

Abstract
Yersinia pestis has caused three worldwide plagues in human history that have led to innumerable deaths. We have completely sequenced the genomes of two strains (D106004 and D182038) of Y. pestis isolated from Yunnan Province of China. The most striking finding of our study is that large amounts of genome rearrangement events exist between the genomes of two Yunnan strains despite being isolated from two foci only 50 kilometers apart. When we compared the genome sequences of the Yunnan strains with six strains (CO92, KIM, 91001, Antiqua, Nepal516, and Pestoides F) of Y. pestis sequenced previously, we found that the genomes of Y. pestis were divided into 61 relatively independent segments. Pairwise comparisons of all 61 segments among eight strains showed that the Yunnan strains were most closely related to strain CO92. We concluded that Y. pestis genomes consist of segments that can change their positions and directions within the genomes caused by genome rearrangements, and our study confirmed the inference that the third plague pandemic originated in Yunnan since the genome sequences of Yunnan strains were closest to the strain CO92 isolated from the United States.
Yersinia pestis, a Gram-negative bacterium, is one of the three pathogenic species in the genus Yersinia. Unlike the other two human pathogenic species (Y. pseudotuberculosis and Y. enterocolitica), Y. pestis causes infectious bubonic and pneumonic plague in humans that are of great importance to public health and biodefense. It is thought that Y. pestis was responsible for three major pandemics throughout history, taking tens of thousands of lives (3, 7, 12). Plague is a zoonotic disease spread by rodents and their fleas and has already been classified as a reemerging disease. Plague has been controlled well in modern times, nevertheless, human plague is sporadic in some places. During the last 15 years of the 20th century, 36,876 plague cases with 2,847 deaths were reported to the World Health Organization (WHO) (9).
Currently, there are active plague foci on all continents except Australia and Antarctica (7). There are complex and diverse natural plague foci in China, and plague among animals is still prevalent in several of these foci, occasionally causing human infections. Furthermore, plague cases reported in 2005 in Yulong, Yunnan, where plague has never been recorded before. Epidemiological studies found high ratios and high titers of anti-F1 antibodies in sera of domestic animals such as cats and dogs. Y. pestis was then isolated from dead wild Rattus nitidus, Apodemus chevrieri, and their fleas, proving the existence of a natural plague focus in this region (10). The newly discovered Yulong natural plague focus is located in the northwest of Yunnan province, between the Marmota himalayana plague focus on the Qinghai-Tibet Plateau and the Rattus flavipectus plague focus in the southern Yunnan province. In addition, there is another natural plague focus less than 50 km apart from the Yulong focus in Jianchuan, Yunnan, that was determined to be the Apodemus chevrieri-Eothenomys miletus plague focus in 1974. Thus far, no human plague case has been reported there. Natural environments and host-vector compositions are very similar between the Yulong focus and the Jianchuan focus; however, the Y. pestis strains isolated from these two regions have distinct biological characteristics. To determine the characteristic of this new plague focus and its potential threat to humans, we chose two representative Y. pestis strains, D106004 isolated from Apodemus chevrieri in the Yulong focus and D182038 isolated from Apodemus chevrieri in the Jianchuan focus and sequenced their whole genomes.
Previous studies showed that there are a large number of insertion sequence (IS) elements in the Y. pestis genome. These IS elements give rise to recombination events that lead to genome rearrangements (including transpositions and inversions) and gene deletions (4). Thus, genome rearrangement is one of the most important genetic features of Y. pestis. Deng et al. (6) compared the genome sequence of Y. pestis strain KIM with that of CO92 by dividing both genomes into 27 segments to calculate the evolutionary processes between the two strains by tracking transpositions and inversions of large DNA segments. The increasing availability of sequences from various Y. pestis strains in NCBI provides the opportunity for an in-depth understanding of the genome organization of Y. pestis. Our study compared the genomes of eight completely sequenced Y. pestis strains (CO92, KIM, 91001, Antiqua, Nepal516, Pestoides F, D106004, and D182038) in an attempt to deduce their genome rearrangement patterns and to assess their potential use for strain identification and phylogenetic relationship determination, especially between D106004 and D182038.
