

Abstract
Deletions and duplications of chromosomal segments (copy-number variations or CNVs) constitute a major source of variation between individual humans, underlying human evolution and many diseases from mental illness and developmental disorders to cancer. CNVs form at rates far outstripping other kinds of mutagenesis, and appear to do so by similar mechanisms in bacteria, yeast, and human. We review models for the mechanisms of formation of CNVs. Whereas non-homologous end-joining mechanisms are well known, recent models focus on perturbation of DNA replication, and replication of non-contiguous DNA segments, including the proposal that repair of broken replication forks switches under stress from high-fidelity homologous recombinational to nonhomologous repair that promotes CNV.
Human populations show extensive polymorphism in the number of copies of chromosomal segments, and of genes included in those segments, consisting of both additions and deletions1-5. This is known as copy number variation (CNV). A high proportion of the genome, currently estimated at up to 12%, is subject to CNV 1-4, 6, which can arise both meiotically and somatically as shown by the finding that identical twins can differ in CNV7 and that different organs and tissues vary in copy number in the same individual8. CNV is at least as important in determining the differences between individual humans as single nucleotide polymorphisms (SNPs)9, and appears to be a major driving force in evolution, especially in the rapid evolution that has occurred, and continues to occur, within the human and great ape lineage10-14. Changes in copy number might change the levels of expression of genes included in the regions of variable copy number, allowing transcription levels higher or lower than those that can be achieved by control of transcription of single copies per haploid genome. Possible adaptive advantages of CNV are discussed in Box 1. However, additional copies of genes also provide redundancy in sequence, so that some copies are free to evolve new or modified functions or patterns of expression, while other copies maintain the original function15, 16. The nonhomologous recombination events that underlie changes in copy number also allow reassorting of exons between different genes by translocation, insertion or deletion17, 18, so that proteins might acquire new domains, and hence new or modified activities.
Box 1. How much CNV is adaptive?
Initially CNV appeared to be advantageous because the genes included are enriched for genes concerned with olfaction and with immunity to disease, and with secreted proteins, that is, with genes relevant to the immediate environment120. These genes were reported to be under recent selection because they contain higher than average frequencies of non-synonymous mutations120. Now alternative explanations have been offered for these features138. The regions where CNVs are found might be regions where copy number is less important than it would be in other regions. In other words, most CNV is rapidly purged from the population, but the selection for balanced genotype is weaker in regions where CNVs are found, so that CNVs in these regions persist for longer. Weakness in purifying selection would also explain the observed higher mutation frequency than in the genome as a whole, because even loss of function alleles in these regions would be purged from the population only slowly138.
Evidence that fewer or more copies of specific genes offer a selective advantage is sparse. The human salivary amylase gene, AMY1, shows CNV in human populations139. The amount of salivary amylase is directly proportional to the copy number of AMY1. The average number of copies of AMY1 is higher in starch consuming cultures than in individuals in cultures that consume little starch139, suggesting that a high copy number of AMY1 is advantageous in starch eating cultures and neutral in cultures with low starch intake139. A second possible example is the correlation between copy number of CCL3L1, which encodes a chemokine, and susceptibility to HIV/AIDS140. An example of reduction in copy number being beneficial is suggested for the α-globin loci. The disadvantages of deletion in homozygotes might be balanced by a resistance to malaria for heterozygotes (reviewed by141). Other examples will be found, but most of the extensive CNV in human seems to be non-adaptive or disadvantageous, and present only because it has not yet been purged. About 75% of CNV is found at a frequency of less than 3% in human populations3, suggesting a stochastic origin and maintenance of most CNV142. Other studies give similar results143-145. Many private CNVs (specific to a family) have been described, and there is limited overlap in lists of CNVs found in different genome-wide searches6. Many CNVs have gone to fixation in the human and other primate lines and are now seen as LCRs 10, 12, 13, 146. These. The low frequency in populations of almost every CNV polymorphism that has been described suggests that few of the CNVs that we see now are tending towards fixation.
However, there is uncertainty in the ascertainment of CNV, caused by differing criteria for length and degree of homology that defines CNV, and use of different techniques. There is a problem concerning the reference genome because of widespread CNV polymorphism, and the fact that the reference was established as a haploid genome rather than the natural diploid state. The relative rarity of specific CNVs described above was challenged by a study showing that 80% of CNVs are found in more than 5% of individuals147.
However, much variation in copy number is disadvantageous. Change in copy number is involved in cancer formation and progression19, 20 and contributes to cancer proneness21. In many situations, a change in copy number of any of a number of specific genes is not well tolerated, and leads to a group of pathological conditions known as genomic disorders22. Because particular gene imbalances are associated with specific clinical syndromes, rare clinical cases of change in copy number are available and have facilitated the study of the chromosomal changes underlying CNV. Further examples have come from studies of complete genomes, and from genome-wide surveys of CNV by such techniques as array comparative genomic hybridization (aCGH)5 comparison of expression levels23 or paired-end mapping2.
Mechanisms of chromosomal structural change have been studied in model organisms, notably baker's yeast, Escherichia coli and Drosophila. By bringing together the findings from model organisms with the characteristics of CNV in human and primate genomes, we can begin to work towards an understanding of the processes that lead to chromosomal structural change, and so gain insights into a major component of the forces that drive human evolution24. Extrapolation from one organism to another is not always reliable, but it has proved very successful in the study of processes acting on DNA. Almost all DNA repair mechanisms acting in human were first described in model organisms, particularly bacteria25. In this review, we describe the properties of CNVs and the mechanisms that lead to change in copy number.
Characteristics of copy number variants
A change in copy number requires a change in chromosome structure, joining two formerly separated DNA sequences. These junctions give important insights into how the structural change has arisen. Many changes show recurrent end-points. That is, most events at a given locus have their end-points confined to a few genomic positions. The junctions of these recurrent CNVs are found to be in low copy repeats (LCRs) that provide extensive homology. LCRs, also called segmental duplications, are sequences that occur twice or a few times in a genome. For practical purposes, the definition is limited by degree of identity (commonly >95%) and length (usually >1 or 5 kilobase pairs). It is likely that recurrent CNVs arose by homologous recombination between repeated sequences. This process is called non-allelic homologous recombination (NAHR), and is discussed further below.
Other structural changes show non-recurrent end-points. Most non-recurrent CNVs occur at sites of very limited homology of 2 to 15 base pairs (bp) (e. g.26-28), much too short to have occurred by homologous recombination (HR) as discussed below. Another characteristic of non-recurrent events is that chromosomal structural changes can be complex, having short sequences from elsewhere inserted at the junctions, and involving a mixture of duplications, triplications, inversions and deletions, and interspersed with lengths of unchanged sequence29-32. An example of a complex rearrangement with microhomology junctions is shown in Figure 1. A third characteristic is that, although the non-recurrent junctions do not coincide with LCRs, they tend to occur in the vicinity of regions that are rich in LCRs, either direct or inverted resulting in complex regional genomic architecture33-35. The origin of LCRs and of regions in which LCRs are prevalent is presumably the same as the origin of the non-recurrent events that we witness today, and hence the mechanisms of non-recurrent copy number change are the mechanisms of evolution of genomes.
Figure 1.
