immunological alterations in KI mice. (A) KI mice were sensitized with DNFB and infected with MV (+) or left uninfected (−) 24 h after sensitization. The mice were given anti-IL-10R MAb (+) or control rat IgG (−) at 3 days p.i. and were challenged with DNFB at 5 days p.i. Ear swelling was measured 48 h after challenge. Five or six mice were analyzed for each group, and the data shown are representative of four independent experiments. **, P < 0.01 (significant differences based on Student's t test). (B and C) Splenocytes were prepared at 5 days p.i. from MV-infected or uninfected KI mice which had been given either anti-IL-10R MAb or control rat IgG at 0 and 3 days p.i. (B) The numbers of total, B220+, and Thy1.2+ cells were determined as described in the legend to Fig. 1B. (C) CD4+ T cells were isolated from the splenocytes described above, stimulated in plates coated with anti-CD3 antibody for 48 h, and pulsed with [3H]thymidine for the last 6 h. [3H]thymidine incorporation was measured in triplicate. The value for each group represents the mean ± standard deviation for three mice, and the data shown are representative of three different experiments.
DISCUSSION
In this study, we have shown that MV-infected KI mice reproduce many immunological alterations observed in human patients, including T lymphopenia, inhibition of T-cell proliferation and antibody production, increased production of the Th2 cytokine IL-4 and the immunosuppressive cytokine IL-10, and suppression of contact hypersensitivity (8, 13, 26). Thus, KI mice serve as a useful small animal model with which to study MV-induced immunosuppression.
Although both T and B cells have been shown to express human-type SLAM and to be susceptible to MV in KI mice (22), only a decrease in splenic T cells, not B cells, was apparent after MV infection. A recent study also showed that in mink infected with canine distemper virus, another morbillivirus causing immunosuppression, T cells were depleted to a higher extent than B cells (21). The reason for the discrepancy between T and B cells in our KI mice is unknown at present, as we do not understand how T cells were reduced in the spleen. Infection-induced cell death cannot explain the decrease, as only a small percentage of cells were infected under the experimental conditions used (22). Redistribution of lymphocytes from the peripheral blood to lymphoid tissues was suspected to account for the observation, but a gross change in lymphocyte distribution was not observed. However, it was notable that lymph node cells in KI mice exhibited enhanced apoptosis at 5 and 8 days p.i., even though the numbers of lymph node cells in KI mice remained almost the same as those in WT mice. The results suggest that lymph nodes in KI mice were replenished with lymphocytes from the peripheral blood to compensate for the loss of cells due to enhanced apoptosis. This may contribute at least partly to the observed lymphopenia in the spleen.
MV-infected KI mice exhibited inhibition of CD4+ T-cell proliferation in response to anti-CD3 antibody as well as antibody production in response to OVA antigen after immunization. Since direct infection of lymphocytes or a decrease in cell numbers cannot explain these findings, there must be indirect mechanisms that cause functional impairment of lymphocytes, presumably through surface contact or soluble factor-mediated means. In humans, lymphopenia and suppression of T-cell proliferation and antibody production last several weeks after infection (8), whereas these manifestations were apparent only around 5 days p.i. in MV-infected KI mice. It should be noted that in our mouse model, MV was first detected in lymph nodes at 2 to 5 days p.i., attained a peak titer at 7 days p.i., and completely disappeared by 11 days p.i. (22). The differences in the courses of MV replication and/or host immune responses may be responsible for the different time courses of immunological alterations between humans and KI mice.
During the past several years, CD4+ FoxP3+ regulatory T cells have been shown to play an important role in the control of autoimmunity as well as in antimicrobial immune responses (18, 25). Regulatory T cells were reported to be increased not only in measles patients (39) but also in MV-infected SLAM transgenic mice (28), suggesting that regulatory T cells are responsible for MV-induced immunosuppression. In the present study, we did not observe an increase in CD4+ FoxP3+ T cells in the spleen and lymph nodes for 8 days after MV infection, during which immunological alterations were observed in mice. Thus, it is unlikely that immunological alterations in our model were caused by the increased activity of regulatory T cells. In the above study with SLAM transgenic mice, the percentages of FoxP3+ regulatory T cells were examined at 11 or 13 days p.i. (28).