MATERIALS AND METHODS
Identification of genome plates and their boundaries.
Preliminary comparison of the eight Y. pestis genomes showed a high degree of gene synteny in certain regions. Large numbers of IS elements that can cause frequent intragenomic transpositions and inversions of large DNA segments are located among these synteny regions, even within them. This phenomenon resembles the Earth's plate tectonics: each plate is highly stable inside while plates can move in relation to one another. Thus, we used the geological term “plate” to describe those DNA segments with similar characteristics in the Y. pestis genome and named them genome plates.
To identify the genome plates, a table of similar coding sequences (CDSs) was first assembled (see supplemental material S1). All CDSs of CO92 were listed in numerical order in the same column of the table. A BLAST search was performed using each CDS of strain CO92 as a query against the DNA sequences of the seven other strains. The gene with the highest percentage identity to the query CDS was retrieved from each strain and added to the table on the same row as the query CDS. It was obvious that except CO92, whose CDSs were completely in numerical order, the CDSs of all seven other strains were arranged in clusters. According to the table of orthologs, a DNA segment with at least 10 continuous sequential CDSs was defined as an independent genome plate. Insertion and deletion of less than 10 continuous genes within an independent genome plate were allowed in our study.
The method described above can only determine the bases between the first and last CDSs of each genome plate. There are gaps between the adjacent plates that should also be included in the plates. Therefore, the boundaries of the genome plates needed to be redefined. In a previously published paper studying genome rearrangement (5), the authors divided each gap regions in half and assigned each half to a neighboring locally colinear block. In the present study, The MegAlign module of DNAStar was used for pair wise alignment of the gap sequences. The assignment of the gap sequence between the second plate (plate 2) and the third plate (plate 3) of CO92 is illustrated below as an example (see Fig. 1). Plate 2 of CO92 whose start-to-end (start/end) location on the chromosome is from bp 19172 to 39262 and plate 56 of KIM whose start/end location on the chromosome of KIM is from bp 4219550 to 4239651 are very similar to each other, but opposite in direction; plate 3 of CO92 whose start/end location on the chromosome is located from bp 41407 to 102380 and plate 3 of KIM whose start/end location on the chromosome of KIM is located from bp 58060 to 119973 are very similar and opposite in direction as well. The gap sequence (from bp 39263 to 41406 on the CO92 chromosome) between plate 2 and plate 3 of CO92 was aligned with the gap sequence (from bp 4217545 to 4219549 on the KIM chromosome) between plate 56 and plate 55 of KIM. The identical sequence that is 1,962 bp was assigned to plate 2 of CO92. The gap sequence between plate 2 and plate 3 of CO92 was then aligned once again with the gap (from bp 119974 to 122055 on the KIM chromosome) between plate 3 and plate 4 of KIM. The segment with an identical sequence that is 2,087 bp was assigned to plate 3 of CO92. Thus, the lower boundary for plate 2 of CO92 and the upper boundary for plate 3 of CO92 were determined. Because there was overlap between the two boundaries, the overlapped region was assigned to the lower numbered plate, plate 2. All other gaps of CO92 and seven other strains were assigned following the above procedure. So the genome position of every plate of each strain was finally determined (data shown in supplemental material S2).
FIG. 1.