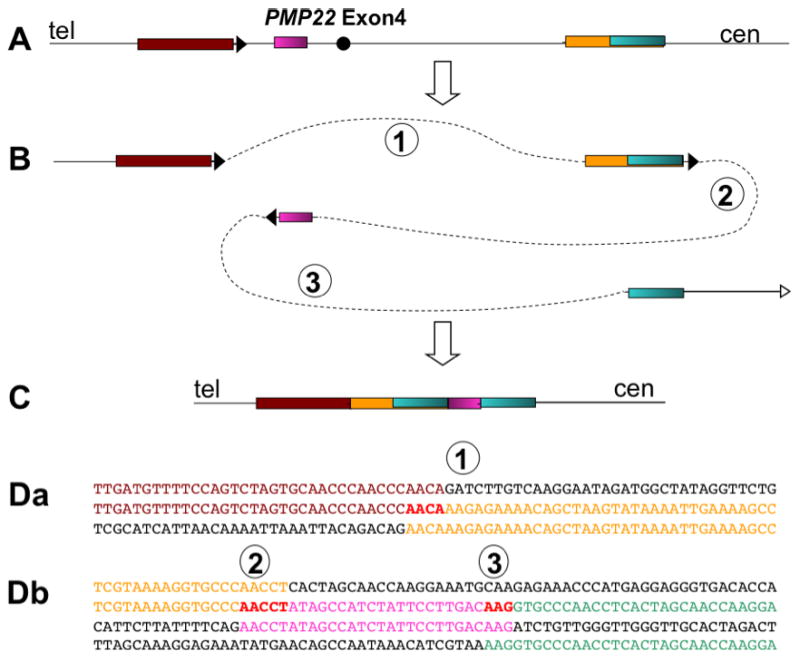
An example of a complex genomic rearrangement with microhomology junctions that deleted about 10 kb including exon 4 of the human PMP22 gene18. A represents a portion of the normal map of part of PMP22. Blocks of sequence are differentiated by colour. B represents a hypothetical series of 3 template switches that would achieve the rearrangements. The switches could have occurred in the opposite order. Numbers correspond to the junctions detailed below. C shows the rearranged chromosomal region with a deletion of exon 4 joining brown sequence to yellow sequence, followed by an inversion of part of the deleted segment (purple) and a direct duplication of part of the sequence (green). Da shows the nucleotide sequences of the coloured segments in the same colours, where the top line is parental sequence, the second line is the sequence of the rearranged chromosome, and the bottom line is the parental interacting sequence from the other side of the deletion. The junction of brown to yellow sequence shows a 4 bp microhomology (red). b shows the sequences that interacted to make junctions 2 and 3. The second line has the new sequence joining yellow to inverted red sequence (third line) with a 5 bp microhomology, while the fourth line show the sequence that interacted in inverted orientation to make junction 3 with a 3 bp microhomology.
Mechanisms of structural change
Change in copy number involves change in the structure of the chromosomes such that previously separated chromosomal regions are now juxtaposed. Because the mechanisms of all structural changes are the same as those that cause CNV, we discuss them here for the understanding that they provide on the mechanisms of copy number change.
Changes in the structure of chromosomes occur by two general mechanisms, homologous recombination (HR) and nonhomologous recombination. HR requires extensive DNA sequence identity (about 50 bp in E. coli36, to as many as 300 bp in mammalian cells and human37, 38) and most mechanisms also require a strand exchange protein, RecA in prokaryotes and its orthologue Rad51 in eukaryotes. The reason for this dual requirement is that an early step in most homologous recombination is the RecA/Rad51 catalyzed invasion of homologous duplex sequence by the 3′ end of single-stranded DNA, that is, the 3′ end replaces the like strand of the duplex. In contrast, nonhomologous recombination mechanisms use only microhomology of a few complementary base pairs or no homology. HR is the basis of several mechanisms of accurate DNA repair that use another identical sequence to repair damaged sequence. Chromosomal structural change can occur by HR, not because the mechanism is inaccurate, but because genomes have tracts of LCRs or segmental duplications. There will be no change in structure if a damaged sequence is repaired using homologous sequence in the same chromosomal position in the sister chromosome or homologue, but repair might utilize homologous sequences in different chromosomal positions. This is called non-allelic or ectopic homologous recombination (NAHR)39. In contrast, any mechanism that repairs a damaged molecule using sequence from a nonhomologous template has the possibility of changing the structure of chromosomes.
Homologous recombination mechanisms
HR underlies many DNA repair processes, and is also responsible for ordered segregation of chromosomes and for generating new combinations of linked alleles at meiosis. HR is used in repair of DNA breaks and gaps. The best studied mechanism of HR is double-strand break (DSB)-induced recombination. Intensive studies of DSB-induced meiotic recombination and of recombination induced by site-specific nucleases have allowed us to understand the mechanisms of DSB repair. However, spontaneous mitotic recombination is probably initiated by other types of DNA lesion such as single-strand DNA gaps.
In the following sections, we describe the HR mechanisms of DSB repair both for situations when two double-stranded ends are present, and when there is only one, and we show how these mechanisms can lead to or avoid generation of copy-number variation. All models are hypotheses based on the evidence available, so that reality might not conform exactly to the mechanisms depicted.
Double Holliday junction and synthesis-dependent strand annealing models of double-strand break repair
Figure 2 illustrates double Holliday junction DSB repair, a mechanism that can lead to gene conversion and crossing over, and synthesis-dependent strand-annealing (SDSA)(reviewed in40, 41), which does not generate crossovers. SDSA seems to be, rather, a mechanism for avoiding crossing-over and loss of heterozygosity (LOH), though still capable of producing change in copy number when the DNA template contains direct repeats (reviewed in41).
Figure 2.
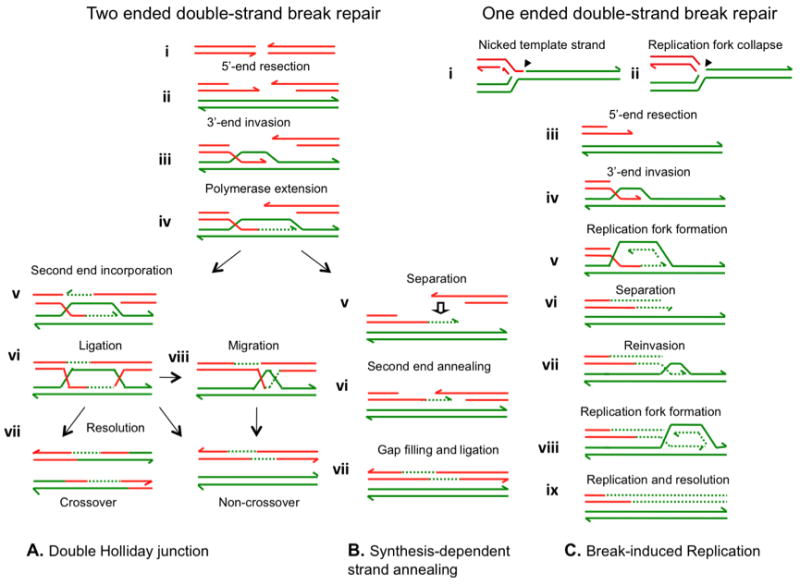
Mechanisms of homologous recombination. A and B Double-strand break repair; C. Double-strand end repair. A. Double Holliday junction recombinational double-strand break repair. At the break (a), 5′ ends are resected (b) to leave 3′ overhanging tails (half arrow heads). These are coated with RecA/Rad51 that catalyzes invasion by one or both ends into homologous sequence forming a D-loop (c). The 3′ end then primes DNA synthesis (dotted lines) that extends it past the position of the original break (d). The second end is incorporated into the D-loop by annealing, and is also extended (e). Following ligation, which forms a double Holliday junction (f), the junctions are resolved by endonuclease (g). The overall effect will be either a non-crossover or a crossover, depending upon whether the two junctions are resolved in the same or different orientations. An alternative resolution pathway is mediated by a helicase and a topoisomerase to converge and undo the double Holliday junction generating only a non-crossover outcome48 (h). B. Synthesis-dependent strand annealing follows the same pathway as in A through polymerase extension (d). At this point the invading end, together with the newly synthesized DNA is separated from the template by a helicase (e). It now encounters the second end from the double-strand break, and anneals with it by complementary base pairing (f) (dotted arrow). The second end is extended by DNA synthesis (g), and ligated, thus completing repair. C. Break -induced replication occurs at collapsed (broken) replication forks that occur when the replicative helicase at a replication fork encounters a nick in a template strand (solid arrowhead) (a and b). Break induced replication can be understood as a modification of SDSA. As before, a 3′ tail invades a homologue (d), usually the sister from which it broke, and is extended by low processivity polymerization that includes both leading and lagging strands104 (e). However, the separated extended 3′ end fails to find a complementary second end to which to anneal (f). The end then reinvades (g), and is extended further by a low processivity replication fork. This process of invasion, extension and separation might be repeated several times until a more processive replication fork is formed (h). The fork can now complete replication to the end of the molecule50 (i). In (g) and (h) we show the Holliday junction following the replication fork, giving conservative segregation of old and new DNA. It is also possible that the Holliday junction is cleaved by an endonuclease, in which case segregation will be semi-conservative. In all parts of the figure, each line shows a single nucleotide chain. Polarity is indicated by half arrows on 3′ ends. New synthesis is shown by dotted lines. The broken DNA molecule is shown in red, a homologue or sister molecule is shown in green.