It is notable that CD4+ T cells from MV-infected KI mice exhibited a Th2 shift of the T-cell response and increased production of IL-10. Similar observations have been seen in humans (9, 19, 39) and can at least partly explain MV-induced immunosuppression. We examined the levels of IL-10 in the blood of MV-infected KI mice, but a significant difference was not found compared with uninfected mice (data not shown). Therefore, if IL-10 is involved in immunological alterations of KI mice, it may act locally, not systemically. The increase of IL-10 production was indeed detected in lymph node cells taken from MV-infected KI mice ex vivo and in KI mouse-derived DCs after MV infection in vitro. Thus, both T cells, although not necessarily regulatory T cells, and DCs could be the source of IL-10. Recently, MV was shown to activate the serine and threonine kinase Raf-1 via the C-type lectin DC-SIGN, which subsequently leads to acetylation of the NF-κB subunit p65 and to increased production of IL-10 mRNA (10). Furthermore, DCs from KI mice produced lower levels of IL-12 mRNA after MV infection than did those from WT mice, consistent with previous studies (17, 30). The reduction in IL-12 may be a mechanism for the Th2 shift in MV-infected KI mice, although the exact mechanism by which MV infection leads to suppression of IL-12 production remains to be determined.
One of the most interesting observations in this study was that blocking of IL-10 by anti-IL-10R antibody ameliorated the suppression of contact hypersensitivity in infected KI mice. IL-10 can suppress cellular immune responses by modulating the function of T cells and DCs (16). IL-10 has been implicated in immunosuppression during persistent infection with lymphocytic choriomeningitis virus (LCMV) (3, 5). Unlike C57BL/6 mice, IL-10-deficient mice maintained robust effector T-cell responses and rapidly eliminated LCMV. Administration of anti-IL-10R antibody to C57BL/6 mice persistently infected with LCMV restored T-cell function and eliminated viral infection. In our model, anti-IL-10R antibody restored only contact hypersensitivity, not T lymphopenia or inhibition of T-cell proliferation. Thus, mechanisms other than IL-10-mediated signaling are also responsible for the immunosuppression observed in MV-infected KI mice.
Other mechanisms proposed include interactions of different MV proteins with cell surface proteins (13, 26). The MV envelope proteins, hemagglutinin and fusion protein, have been shown to induce surface contact-mediated signaling, resulting in the disruption of Akt kinase activation and in inhibition of T-cell proliferation (2). The interaction of the nucleoprotein with Fcγ receptor II on antigen-presenting cells or with an unidentified nucleoprotein receptor on various types of cells has been shown to affect cell functions (14, 15, 24). MV-induced ceramide accumulation is also implicated in MV-induced immunosuppression by preventing actin cytoskeletal dynamics (7, 26). Our KI mice, in conjunction with recombinant MVs containing a variety of mutant MV proteins, have the potential to be a powerful tool in unraveling MV-induced immunosuppression mechanisms.
Acknowledgments
We thank T. Iwaki and H. Kishimoto for invaluable comments and Schering-Plough Biopharma for providing rat anti-mouse IL-10R MAb YL03.1B1.3.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology and the Ministry of Health, Labor and Welfare of Japan.
Footnotes
Published ahead of print on 3 March 2010.
REFERENCES
- 1.Auwaerter, P. G., H. Kaneshima, J. M. McCune, G. Wiegand, and D. E. Griffin. 1996. Measles virus infection of thymic epithelium in the SCID-hu mouse leads to thymocyte apoptosis. J. Virol. 70:3734-3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avota, E., A. Avots, S. Niewiesk, L. Kane, U. Bommhardt, V. ter Meulen, and S. Schneider-Schaulies. 2001. Disruption of Akt kinase activation is important for immunosuppression induced by measles virus. Nat. Med. 7:725-731. [DOI] [PubMed] [Google Scholar]