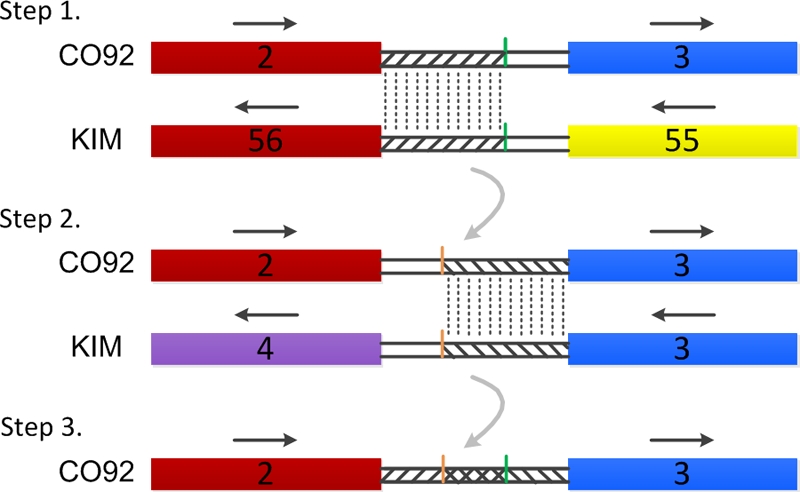
Method used for gap sequences assignment. Rectangle indicates the genome plate, and double black line indicates the gap sequence. The number in rectangle indicates the serial number of genome plate and the name of Y. pestis strains are placed forward. Two genome plates that have a high degree of similarity are shown by rectangles with the same color. Straight arrow indicates the direction of genome plate. Gap sequences linked by the dotted line are 100% identity between CO92 and KIM. The overlapped region is marked by a cross-slash.
Similarity analysis of related genome plates.
The related genome plates of each strain were pairwise aligned by using BLAST. The means and standard deviations of the sequence identities were calculated for each genome plate of the eight completely sequenced Y. pestis strains. In addition, the values of sequence identity for all of the genome plates from each strain, obtained by pairwise comparison with related genome plates from the other seven, were summed up, respectively, to generate a sequence identity matrix for each strain versus the other seven. Phylogenic relationship analysis of the eight strains was performed by using BioNumerics v4.0 software based on the identity matrix.
Genome rearrangement diversity of Y. pestis strains.
The order of the genomic plates was compared pairwise among strains, and a breakpoint was defined wherever there was a difference. The number of breakpoints between two strains may reflect the phylogenic relationships of Y. pestis with respect to genome rearrangement. A dendrogram was generated by UPGMA (unweighted pair-group method with arithmetic averages) clustering using BioNumerics v4.0 software according to the breakpoint matrix.
RESULTS
Genome plates of Y. pestis.
All plate boundaries in the genome of eight strains were assigned according to the procedure described in Materials and Methods. The defined genome plates were continuous with no intervening gap sequences (see supplemental material S2). It was demonstrated that the genomes of all eight Y. pestis strains could be divided into 61 plates, 58 of which were shared by all strains. Plates 11 and 16 existed in all strains except Pestoides F. Plate 30 was only absent from Nepal516.
Generally, the Y. pestis genome carries three different plasmids: pPCP, pCD, and pMT. Among the eight strains examined in our study, Pestoides F lacks pPCP. The sequence of Nepal516 plasmid pCD is not available on NCBI. In addition, 91001 has an extra copy of pCRY. Sequence analyses of the CDSs showed that the gene order in pPCP was identical in all tested Y. pestis strains except Pestoides F and belonged to the same plate. pCD was divided into three (see supplemental material S2) and pMT into four plates (see supplemental material S2), respectively.
Sequence identity of genome plates among Y. pestis strains.
On the genomic level, Y. pestis strains from different sources and hosts share very high percentage identity. However, the level of sequence identity is not consistent among different genome regions. A pairwise comparison of all 61 genome plates of the eight Y. pestis strains was performed by using BLAST to determine the sequence identity defined by the percentage of identical nucleotides in the longer plate. The mean sequence identities and standard deviations for each corresponding genome plate from all strains were determined (Table 1).
TABLE 1.