Crossing-over between homologous chromosomes can lead to LOH if the chromatids carrying the same alleles segregate together at mitosis. If a crossover forms when the interacting homologies are in non-allelic positions on the same chromosome (NAHR), this will result in duplication and deletion of sequence between the repeats (Figure 3Aa). Crossing-over during intrachromosomal recombination between direct or inverted repeats leads to deletion or inversion respectively. In all organisms tested including human there is a bias in vegetative cells towards the non-crossover outcome (e.g.42, 43). The differences in crossing-over frequency can be explained if HR often proceeds according to the double Holliday junction model (Figure 2A) in meiotic cells and via SDSA (Figure 2B) in mitotic cells. Several different DNA helicases and topoisomerases can channel the DSB repair into a noncrossover pathway either by unwinding the D-loop after an invading strand primed new DNA synthesis44-47 (Figure 2Be) or by resolving a double Holliday junction into a noncrossover (Figure 2Ah)48. Also crossovers are less likely to form during HR between short repeats probably due to decreased ability to form an intermediate of crossing-over – a double Holliday junction49.
HR is used not only to repair two-ended double-strand breaks (Figure 2A and B), but also to repair collapsed, or broken, replication forks (Figure 2C) in a process called break-induced replication (BIR). This process is normally faithful and leaves no trace, except that, if the broken end invades a homologue instead of a sister molecule, it can lead to LOH. If the repair involves homologous sequence in a different chromosomal position, translocation50, duplication or deletion can result, thus constituting an alternative mechanism for NAHR (Figure 3Ab). BIR has been suggested as a mechanism for achieving chromosomal structural change by several authors31, 51-55, and we suggest below that it underlies a major mechanism for change in copy number.
Small deletions can form by a mechanism of break repair acting at directly repeated sequences. Single-strand annealing (SSA) was first described in mammalian and amphibian cells56, 57. SSA happens when neither end at a two-ended double-strand break invades homologous sequence. In this case, erosion of the 5′ ends continues, exposing substantial lengths of single-stranded 3′ ends (Figure 3B). If this process exposes complementary sequences in the two single strands, annealing can occur. After removal of the flaps and ligation, the broken molecule has been repaired, but all sequence between the repeat sequence and one of the repeats themselves have been deleted. Because there is no invasion step, SSA does not require RecA/Rad51, but requires the annealing protein Rad52. SSA has been studied in yeast58 where it has been found to be limited in most situations to deletions of up to a few tens of kilobase pairs. The longer the sequence separating the repeats, the less likely is it that resection reaches both repeats so that the break is repaired by SSA. This length restriction means that SSA is likely to be only a minor player in CNV formation. In human, DSB-induced SSA was observed between identical Alu repeats separated by few hundred base pairs59.
Figure 3.
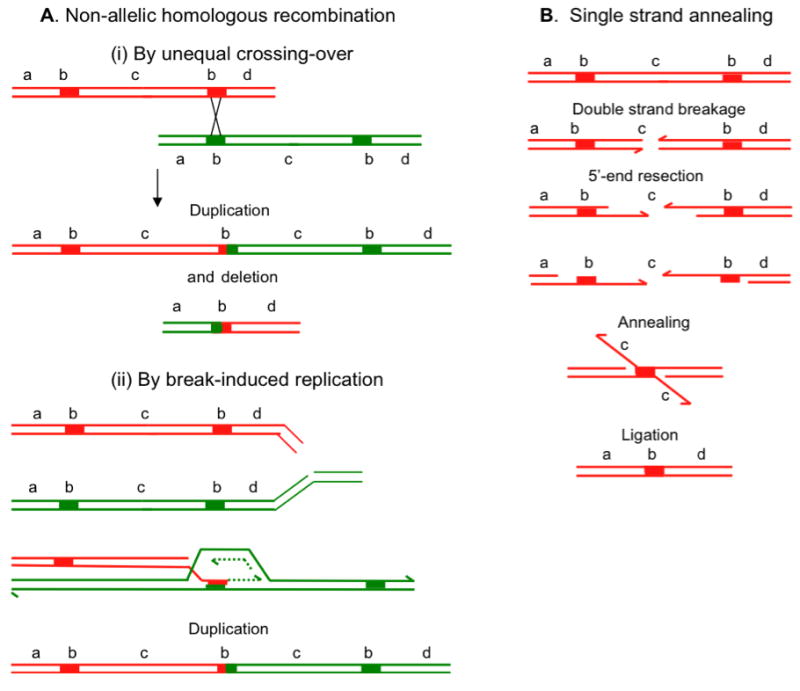
Change in copy number by homologous recombination. A. Non-allelic homologous recombination (NAHR). (Aa) If a recombination repair event uses a direct repeat (b) as homology, a crossover outcome (shown as an “x”) leads to products that are reciprocally duplicated and deleted for sequence (c) between the repeats. These might segregate from each other at the next cell division, thus changing the copy number in both daughter cells. NAHR can also occur by BIR when the broken molecule uses ectopic homology to restart the replication fork (Ab). BIR will form duplications and deletions in separate events. B. Single-strand annealing. When 5′ end resection on either side of a double-strand break does not lead to invasion of homologous sequence, resection continues. If this resection reveals complementary single-stranded sequence (b) shown by thickened lines, these can anneal. Removal of flaps, gap-filling and ligation complete repair of the double-strand break with deletion of the sequence between the repeats (c) and of one of the repeats. Each line represents a single DNA strand, polarity is indicated by half arrows on 3′ ends, and specific sequences are identified by letters “a” to “d”.
Correct choice of recombination partner prevents chromosomal structural change
Many of the pathways of chromosomal structural change described here result from a choice of a nonallelic partner for repair. Cells regulate the choice of partner for repair in several different ways. Mismatch repair provides the first barrier to choice of homeologous sequence for repair. In E. coli this is presumably because MutS and MutL together undo base-paired DNA molecules that are imperfectly matched60. Homeologous sequences are similar sequences that share less than about 95% identity. Mismatch repair also prevents interaction with very short lengths of homology. Second, a sister chromatid is the preferred partner for recombinational repair. The proteins that hold two sister chromatids together are called cohesins. Cohesins are assembled at DSBs in both yeast and human61-63 and facilitate DSB repair64. Cohesins restrict the opportunity to utilize either intrachromosomal or interchromosomal nonallelic recombination templates. In yeast, cohesins regulate copy number of tandem ribosomal RNA gene repeats (rDNA)65. rDNA repeats are susceptible to deletions and insertions. Transcription within rDNA has been suggested to disrupt cohesins locally, thus leading to the choice of nonallelic repeats for repair in rDNA, resulting in copy number change66. However, cells respond to such changes by regulating recombination to bring the number of the repeats back to the normal level. It seems likely that loss of cohesion may induce copy number change at other loci.
Besides having two sister chromatids held together upon DNA damage, yeast and human cells also keep two ends of a single DSB together67, 68. Sgs1, an orthologue of human BLM helicase is one of the proteins that coordinate template choice by two ends of a DSB69, 70. However, multiple reports demonstrate that two ends of single DSB can engage in recombination with different homologous templates. Copying diverse sequences by two ends of single DSB from different templates will lead to rearrangements.
Although HR provides vital repair mechanisms, it is also hazardous, as revealed by the many ways in which it can effect chromosomal structural change including copy-number variation. HR repair mechanisms minimize this by avoiding crossing-over, by regulating partner choice, and by requiring substantial lengths of perfect homology. However, meiosis requires crossing-over, and we see a possible impact of this in the elevated frequency of CNV arising in meiosis (Box 2).