- 3.Brooks, D. G., M. J. Trifilo, K. H. Edelmann, L. Teyton, D. B. McGavern, and M. B. Oldstone. 2006. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 12:1301-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Swart, R. L., M. Ludlow, L. de Witte, Y. Yanagi, G. van Amerongen, S. McQuaid, S. Yuksel, T. B. Geijtenbeek, W. P. Duprex, and A. D. Osterhaus. 2007. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog. 3:e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ejrnaes, M., C. M. Filippi, M. M. Martinic, E. M. Ling, L. M. Togher, S. Crotty, and M. G. von Herrath. 2006. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J. Exp. Med. 203:2461-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esolen, L. M., S. W. Park, J. M. Hardwick, and D. E. Griffin. 1995. Apoptosis as a cause of death in measles virus-infected cells. J. Virol. 69:3955-3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gassert, E., E. Avota, H. Harms, G. Krohne, E. Gulbins, and S. Schneider-Schaulies. 2009. Induction of membrane ceramides: a novel strategy to interfere with T lymphocyte cytoskeletal reorganisation in viral immunosuppression. PLoS Pathog. 5:e1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffin, D. E. 2007. Measles virus, p. 1551-1585. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 9.Griffin, D. E., and B. J. Ward. 1993. Differential CD4 T cell activation in measles. J. Infect. Dis. 168:275-281. [DOI] [PubMed] [Google Scholar]
- 10.Gringhuis, S. I., J. den Dunnen, M. Litjens, B. van Het Hof, Y. van Kooyk, and T. B. Geijtenbeek. 2007. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 26:605-616. [DOI] [PubMed] [Google Scholar]
- 11.Hahm, B., N. Arbour, D. Naniche, D. Homann, M. Manchester, and M. B. Oldstone. 2003. Measles virus infects and suppresses proliferation of T lymphocytes from transgenic mice bearing human signaling lymphocytic activation molecule. J. Virol. 77:3505-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahm, B., N. Arbour, and M. B. Oldstone. 2004. Measles virus interacts with human SLAM receptor on dendritic cells to cause immunosuppression. Virology 323:292-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerdiles, Y. M., C. I. Sellin, J. Druelle, and B. Horvat. 2006. Immunosuppression caused by measles virus: role of viral proteins. Rev. Med. Virol. 16:49-63. [DOI] [PubMed] [Google Scholar]
- 14.Laine, D., J. M. Bourhis, S. Longhi, M. Flacher, L. Cassard, B. Canard, C. Sautes-Fridman, C. Rabourdin-Combe, and H. Valentin. 2005. Measles virus nucleoprotein induces cell-proliferation arrest and apoptosis through NTAIL-NR and NCORE-FcgammaRIIB1 interactions, respectively. J. Gen. Virol. 86:1771-1784. [DOI] [PubMed] [Google Scholar]
- 15.Laine, D., M. C. Trescol-Biemont, S. Longhi, G. Libeau, J. C. Marie, P. O. Vidalain, O. Azocar, A. Diallo, B. Canard, C. Rabourdin-Combe, and H. Valentin. 2003. Measles virus (MV) nucleoprotein binds to a novel cell surface receptor distinct from FcgammaRII via its C-terminal domain: role in MV-induced immunosuppression. J. Virol. 77:11332-11346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li, M. O., and R. A. Flavell. 2008. Contextual regulation of inflammation: a duet by transforming growth factor-beta and interleukin-10. Immunity 28:468-476. [DOI] [PubMed] [Google Scholar]
- 17.Marie, J., J. Kehren, M. Trescol-Biemont, A. Evlashev, H. Valentin, T. Walzer, R. Tedone, B. Loveland, J. Nicolas, C. Rabourdin-Combe, and B. Horvat. 2001. Mechanism of measles virus-induced suppression of inflammatory immune responses. Immunity 14:69-79. [DOI] [PubMed] [Google Scholar]
- 18.Mills, K. H. 2004. Regulatory T cells: friend or foe in immunity to infection? Nat. Rev. Immunol. 4:841-855. [DOI] [PubMed] [Google Scholar]
- 19.Moss, W. J., J. J. Ryon, M. Monze, and D. E. Griffin. 2002. Differential regulation of interleukin (IL)-4, IL-5, and IL-10 during measles in Zambian children. J. Infect. Dis. 186:879-887. [DOI] [PubMed] [Google Scholar]
- 20.Nakano, Y. 1977. Antigenic competition in the induction of contact sensitivity in mice. Immunology 33:167-178. [PMC free article] [PubMed] [Google Scholar]
- 21.Nielsen, L., M. Sogaard, T. H. Jensen, M. K. Andersen, B. Aasted, and M. Blixenkrone-Moller. 2009. Lymphotropism and host responses during acute wild-type canine distemper virus infections in a highly susceptible natural host. J. Gen. Virol. 90:2157-2165. [DOI] [PubMed] [Google Scholar]