Mean identity of each genome plate among eight Y. pestis strains
| Plate no. | Mean identity ± SD |
|---|---|
| 1 | 0.926 ± 0.043 |
| 2 | 0.866 ± 0.106 |
| 3 | 0.966 ± 0.019 |
| 4 | 0.880 ± 0.067 |
| 5 | 0.954 ± 0.024 |
| 6 | 0.966 ± 0.047 |
| 7 | 0.973 ± 0.017 |
| 8 | 0.983 ± 0.011 |
| 9 | 0.947 ± 0.088 |
| 10 | 0.933 ± 0.055 |
| 11 | 0.924 ± 0.041 |
| 12 | 0.888 ± 0.153 |
| 13 | 0.983 ± 0.011 |
| 14 | 0.990 ± 0.007 |
| 15 | 0.941 ± 0.031 |
| 16 | 0.929 ± 0.045 |
| 17 | 0.934 ± 0.068 |
| 18 | 0.948 ± 0.028 |
| 19 | 0.787 ± 0.170 |
| 20 | 0.937 ± 0.069 |
| 21 | 0.986 ± 0.012 |
| 22 | 0.977 ± 0.014 |
| 23 | 0.935 ± 0.035 |
| 24 | 0.972 ± 0.047 |
| 25 | 0.916 ± 0.048 |
| 26 | 0.980 ± 0.014 |
| 27 | 0.957 ± 0.027 |
| 28 | 0.799 ± 0.191 |
| 29 | 0.978 ± 0.025 |
| 30 | 0.975 ± 0.017 |
| 31 | 0.925 ± 0.072 |
| 32 | 0.950 ± 0.048 |
| 33 | 0.957 ± 0.028 |
| 34 | 0.965 ± 0.024 |
| 35 | 0.953 ± 0.047 |
| 36 | 0.936 ± 0.033 |
| 37 | 0.970 ± 0.029 |
| 38 | 0.969 ± 0.013 |
| 39 | 0.966 ± 0.015 |
| 40 | 0.953 ± 0.064 |
| 41 | 0.942 ± 0.030 |
| 42 | 0.882 ± 0.150 |
| 43 | 0.977 ± 0.018 |
| 44 | 0.909 ± 0.090 |
| 45 | 0.970 ± 0.031 |
| 46 | 0.965 ± 0.017 |
| 47 | 0.963 ± 0.017 |
| 48 | 0.993 ± 0.010 |
| 49 | 0.972 ± 0.022 |
| 50 | 0.969 ± 0.022 |
| 51 | 0.933 ± 0.079 |
| 52 | 0.978 ± 0.014 |
| 53 | 0.993 ± 0.008 |
| 54 | 0.907 ± 0.054 |
| 55 | 0.863 ± 0.079 |
| 56 | 0.883 ± 0.108 |
| 57 | 0.977 ± 0.018 |
| 58 | 0.949 ± 0.053 |
| 59 | 0.935 ± 0.045 |
| 60 | 0.919 ± 0.081 |
| 61 | 0.946 ± 0.076 |
The data in Table 1 suggested close phylogenetic relationships among Y. pestis species. Each genome plate can change its location and orientation in the genomes of Y. pestis. However, the gene content between the plate and its counterpart is almost consistent. Most plates have identity values greater than 0.9. The most stable plates were plates 48 and 53 with sequence identity values of 0.993. According to gene functional analyses using the COG database, these two plates are composed mainly of genes involved in inorganic ion transport and metabolism, cell envelope biogenesis, transcription, and carbohydrate transport and metabolism, which are presumably critical for Y. pestis survival and pathogenicity. Plates 19 and 28 were the least stable, with sequence identity values of <0.8. These two plates contain genes related to coenzyme metabolism and DNA replication and recombination or are annotated as function unknown. Their instability was probably caused by gene mutation or deletion under selective pressure for Y. pestis to adapt to different niches.
It was noticed that there are different degrees of similarity among the eight sequenced Y. pestis strains. All of the plate sequence identity values of each strain compared to those of the seven other strains were summed up first, then the sum was divided by 61 (Table 2), and a dendrogram was generated accordingly (Fig. 2).
TABLE 2.