Box 2. When and how frequently do changes in CNV occur?
Much CNV occurs as inherited polymorphism, but it also arises de novo at a significant rate. It is apparent that CNV arises both in the germline and in somatic cells. A study of four hotspots at which CNV occurs by NAHR148 found a frequency of CNV of 10-6 to 5×10-5 per gamete, as determined from sperm cell analysis. A study using similar methods to analyze blood and sperm from 2 individuals for NAHR-mediated deletions in α-globin genes reported a frequency of over 10-6 in blood cells, and an order of magnitude higher in sperm cells149. Similar results were obtained for duplications at the α-globin genes150. Analysis of blood for three specific chromosomal inversions arising by NAHR between inverted LCRs found very high frequencies ranging from 10-4 to 100. Newborns carried much less NAHR product, suggesting that somatic structural changes accumulate during life151. There are few comparable data for CNV caused by non-recurrent changes. Extensive somatically generated CNV between different embryonic stem-cell lines derived from the same inbred laboratory strains of mice has been reported152. Most, but not all of the variants were associated with LCRs, suggesting NAHR. Thus the rate of arising of CNV is several orders of magnitude higher than that of point mutations, and is especially high during meiosis.
Data on the occurrence of sporadic genomic disorders have recently been reviewed153, reporting frequencies in the range of 10-6 to 10-4 CNVs per gamete, including nonrecurrent rearrangements at the DMD locus154. This is two to 4 orders of magnitude more than for point mutation153, and is in good agreement with the estimate from sperm cells (above148). There is, however, a notable difference in that deletions were about twice as common as duplications in sperm cells, while in healthy individuals deletions and duplications are about equally common4. This suggests that there is less selection against duplication CNV than against deletions, sperm cells not being subject to the selective pressures during development.
It is interesting to speculate that a high rate of generation of CNV has played a role in the rapid evolution of the primate lineage12. Rat and mouse have fewer LCRs than human155. Mouse lab strains show CNV, and it co-localizes with LCRs, suggesting NAHR156. So with a low occurrence of LCR, de novo CNV formation will be substantially lower than that in human. In contrast, CNV and LCRs in the chimpanzee appear to be evolving at a rate comparable to that in human10-13.
Nonhomologous repair
In addition to HR pathways, there are mechanisms of DNA repair that use very limited or no homology. When homology is not used to ensure that molecules are rejoined in the correct positions, there is some probability that genetic change including CNV will result. These mechanisms that do not use HR can be divided into replicative and non-replicative mechanisms.
Nonhomologous repair: non-replicative mechanisms
Nonhomologous end joining
There are two pathways of DSB repair that either do not require homology or need very short microhomologies for repair: nonhomologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ). These pathways have recently been described in detail elsewhere71-73. NHEJ rejoins DSB ends accurately or leads to small 1-4 bp deletions, and also in some cases to insertion of free DNA, often from mitochondria or retrotransposons74, 75. MMEJ uses 5 to 25 bp long homologies to anneal to ends of DSBs and, like SSA, leads to deletions of sequences between annealed microhomologies. The second distinction between these pathways is that they require different proteins. Key proteins involved in NHEJ (Ku70/Ku80) are not required for MMEJ. Also the strand-annealing protein Rad52 is not required for MMEJ, which distinguishes this pathway from SSA.
It is likely that NHEJ and MMEJ contribute to some chromosomal rearrangements by joining nonhomologous sequences. This is possible during repair of two ended DSBs such as endonuclease-induced breaks, damage by exogenous agents including chemotherapeutic agents, or when two converging replication forks encounter a nick in the DNA. Programmed two-ended DSBs occur in the immune system, and their repair might relate to the formation of some translocations seen in leukemia patients (reviewed by76). Programmed two-ended DSBs also occur in cells undergoing meiosis where they initiate HR. As discussed below, single-ended DSB are likely to be a more frequent spontaneous lesion and to be repaired replicatively.
Breakage-fusion-bridge cycle
Upon replication of a chromosome that has lost its telomere due to a DSB, there will be two sister chromatids that lack telomeres. McClintock77 proposed that sister chromatids that lack telomeres will fuse, creating a dicentric chromosome (Figure 4). During anaphase, the two centromeres will be pulled to separate nuclei, causing eventual breakage of the dicentric chromosome. The break will lead, after replication, to new ends that lack telomeres, so that these ends will again fuse, forming a new dicentric chromosome and a cycle is established. Random breakage causes large inverted duplications, and repeated cycles could lead to amplification of the inverted repeat. The cycle will cease when the chromosome acquires a telomere. This process, the breakage-fusion-bridge cycle, has been linked to the formation of amplification in mammalian cells (reviewed by78), and it is believed to play a major role in amplification in cancer. The random breakage of the dicentric chromosome formed by fusion of the ends of sister chromatids provides a ready explanation for the occurrence of large inverted repeats in human cancer cells79. The breakage-fusion-bridge cycle can be induced by enzymatic breakage of chromosomes and by inhibition of DNA synthesis80, and the bridges formed can be observed microscopically.
Figure 4.
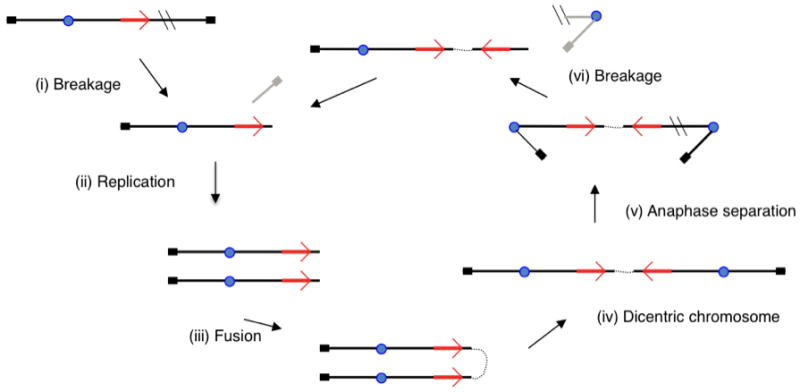
The Breakage-fusion-bridge cycle. (a) An unreplicated chromosome suffers a double-strand break so that it loses a telomere. (b) Upon replication, both sister chromatids lack telomeres. (c) These two ends are proposed to fuse, (d) forming a dicentric chromosome. (e) At anaphase, the two centromeres of the dicentric chromosome are pulled apart, initially forming a bridge between the telophase nuclei. (f) Eventually the bridge is broken in a random position. This inevitably leads to the formation of a large inverted duplication. The chromosome once again has an unprotected end, and upon replication will form two sisters that can fuse to form anew a dicentric chromosome, and so the process is repeated until the end acquires a telomere from another source. Amplification of the large inverted duplication can occur by random breakage in later cycles (not shown). Centromeres are indicated by a blue circle, telomeres by a black block and genomic sequence as red arrows showing orientation. Breakage points are shown as double black lines and fragments that are lost are in grey.
Some events that change chromosome structure that have been attributed to the breakage-fusion-bridge cycle could also be caused by any repeated nonhomologous recombination process without repeated breakage and fusion55. Clearly, when a translocation forms in inverted orientation creating a dicentric chromosome, a second event will be required to restore stability to the genome. This second event might be part of the first event as described in replicative models below, rather than being a result of anaphase bridge formation.
Non-homologous repair: replicative mechanisms
The presence of microhomology at a site of nonhomologous recombination has been regarded as the signature of NHEJ 34, 81. However, as evidence has accumulated that the formation of microhomology junctions is linked in some cases to DNA replication, replicative mechanisms are more often cited, and specifically BIR is suggested as the mechanism24, 31, 51, 52, 54, 55. The evidence for the involvement of replication in at least some chromosomal structural change was recently reviewed24. There is growing evidence that replicative stress might underlie copy number change. Aphidicolin, an inhibitor of replicative DNA polymerases, induces CNV both at chromosomal fragile sites, and throughout the genome80, 82-85. This suggests a replication-based mechanism involving DNA double-strand ends, because these are known to result from replication inhibition86. In one study, the new aphidicolin-induced CNVs were found to have microhomology (65%) or no homology at their endpoints, showing that they did not arise by HR82. In the following sections we explore the replicative mechanisms that have been proposed as the origin of CNV.