- 22.Ohno, S., N. Ono, F. Seki, M. Takeda, S. Kura, T. Tsuzuki, and Y. Yanagi. 2007. Measles virus infection of SLAM (CD150) knock-in mice reproduces tropism and immunosuppression in human infection. J. Virol. 81:1650-1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ono, N., H. Tatsuo, Y. Hidaka, T. Aoki, H. Minagawa, and Y. Yanagi. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J. Virol. 75:4399-4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ravanel, K., C. Castelle, T. Defrance, T. F. Wild, D. Charron, V. Lotteau, and C. Rabourdin-Combe. 1997. Measles virus nucleocapsid protein binds to FcgammaRII and inhibits human B cell antibody production. J. Exp. Med. 186:269-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakaguchi, S., T. Yamaguchi, T. Nomura, and M. Ono. 2008. Regulatory T cells and immune tolerance. Cell 133:775-787. [DOI] [PubMed] [Google Scholar]
- 26.Schneider-Schaulies, S., and J. Schneider-Schaulies. 2009. Measles virus-induced immunosuppression. Curr. Top. Microbiol. Immunol. 330:243-269. [DOI] [PubMed] [Google Scholar]
- 27.Sellin, C. I., N. Davoust, V. Guillaume, D. Baas, M. F. Belin, R. Buckland, T. F. Wild, and B. Horvat. 2006. High pathogenicity of wild-type measles virus infection in CD150 (SLAM) transgenic mice. J. Virol. 80:6420-6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sellin, C. I., J. F. Jegou, J. Renneson, J. Druelle, T. F. Wild, J. C. Marie, and B. Horvat. 2009. Interplay between virus-specific effector response and Foxp3 regulatory T cells in measles virus immunopathogenesis. PLoS One 4:e4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Servet-Delprat, C., P. O. Vidalain, O. Azocar, F. Le Deist, A. Fischer, and C. Rabourdin-Combe. 2000. Consequences of Fas-mediated human dendritic cell apoptosis induced by measles virus. J. Virol. 74:4387-4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Servet-Delprat, C., P. O. Vidalain, H. Bausinger, S. Manie, F. Le Deist, O. Azocar, D. Hanau, A. Fischer, and C. Rabourdin-Combe. 2000. Measles virus induces abnormal differentiation of CD40 ligand-activated human dendritic cells. J. Immunol. 164:1753-1760. [DOI] [PubMed] [Google Scholar]
- 31.Shingai, M., N. Inoue, T. Okuno, M. Okabe, T. Akazawa, Y. Miyamoto, M. Ayata, K. Honda, M. Kurita-Taniguchi, M. Matsumoto, H. Ogura, T. Taniguchi, and T. Seya. 2005. Wild-type measles virus infection in human CD46/CD150-transgenic mice: CD11c-positive dendritic cells establish systemic viral infection. J. Immunol. 175:3252-3261. [DOI] [PubMed] [Google Scholar]
- 32.Takeda, M., K. Takeuchi, N. Miyajima, F. Kobune, Y. Ami, N. Nagata, Y. Suzaki, Y. Nagai, and M. Tashiro. 2000. Recovery of pathogenic measles virus from cloned cDNA. J. Virol. 74:6643-6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tatsuo, H., N. Ono, K. Tanaka, and Y. Yanagi. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893-897. [DOI] [PubMed] [Google Scholar]
- 34.Thompson-Snipes, L., V. Dhar, M. W. Bond, T. R. Mosmann, K. W. Moore, and D. M. Rennick. 1991. Interleukin 10: a novel stimulatory factor for mast cells and their progenitors. J. Exp. Med. 173:507-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Pirquet, C. 1908. Das Verhalten der kutanen Tuberkulin-reaktion wahrend der Masern. Dtsch. Med. Wochenschr. 30:1297-1300. [Google Scholar]
- 36.Welstead, G. G., C. Iorio, R. Draker, J. Bayani, J. Squire, S. Vongpunsawad, R. Cattaneo, and C. D. Richardson. 2005. Measles virus replication in lymphatic cells and organs of CD150 (SLAM) transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 102:16415-16420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whittle, H. C., A. Bradley-Moore, A. Fleming, and B. M. Greenwood. 1973. Effects of measles on the immune response of Nigerian children. Arch. Dis. Child. 48:753-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yanagi, Y., M. Takeda, S. Ohno, and T. Hashiguchi. 2009. Measles virus receptors. Curr. Top. Microbiol. Immunol. 329:13-30. [DOI] [PubMed] [Google Scholar]
- 39.Yu, X. L., Y. M. Cheng, B. S. Shi, F. X. Qian, F. B. Wang, X. N. Liu, H. Y. Yang, Q. N. Xu, T. K. Qi, L. J. Zha, Z. H. Yuan, and R. Ghildyal. 2008. Measles virus infection in adults induces production of IL-10 and is associated with increased CD4+ CD25+ regulatory T cells. J. Immunol. 181:7356-7366. [DOI] [PubMed] [Google Scholar]