Sequence identity matrix among eight Y. pestis strains
| Strain | Sequence identity |
|||||||
|---|---|---|---|---|---|---|---|---|
| CO92 | KIM | 91001 | Nepal516 | Antiqua | Pestoides F | D182038 | D106004 | |
| CO92 | 1.000 | |||||||
| KIM | 0.951 | 1.000 | ||||||
| 91001 | 0.916 | 0.916 | 1.000 | |||||
| Nepal516 | 0.955 | 0.956 | 0.908 | 1.000 | ||||
| Antiqua | 0.946 | 0.939 | 0.907 | 0.936 | 1.000 | |||
| Pestoides F | 0.946 | 0.914 | 0.909 | 0.925 | 0.915 | 1.000 | ||
| D182038 | 0.967 | 0.958 | 0.937 | 0.954 | 0.942 | 0.959 | 1.000 | |
| D106004 | 0.959 | 0.947 | 0.934 | 0.960 | 0.926 | 0.950 | 0.975 | 1.000 |
FIG. 2.

Phylogenetic relationships between eight Y. pestis genomes inferred by UPGMA using BioNumerics v4.0 software based on the average sequence identity of the genome plates that was listed in Table 2.
Genome rearrangement diversity among Y. pestis strains.
Using Y. pestis CO92 as a reference strain, the genome plate order and orientation of the other seven strains were listed in Fig. 3. Many inversion and transposition events occurred in the genome of Y. pestis strains. It should be noted that each chromosome represented by plate of CO92 could not include all of the genes of the corresponding strain except CO92, because some genes in other strains could not be found in CO92. When the genome plate order of CO92 was compared to that of KIM, 25 breakpoints were identified. When strain 91001 was included in the comparison, the number of breakpoints increased to 45. If the other six strains were all considered, the total breakpoints number increased to 60. Based on this tendency, we proposed that the genome plates in Y. pestis are relatively stable. The genome cannot be divided unrestrictedly, forming unlimited plates.
FIG. 3.

Arrangement patterns of genome plates of each chromosome from eight completely sequenced Y. pestis strains. Each chromosome has been laid out horizontally, and the origin of replication is located in the left-most. Each rectangle indicates a plate (scale is not shown), and the number in the rectangle indicates the serial number of plate of strain CO92, which acts as reference strain. For example, the number “1” in the rectangle represents the plate 1 of CO92. Plates that are inverted relative to strain CO92 are shifted below a genome's center axis shown by a strikethrough line.
Among all 60 breakpoints, 21 belonged exclusively to a single Y. pestis strain (35%). Seven were shared by two strains (11.67%). Ten were shared by three strains (16.67%). Six were shared by four strains (10%). Seven were shared by five strains (11.67%). One was shared by six strains (1.67%), and eight were shared by all strains except CO92, which acted as the reference strain (13.33%).
The number of breakpoints indicates the frequency of genome rearrangement. It is characteristic for a strain and an indication of the phylogenic relationship among species. Based on the breakpoint matrix obtained by pairwise comparison, CO92 is closest to D182038 and D106004, and they cluster together in the tree. KIM and Nepal516 are on the same branch, while strain 91001 is not as close to them (Fig. 4).
FIG. 4.

Phylogenetic relationships among eight Y. pestis genomes inferred from the breakpoint distance matrix using BioNumerics v4.0 software.
DISCUSSION
Y. pestis cannot be subclassified based on serotype and phage type. Thus, according to the ability to ferment glycerol and to reduce nitrate, they are classified into three biotypes, Antiqua, Medievalis, and Orientalis, which are believed to be responsible for the three plague pandemics in history. However, recent studies show that this classification system is insufficient to reflect phylogenetic relationships among Y. pestis species, which also rouse skepticism about the previous hypothesis. Achtman et al. (2) have determined an evolutionary branch order within Y. pestis using three different multilocus molecular methods. The result was that there are three major branches on the tree, all of the Orientalis and African Antiqua types belong to branch 1, All of the Medievalis and Asian Antiqua types belong to branch 2, All pestoide isolates and the Microtus isolate 91001 belong to branch 0. Nepal516 and Antiqua are two strains of the classical Antiqua biovar, and their genomes were completely sequenced in 2006 (4). It has been noticed that although strains Antiqua and Nepal516 are grouped into the same biovar, they represent different lineages by SNP analysis (strains Antiqua and CO92 belong to one branch, strains Nepal516 and KIM belong to another branch). Darling et al. (5) compared the genomes of eight Yersinia stains (six Y. pestis strains and two Y. pseudotuberculosis strains) using the Mauve software and determined 78 locally colinear blocks. These authors analyzed the phylogenetic relationships of the eight strains based on inversion rates and reported that KIM and Nepal516 belong to the same branch. In our study, we analyzed genome rearrangements of eight Y. pestis strains, six of which have genome sequences available in public databases and two of which were recently sequenced in our lab. Among the eight strains, CO92 belongs to Orientalis, KIM belongs to Medievalis, 91001 belongs to Microtus, and the other five belong to Antiqua. According to sequence identity and rearrangement diversity analyses, we obtained similar results: KIM and Nepal516 have the closest phylogenetic relationship; CO92, D182038, and D106004 belong to the same branch; and 91001 and the Antiqua strains are not closely related to the other strains. It has been postulated that the third plague pandemic at the end of the nineteenth century originated in Yunnan, China, and then spread to other countries through Hong Kong (2). Our result confirms this inference, providing that CO92 is more closely related to Yunnan strains D182038 and D106004 than to the others.