Replication slippage or template switching
When short lengths of DNA sequence identity occur within the length of genome that is expected to be single-stranded during replication, i.e. the length of an Okazaki fragment, 1 or 2 Kb or shorter in human, the sequence between the homologous lengths is often deleted or duplicated. This has been attributed to a mechanism of replication slippage along the exposed template during DNA replication 87, 88 (see Figure 5A). In E. coli, replication slippage can occur in the absence of RecA87-90, and is strongly dependent on the length of homology87, 91, 92 and the distance between the repeat units91, 93, 94. The frequency of these events is increased by mutations in genes encoding components of the replicative DNA polymerase holoenzyme, presumably because perturbation of replication promotes the slippage95-97. When the repeat sequences are not identical (i. e., they contain mismatches), the frequency of replication slippage is higher in mutants carrying mutation in the mismatch-repair system98. Taken together with a failure to find genetic requirements for short homology deletion97 (suggesting that essential functions are involved, so that most mutations in these genes would render the cells inviable), and the absence of any requirement for HR functions97, the evidence all points to a replicative mechanism. Because of the very strong distance limitation, the replication slippage mechanism is proposed to operate within a replication fork and so cannot account for most of the events that change copy number in human, where distances of tens of kilobases to megabases are involved.
Figure 5.
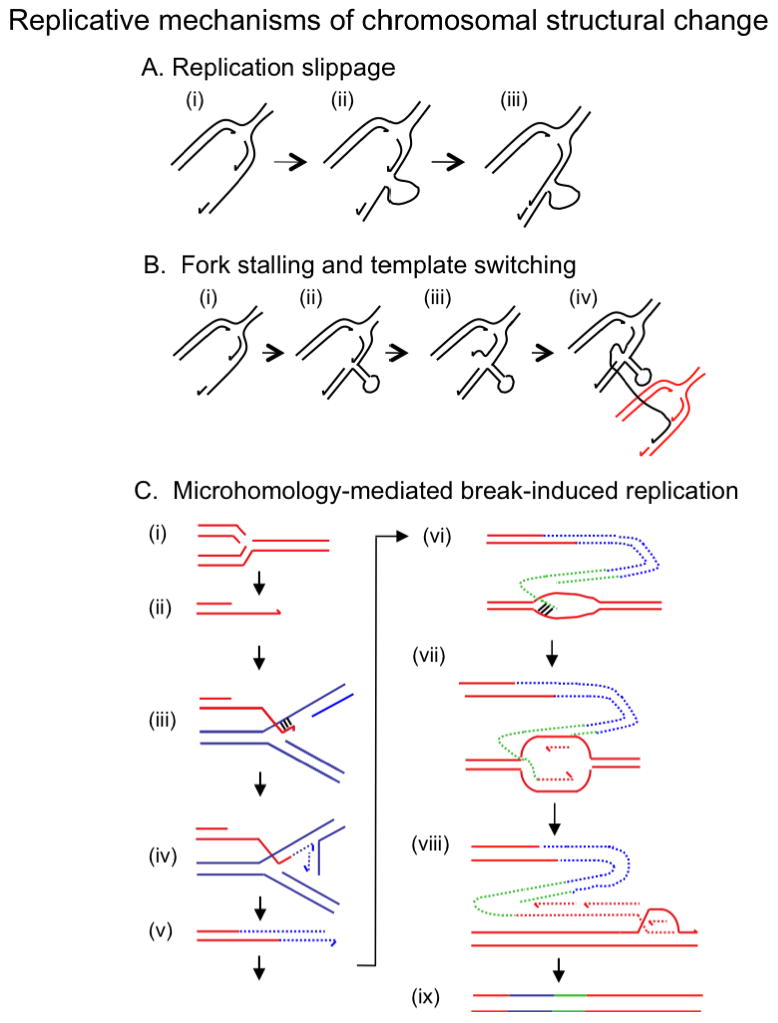
Replicative mechanisms for nonhomologous structural change. A. Replication slippage. (a), during replication, a length of lagging-strand template becomes exposed as a single strand. (b) Whether or not due to secondary structures in the lagging-strand template, the 3′ primer end can move to another sequence showing a short length of homology on the exposed template and (c) continue synthesis after having failed to copy part of the template. As shown, this will produce a deletion. Several variations on this mechanism can also produce a duplication of a length of DNA sequence with or without sister chromatid exchange (reviewed by157). Events occurring by this mechanism are confined to the length of genome to be found in a single replication fork (1 to 2 Kb). B. Fork stalling and template switching (FoSTeS)26, 100. Exposed single-stranded lagging strand template (a) might acquire secondary structures (b), which can block the progress of the replication fork. The 3′ primer ends then become free from their templates (c), and might then alight on other exposed single-stranded-template sequence on another replication fork that shares microhomology (d), thus causing duplication, deletion, inversion or translocation depending on the relative position of the other replication fork. Fork stalling can be caused by other situations, such as lesions in the template strand or shortage of deoxynucleotide triphosphates. C. Microhomology-mediated break-induced replication (MMBIR). (a) Replication fork collapse, in which one arm breaks off a replication fork, can occur because the fork encounters a nick on a template strand, or can be caused by endonuclease. (b) the 5′ end of the broken molecule (red) will be recessed from the break, exposing a 3′ tail. When insufficient RecA or Rad51 is present to allow invasion of homologous duplex as shown in Figure 2, the 3′ tail will anneal to any exposed single stranded DNA that shares microhomology. (c) shows the 3′ tail annealing to the lagging-strand template of another replication fork (blue). (d) shows the establishment of a replication fork with both leading and lagging strand synthesis from the microhomology junction. (e), The replication is of low processivity, and the broken end, now extended by a length of a different sequence, shown in blue, is separated from the template and again processed to a 3′ tail, which will then anneal to another single-stranded microhomology sequence. (f), the extended broken end now carrying both the sequence identified in blue, and a length of different sequence identified in green, anneals with single-stranded sequence back onto the red molecule. In this case the single-stranded sequence is shown as a locally melted length of DNA. (g), Another short-processivity fork is established, but this one becomes a fully processive replication fork (h) that can continue to the end of the chromosome or replicon. (i) shows the molecule produced, carrying short sequences from other genomic locations. Whether or not a length of red sequence is duplicated or deleted depends on the position at which synthesis returns to the red chromosome relative to where the initial fork collapse occurred. If the second black sequence is a homologous chromosome instead of the sister chromatid, there will be extensive LOH downstream from the event. Each line represents a single DNA strand, polarity is indicated by half arrows on 3′ ends, and arrowheads show the position of nicks and breaks. Microhomology junctions are indicated by black crosshatching.
Fork stalling and template switching
Study of stress-induced amplification of the lac genes, using the E. coli Lac system of Cairns and Foster99 led Slack et al.100 to propose that template switching was not confined to single replication forks, but could also occur between different replication forks. This model, now called fork stalling and template switching (FoSTeS)26, illustrated in Figure 5B, proposes that when replication forks stall in cells under stress, the 3′ primer end of a DNA strand can change templates to single-stranded DNA templates in other nearby replication forks. This hypothesis was necessary because the mean length of amplified units (amplicons) in that study was about 20 kb100, which is too long to have occurred within a replication fork. The evidence that the mechanism was replicative was first, that the junctions between amplicons showed only microhomology of 4 to 15 basepairs100, 101, showing that HR is not involved. Second, that there was a requirement for DNA polymerase I, specifically for the 5′ flap endonuclease domain. This implicated lagging strands at replication forks because the other functions of this nuclease in excision repair were not involved100. Third, overproduction of the main 3′ single-strand DNA exonuclease, ExoI, decreased the frequency of rearrangements, implying that 3′ DNA ends promote the amplification, and also suggesting that DNA synthesis is being primed from 3′ ends during amplification100. The reciprocal increase was seen for deletion of the gene for ExoI in short- range deletion events102, 103, and was interpreted in the same way. The physical properties of the amplicons, microhomology at the boundaries, and complexity in the structure of amplicons in E. coli100, 101, have also been found to be properties of human duplications and deletions, as discussed above. This led to the proposal26, 100 that the same mechanism was involved in the formation of some human chromosomal rearrangements.
Microhomology-mediated break-induced replication
Other authors have proposed that BIR can be mediated by microhomology, notable Payen et al.54, who demonstrated the involvement of BIR in microhomology-mediated non-homologous recombination by showing a requirement for Pol32. Pol32 is a non-essential DNA polymerase that has been shown to be required for BIR in yeast104. Bauters et al.51 invoked microhomology mediated BIR to explain non-recurrent copy number changes in human. Both these teams proposed that there was a microhomology-mediated invasion of double-stranded DNA. We, however, propose that invasion will not occur without extensive homology, so that we must seek a mechanism other than invasion when only microhomology is involved.
The FoSTeS model does not propose mechanistic molecular detail, does not involve DNA double-strand ends and is not readily testable. FoSTeS is now superseded by a new model, the microhomology-mediated break-induced replication (MMBIR) model, based on the mechanism of BIR repair of single double-strand ends24 (Figure 5C). The proposal is that when a single double-strand end results from replication fork collapse in a cell under stress, RecA/Rad51 is down-regulated as part of the stress response, so that classical BIR repair of the double-strand end cannot occur. BIR is strongly RecA/Rad51-dependent because it includes an invasion by a 3′ DNA end into double-stranded DNA of the repair partner. However, BIR is known to occur at a low rate in the absence of Rad51105, 106. MMBIR postulates that, because strand invasion is limited or not possible when RecA/Rad51 is down regulated, the 3′ end from the collapsed fork will anneal to any single-stranded template with which it shares microhomology and that occurs in physical proximity to the 3′ DNA end, and initiate DNA synthesis and a low-processivity replication fork. Single-stranded DNA will occur in other replication forks, especially in the lagging-strand template, at excision repair tracts, at sites of transcription and at secondary structures in DNA. This annealing reaction does not require RecA/Rad51, and requires very little homology, so that annealing will occur with the sister molecule either in front of or behind the position of the fork collapse, leading to deletion or duplication respectively, and in either orientation, giving the opportunity to form an inversion. Microhomology might also be found in a different chromosome, leading to translocation. Annealing with the homologous chromosome instead of the sister could be a cause of extensive LOH. The repeated extension and separation from the template that are characteristic of BIR50 will cause several of these changes to occur in the same event, leading to the observed complexity. The ability of this mechanism to explain easily the complexity of multiple junctions in close proximity is a major attractive feature of this model.
Supporting the idea that chromosomal structural change results from an insufficiency of RecA/Rad51 is the observation that Drosophila in which one copy of the gene homologous to RAD51 has been deleted gives a mixture of homologous and nonhomologous junctions in the repair of double-strand breaks107. RecA/Rad51 is postulated to be down regulated when MMBIR is occurring because the cells are under stress. There are two lines of evidence supporting this concept. First, hypoxic stress in human cancer cell lines leads to repression of RAD51 and to reduced homologous recombination (reviewed by108, 109, 121). This has been interpreted as a switch from high-fidelity HR to lower fidelity NHEJ caused by stress110. However, in the case of a collapsed replication fork, NHEJ is not possible because there is only one end. Such a switch in double-strand-end repair would therefore, we suggest, be expected to lead to a BIR-based mechanism such as MMBIR. Hypoxia is known to induce gene amplification in cancer cell lines by activating fragile sites leading to DSBs111, which are also activated by DNA synthesis inhibition85, presumably leading to single double-strand ends. Second, amplification in E. coli that involves the formation of microhomology junctions, discussed above100, 101, is induced by the stress of starvation. This is demonstrated by the observation that amplification does not begin to appear until the cells are starved112, and that it requires induction of the general and starvation stress response by the RpoS transcriptional activator113. However, it has not been shown that this relates to down-regulation of the recA gene.
Another switch from high-fidelity to error-prone double-strand-break repair seen in starved E. coli, depends on the expression of the cells' major general stress response114. Even artificially inducing the stress response in the absence of stress causes this switch114.
Because, during BIR, either homologue may be copied, it is a clear prediction of the MMBIR model that structural change will often be accompanied by extensive LOH, and in some case by loss of imprinting24. Many instances of deletions associated with extensive LOH have been reported from cells from patients with acute lymphoblastic leukemia115.
As described, both end-joining and MMBIR mechanisms can show microhomology junctions and insertion of other sequences at the junction, so that we do not yet know a diagnostic criterion by which to distinguish them after the fact, though the presence of complexity is more characteristic of MMBIR than of NHEJ. Because most insertions at end-points are insertions of nearby sequence, because evidence is extensive and increasing that replication plays a role in the generation of non-recurrent CNV and because it is expected that one-ended DSBs will be much more common than two-ended DSBs, we favour mechanisms such as MMBIR over end-joining mechanisms as being responsible for most non-recurrent CNV generation. This is particularly true for duplications, triplications and complex rearrangements.
Effects of chromosome architecture on CNV
CNVs are not randomly distributed in the human genome, but tend to be clustered in regions of complex genomic architecture, consisting of complex patterns of direct and inverted LCRs as illustrated in Figure 1. Some of this clustering might relate to the absence of dosage-sensitive genes in some regions, but there is ample evidence that specific chromosomal architectural features are also involved.
The most obvious effect of architecture is that changes mediated by NAHR occur where there are pre-existing LCRs that provide the homology needed for recombination (discussed above). More subtle influences are the preferential occurrence of copy number variation in regions of heterochromatin near telomeres116, 117 and centromeres118-120, the association with sequence-specific structures such as replication origins and terminators54, and scaffold attachment sequences27, 121, the occurrence of nonrecurrent changes in regions carrying multiple LCRs26, 33, including inverted repeats and palindromic sequences (reviewed by78), and the role of highly repeated sequences, LINES and SINES in generating structural change122. The ability of DNA sequences to adopt a non-B conformation, such as cruciforms, affects chromosomal structural change in a way that depends upon the structures rather than the specific sequences that generate them123-125. Finally, there are reports of specific consensus sequences associated with CNV30, 100, 121, 126. The preferential occurrence of non-recurrent structural changes close to LCRs has been recognized for some time33, 34, 127. This has been explained as a tendency for secondary structures in DNA to cause replication fork stalling26, and also to provide single-stranded regions that can facilitate the formation of microhomology junctions24. SINEs, predominantly Alu sequences, and LINEs are retrotransposons that account for a large proportion of the human genome. These elements cause mutation by insertion into coding sequences, cause CNV by NAHR and also provide a focus for non-recurrent changes in copy number128 in a way that is not understood. For example, one study found Alu elements at 13 out of 40 microhomology deletion endpoints122. The association of LINEs and SINEs with CNV could be caused either by DNA breakage being frequent in these areas (because of live transposon activity for example), which could initiate non-homologous recombination, or by persistent single-strandedness in these regions (due to extensive transcription, secondary structures, or replication pausing), which could make them preferred sites for annealing by single-strand DNA ends.
Thus one concludes that copy number changes are not randomly distributed, but that multiple genomic features can affect the probability of their occurrence. Detailed mechanistic explanations for the impact of these architectural characteristics on CNV formation await further work.