Y. pestis is a very young species. It evolved from the enteric pathogen Yersinia pseudotuberculosis serotype O:1b around 1,500 to 20,000 years ago according to conventional microbiology, bacterial population genetics, and genome sequence data (1, 9). One important characteristic that distinguishes Y. pestis from its ancestor is the large number of insertion sequence elements, which account for 3.7% of its genome. IS elements can induce transpositions, inversions, and deletions of large DNA segments and lead to different gene orders among strains. Although sequence similarity among Y. pestis strains is high, the frequent occurrence of genome rearrangement indicates intense gene flow (8). With more genome sequences available, large amounts of genome rearrangements are also observed in other prokaryotes and eukaryotes (5). However, the relationships between genome rearrangement and biological characteristics and virulence of pathogens remain unclear. In the present study, we analyzed the genome rearrangements of Y. pestis and determined 61 genome plates that can shift relative to each other. We concluded that the plate number and patterns are characteristic for Y. pestis. The arrangement of the plates can be random, which does not affect Y. pestis survival but could affect its pathogenic characteristics. Rearrangements in the Y. pestis genome accumulate during evolution, as other mutations do. The evolutionary distance can be reliably determined through genome rearrangement. Thus, it can be utilized in systemic classification and phylogenetic analysis of Y. pestis species if the genome plate composition of a Y. pestis strain can be easily deduced without genome sequencing when the specific sequences at the junctions of two neighboring plates can be amplified. The recently discovered Yunnan Yulong natural plague focus is located close to the Yunnan Jianchuan plague focus. Both foci share similar natural environments and host-vector compositions. However, the Y. pestis strains isolated from the two foci show very different genome rearrangement patterns, indicating the relative independence of these two plague foci. The genotype analyses of D182038 and D106004 by pulsed-field gel electrophoresis indicated that the genomic variability of the Y. pestis strains from different foci were caused by genome rearrangement, which may provide a positive selective advantage for Y. pestis to adapt to its host environments (11). The two strains are possibly the remnants of the Y. pestis that formed the Yunnan Xenopsylla cheopis plague focus when it traveled south from the Qinghai-Tibet Plateau. The two strains generally do not frequently spread between the two foci. According to our analyses, although the genomes of the two strains have very different syntenic structures due to rearrangement, they share high similarity between plates, which may be an indication of similar pathogenicity. It has been proven that Y. pestis of the Yunnan Yulong focus can infect humans, causing severe and lethal plague. No plague cases have been reported in Jianchuan thus far. Factors other than the pathogen may play a role. Nevertheless, vigilance should be maintained with tightened surveillance in the Jianchuan focus in case of human infection.
Acknowledgments
We gratefully acknowledge the data analysis support by Qi Wang (Beijing Institute of Genomics, Chinese Academy of Sciences).
We also acknowledge financial support from the 10th Five Years Key Programs for Science and Technology Development of China(2004BA718B07), Major Projects from Department of Science and Technology (2008ZX10004-008), and a grant of the Special Fund for Health Sector, People's Republic of China (award 200802016).