Conclusions and ramifications
There are at least two main mechanisms for change in copy number: NAHR and microhomology-mediated events. NAHR can be formed either by classical HR-mediated DSB repair via a double Holliday junction, or from BIR, which restarts broken replication forks by HR. However, the LCRs that mediate NAHR were presumably formed predominantly by the same mechanism as non-recurrent copy-number changes that are being formed now. Thus the mechanisms of microhomology-mediated copy number change underlie most copy-number change. Based on the evidence favouring replicative mechanisms, the known enzymology of DNA transactions in model organisms, and the evidence presented above concerning the potential involvement of stress responses in altering the availability of DNA repair proteins, we suggest that a mechanism like MMBIR presently constitutes our best working hypothesis for most events of copy number change. It is also likely that end-joining mechanisms including NHEJ, MMEJ and SSA will play a role, especially in cells of the immune system. The breakage-fusion-bridge cycle has been shown to operate in experimental systems and appears to be important in amplification in some cancers. However, we need not assume that only one mechanism acts in any given event. Microhomology-mediated events might trigger the breakage-fusion-bridge cycle by forming a dicentric chromosome, which must eventually be resolved to a more stable genotype. This could also happen when NAHR causes formation of a dicentric chromosome. Similarly, end-joining mechanisms might have a role to play in cleaning up loose ends that result from other events. Notably, the fusion step of the breakage-fusion-bridge cycle might be mediated by any of these end-joining mechanisms.
The molecular events proposed in MMBIR have not been demonstrated experimentally. Because of its molecular detail, several aspects of the model are testable. The hypothesized involvement of stress responses will also be important to test. The potential for extensive LOH downstream from the initiating event has already been seen in some systems, and further testing of this correlation should be available from the study of genome-wide single nucleotide polymorphism data. This LOH might extend as far as the next replication fork travelling in the opposite direction, or it might process to the telomere.
If it can be substantiated that CNV stems from stress response, this has interesting implications for physiology, evolution and disease. First, stress-inducible chromosomal structural variation suggests that cells have an inducible ability to evolve (“evolvability”). If the mechanism is indeed stress-inducible, then cells and organisms will be predisposed to genome rearrangement when they are stressed and activate stress responses. This is specifically when they are maladapted to their environments. If stress fuels CNV formation, then the generation of genetic diversity upon which natural selection acts will be maximal specifically when a population will benefit most from such diversity, potentially fueling evolution at that time. This idea was developed to explain mutagenesis affecting evolution of bacterial populations (reviewed by129, 130) including generation of antibiotic resistance upon antibiotic exposure131, 132. If stress inducibility applies to CNV formation, then CNVs may not only be important promoters of evolutionary divergence12 but, in addition, their formation may be an “evolvability” enhancing mechanism. Although human cells show stress-inducible genetic change109, 133, 134, it is not yet known in any organism with a differentiated germline whether such stress-inducible genome instability mechanisms can contribute to the germline and so to evolution. This is an important topic for future study.
Second, similarly, the predicted occurrence of LOH with CNV formation is likely to be important in human cancer in which both mutations and LOH drive tumor progression and resistance to therapies. The same mechanism is proposed to produce translocations when the microhomology used in repair resides in a different chromosome24, further exacerbating the problem. The problem of stress-inducible cancer progression and resistance mechanisms are discussed elsewhere108, 129.
Third, given the large and ever increasing number of developmental, neurological, and psychiatric syndromes linked to CNV, we wish to propose an important analogy or corollary from cancer biology to these other CNV-based diseases. It is well known that mutations that increase mutation rate, “mutator mutations”, promote cancer predisposition because cancer is a genetic disease fueled by mutagenesis and genome instability135-137. Mutator mutations are common in microbes and many are known in a wide variety of cancer-predisposition and aging syndromes25. We propose that in addition to CNVs' directly promoting various syndromes, that there will be human variants with increased rates of CNV change, because of mutations in any of many possible genes affecting DNA-damage processing and repair (among other processes). We expect that such alleles will predispose families and individuals to the mental, developmental, neurological and other syndromes caused by CNVs. Future work should seek to identify such modifier-locus mutations that affect human developmental and other disorders, some of which, we predict, will do so by elevating CNV-formation rates. Individuals with such mutations might also be cancer prone. Moreover, in cases where the disease can be caused by a CNV present as a mosaic, therapies to reduce CNV genesis might reduce penetrance of such disease. Drugs that do this are neither known nor on the drawing board. Identification of the many genes, proteins and pathways affecting CNV formation rates and understanding their mechanisms of action are prerequisites to considering any potential therapeutic approaches.
Acknowledgments
This work was supported by grants from the National Institutes of Health, R01 GM64022 to PJH, R01 NS59529 to J.R.L., R01 GM53158 and R01 CA85777to SMR, and R01 GM80600 to GI.
Footnotes
Summary
- Copy number variants (CNVs) arise by homologous recombination (HR) between repeated sequences (recurrent CNVs). Or by non-homologous mechanisms that occur throughout the genome (non-recurrent CNVs).
- Non-recurrent CNVs frequently show microhomology at their end-points, and can have complex structure.
- Locus-specific mutation frequency for CNV and other structural changes are 2 to 4 orders of magnitude greater than for point mutations.
- HR mechanisms generally achieve accurate repair of DNA damage.
- Double-strand breaks are repaired by HR or by end-joining mechanisms, which are generally non-homologous.
- Broken replication forks with single double-strand ends are also repaired by HR.
- There is evidence that repair of broken replication forks underlies some non-homologous recombination.
- Repair of broken forks in stressed cells could cause non-homologous repair because of stress-induced down-regulation of HR proteins.
- Models are presented for mechanisms by which stress might induce non-homologous events leading to CNV.
References
- 1.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Korbel JO, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redon R, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]; A survey by SNP array and by aCGH of 270 individuals from the human HapMap samples.
- 4.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 5.Wong KK, et al. A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kidd JM, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruder CEG, et al. Phenotypically concordant and discordant monozygotic twins display different DNA copy-number-variation profiles. Am J Hum Genet. 2008;82:1–9. doi: 10.1016/j.ajhg.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piotrowski A, et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- 9.Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8:639–646. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- 10.Dumas L, et al. Gene copy number variation spanning 60 million years of human and primate evolution. Genome Res. 2007;17:1266–1277. doi: 10.1101/gr.6557307. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper traces the history of copy number variation through the evolution of the primate lineage.
- 11.Nahon JL. Birth of ‘human-specific’ genes during primate evolution. Genetica. 2003;118:193–208. doi: 10.1023/a:1024157714736. [DOI] [PubMed] [Google Scholar]
- 12.Bailey JA, Eichler EE. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat Rev Genet. 2006;7:552–564. doi: 10.1038/nrg1895. [DOI] [PubMed] [Google Scholar]
- 13.Stankiewicz P, Shaw CJ, Withers M, Inoue K, Lupski JR. Serial segmental duplications during primate evolution result in complex human genome architecture. Genome Res. 2004;14:2209–2220. doi: 10.1101/gr.2746604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marques-Bonet T, et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature. 2009;457:877–881. doi: 10.1038/nature07744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohno S. Evolution by Gene Duplication. Springer-Verlag; Berlin, New York: 1970. [Google Scholar]
- 16.Inoue K, et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36:361–9. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
- 17.Rotger M, et al. Partial deletion of CYP2B6 owing to unequal crossover with CYP2B7. Pharmacogenet Genomics. 2007;17:885–890. doi: 10.1097/FPC.0b013e3282ef5cd1. [DOI] [PubMed] [Google Scholar]
- 18.Zhang F, Khajavi M, Towne CF, Connolly AM, Batish SV, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate human genomic, genic, and exon shuffling rearrangements. Nature genetics. 2009 doi: 10.1038/ng.399. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volik S, et al. Decoding the fine-scale structure of a breast cancer genome and transcriptome. Genome Res. 2006;16:394–404. doi: 10.1101/gr.4247306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brodeur GM, Hogarty MD. In: The Genetic Basis of Human Cancer. Vogelstein B, Kinzler KW, editors. McGraw-Hill; New York: 1998. pp. 161–172. [Google Scholar]
- 21.Frank B, et al. Copy number variant in the candidate tumor suppressor gene MTUS1 and familial breast cancer risk. Carcinogenesis. 2007;28:1442–1445. doi: 10.1093/carcin/bgm033. [DOI] [PubMed] [Google Scholar]
- 22.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangement and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 23.Stranger BE, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hastings PJ, Ira G, Lupski JR. A Microhomology-mediated Break-Induced Replication Model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. doi: 10.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review presents the MMBIR model for chromosomal rearrangement with detail of the evidence on which it is based.