Footnotes
Published ahead of print on 3 March 2010.
Supplemental material for this article may be found at http://jcm.asm.org/.
REFERENCES
- 1.Achtman, M., K. Zurth, G. Morelli, G. Torrea, A. Guiyoule, and E. Carniel. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 96:14043-14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Achtman, M., G. Morelli, P. Zhu, T. Wirth, I. Diehl, B. Kusecek, A. J. Vogler, D. M. Wagner, C. J. Allender, W. R. Easterday, V. C. Francisque, P. Worsham, N. R. Thomson, J. Parkhill, L. E. Lindler, E. Carniel, and P. Keim. 2004. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl. Acad. Sci. U. S. A. 101:17837-17842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auerbach, R. K., A. Tuanyok, W. S. Probert, L. Kenefic, A. J. Vogler, D. C. Bruce, C. Munk, T. S. Brettin, M. Eppinger, J. Ravel, D. M. Wagner, and P. Keim. 2007. Yersinia pestis evolution on a small timescale: comparison of whole genome sequences from North America. PLoS One 2:e770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chain, P. S. G., P. Hu, S. A. Malfatti, L. Radnedge, F. Larimer, L. M. Vergez, P. Worsham, M. C. Chu, and G. L. Andersen. 2006. Complete genome sequence of Yersinia pestis strains Antiqua and Nepal516: evidence of gene reduction in an emerging pathogen. J. Bacteriol. 188:4453-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darling, A. E., I. Miklós, and M. A. Ragan. 2008. Dynamics of genome rearrangement in bacterial populations. PLoS Genet. 4:e1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng, W., V. Burland, G. Plunkett III, A. Boutin, G. F. Mayhew, P. Liss, N. T. Perna, D. J. Rose, B. Mau, S. Zhou, D. C. Schwartz, J. D. Fetherston, L. E. Lindler, R. R. Brubaker, G. V. Plano, S. C. Straley, K. A. McDonough, M. L. Nilles, J. S. Matson, F. R. Blattner, and R. D. Perry. 2002. Genome sequence of Yersinia pestis KIM. J. Bacteriol. 184:4601-4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry, R. D., and J. D. Fetherston. 1997. Yersinia pestis: etiologic agent of plague. Clin. Microbiol. Rev. 10:35-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pevzner, P., and G. Tesler. 2002. Genome rearrangements in mammalian evolution: lessons from human and mouse genomes. Genome Res. 13:37-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prentice, M. B., and L. Rahalison. 2007. Plague. Lancet 369:1196-1207. [DOI] [PubMed] [Google Scholar]
- 10.Song, Z., L. Xia, Y. Liang, L. Lu, G. Wang, W. Cai, Z. Zhang, Y. He, F. Zhang, X. Dong, G. Yu, J. Wang, and D. Yu. 2008. Confirmation and study of plague natural foci for Yulong county and Guchengqu in Yunnan province. Chinese J. Control Endemic Dis. 23:3-7. [Google Scholar]
- 11.Zhang, Z., R. Hai, Z. Song, L. Xia, Y. Liang, H. Cai, Y. Liang, X. Shen, E. Zhang, J. Xu, D. Yu, and X. Yu. 2009. Spatial variation of Yersinia pestis from Yunnan Province of China. Am. J. Trop. Med. Hyg. 81:714-717. [DOI] [PubMed] [Google Scholar]
- 12.Zhou, S., W. Deng, T. S. Anantharaman, A. Lim, E. T. Dimalanta, J. Wang, T. Wu, C. Tao, R. Creighton, A. Kile, E. Kvikstad, M. Bechner, G. Yen, A. G. Stankovic, J. Severin, D. Forrest, R. Runnheim, C. Churas, C. Lamers, N. T. Perna, V. Burland, F. R. Blattner, B. Mishra, and D. C. Schwartz. A whole-genome shotgun optical map of Yersinia pestis strain KIM. Appl. Environ. Microbiol. 68:6321-6331. [DOI] [PMC free article] [PubMed]