- 25.Friedberg EC, et al. DNA Repair and Mutagenesis. ASM Press; Washington, D. C.: 2005. [Google Scholar]
- 26.Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]; Description of the structure of duplications seen in genomic disorder patients with complex structure and microhomology.
- 27.Nobile CT, Rizzi L, Simionati F, Nigro B, Cardazzo V, Patarnello B, Valle T, Danieli G, A G. Analysis of 22 deletion breakpoints in dystrophin intron 49. Hum Genet. 2002;110:418–421. doi: 10.1007/s00439-002-0721-7. [DOI] [PubMed] [Google Scholar]
- 28.Carvalho CM, et al. Complex rearrangements in patients with duplications of MECP2 can occur by Fork Stalling and Template Switching. Hum Mol Genet. 2009 doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. Intrachromosomal serial replication slippage in trans gives rise to diverse genomic rearrangements involving inversions. Hum Mutat. 2005;26:362–373. doi: 10.1002/humu.20230. [DOI] [PubMed] [Google Scholar]
- 30.Gajecka M, et al. Unexpected complexity at breakpoint junctions in phenotypically normal individuals and mechanisms involved in generating balanced translocations t(1;22)(p36;q13) Genome Res. 2008;18:1733–1742. doi: 10.1101/gr.077453.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheen CR, et al. Double complex mutations involving F8 and FUNDC2 caused by distinct break-induced replication. Hum Mutat. 2007;28:1198–2006. doi: 10.1002/humu.20591. [DOI] [PubMed] [Google Scholar]
- 32.Vissers LE, et al. Complex chromosome 17p rearrangements associated with low-copy repeats in two patients with congenital anomalies. Hum Genet. 2007;121:697–709. doi: 10.1007/s00439-007-0359-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stankiewicz P, et al. Genome architecture catalyzes nonrecurrent chromosomal rearrangements. Am J Hum Genet. 2003;72:1101–1116. doi: 10.1086/374385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JA, et al. Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet. 2006;15:2250–2265. doi: 10.1093/hmg/ddl150. [DOI] [PubMed] [Google Scholar]
- 35.Lee JA, et al. Spastic paraplegia type 2 associated with axonal neuropathy and apparent PLP1 position effect. Ann Neurol. 2006;59:398–403. doi: 10.1002/ana.20732. [DOI] [PubMed] [Google Scholar]
- 36.Lovett ST, Hurley RL, Sutera VA, Jr, Aubuchon RH, Lebedeva MA. Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways. Genetics. 2002;160:851–859. doi: 10.1093/genetics/160.3.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liskay RM, Letsou A, Stachelek JL. Homology requirement for efficient gene conversion between duplicated chromosomal sequences in mammalian cells. Genetics. 1987;115:161–167. doi: 10.1093/genetics/115.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reiter LT, et al. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am J Hum Genet. 1998;62:1023–1033. doi: 10.1086/301827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 40.Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- 41.Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esposito MS. Evidence that spontaneous mitotic recombination occurs at the two-strand stage. Proc Natl Acad Sci U S A. 1978;75:4436–4440. doi: 10.1073/pnas.75.9.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stark JM, Jasin M. Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol Cell Biol. 2003;23:733–743. doi: 10.1128/MCB.23.2.733-743.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dupaigne P, et al. The Srs2 helicase activity is stimulated by Rad51 filaments on dsDNA: implications for crossover incidence during mitotic recombination. Mol Cell. 2008;29:243–254. doi: 10.1016/j.molcel.2007.11.033. [DOI] [PubMed] [Google Scholar]
- 45.Sun W, et al. The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol Cell. 2008;32:118–128. doi: 10.1016/j.molcel.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ira G, Malkova A, Liberi G, Foiani M, Haber JE. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prakash R, et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu L, Hickson ID. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 49.Prado F, Cortes-Ledesma F, Huertas P, Aguilera A. Mitotic recombination in Saccharomyces cerevisiae. Curr Genet. 2003;42:185–198. doi: 10.1007/s00294-002-0346-3. [DOI] [PubMed] [Google Scholar]
- 50.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]; This paper shows the experimental evidence from yeast on the nature of BIR, specifically showing template switching between homologues and sometimes with non-homologous chromosomes causing translocation, and then showing replication out to the telomere.
- 51.Bauters M, et al. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome Res. 2008;18:847–858. doi: 10.1101/gr.075903.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deem A, et al. Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics. 2008;179:1845–1860. doi: 10.1534/genetics.108.087940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Narayanan V, Lobachev KS. Intrachromosomal gene amplification triggered by hairpin-capped breaks requires homologous recombination and is independent of nonhomologous end-joining. Cell Cycle. 2007;6:1814–1818. doi: 10.4161/cc.6.15.4522. [DOI] [PubMed] [Google Scholar]
- 54.Payen C, Koszul R, Dujon B, Fischer G. Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence from yeast that LCRs arrise by a replicative mechanism, specifically one involving BIR.
- 55.Schmidt KH, Wu J, Kolodner RD. Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol Cell Biol. 2006;26:5406–5420. doi: 10.1128/MCB.00161-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin FL, Sperle K, Sternberg N. Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol Cell Biol. 1984;4:1020–1034. doi: 10.1128/mcb.4.6.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sweigert SE, Carroll D. Repair and recombination of X-irradiated plasmids in Xenopus laevis oocytes. Mol Cell Biol. 1990;10:5849–56. doi: 10.1128/mcb.10.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haber JE. Exploring the pathways of homologous recombination. Curr Opin Cell Biol. 1992;4:401–412. doi: 10.1016/0955-0674(92)90005-w. [DOI] [PubMed] [Google Scholar]
- 59.Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–94. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 60.Rayssiguier C, Thaler DS, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- 61.Unal E, et al. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 62.Strom L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 63.Kim JS, Krasieva TB, LaMorte V, Taylor AM, Yokomori K. Specific recruitment of human cohesin to laser-induced DNA damage. J Biol Chem. 2002;277:45149–45153. doi: 10.1074/jbc.M209123200. [DOI] [PubMed] [Google Scholar]
- 64.Sjogren C, Nasmyth K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol. 2001;11:991–995. doi: 10.1016/s0960-9822(01)00271-8. [DOI] [PubMed] [Google Scholar]
- 65.Kobayashi T, Horiuchi T, Tongaonkar P, Vu L, Nomura M. SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell. 2004;117:441–453. doi: 10.1016/s0092-8674(04)00414-3. [DOI] [PubMed] [Google Scholar]
- 66.Kobayashi T, Ganley AR. Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science. 2005;309:1581–1584. doi: 10.1126/science.1116102. [DOI] [PubMed] [Google Scholar]
- 67.Kaye JA, et al. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol. 2004;14:2096–2106. doi: 10.1016/j.cub.2004.10.051. [DOI] [PubMed] [Google Scholar]
- 68.Soutoglou E, et al. Positional stability of single double-strand breaks in mammalian cells. Nat Cell Biol. 2007;9:675–682. doi: 10.1038/ncb1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh SD, et al. BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell. 2007;130:259–272. doi: 10.1016/j.cell.2007.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jain S, et al. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev. 2009;23:291–303. doi: 10.1101/gad.1751209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lieber MR. The mechanism of human nonhomologous DNA end joining. J Biol Chem. 2008;283:1–5. doi: 10.1074/jbc.R700039200. [DOI] [PubMed] [Google Scholar]
- 73.Daley JM, Palmbos PL, Wu D, Wilson TE. Nonhomologous end joining in yeast. Annu Rev Genet. 2005;39:431–451. doi: 10.1146/annurev.genet.39.073003.113340. [DOI] [PubMed] [Google Scholar]
- 74.Haviv-Chesner A, Kobayashi Y, Gabriel A, Kupiec M. Capture of linear fragments at a double-strand break in yeast. Nucleic Acids Res. 2007;35:5192–5202. doi: 10.1093/nar/gkm521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yu X, Gabriel A. Ku-dependent and Ku-independent end-joining pathways lead to chromosomal rearrangements during double-strand break repair in Saccharomyces cerevisiae. Genetics. 2003;163:843–856. doi: 10.1093/genetics/163.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nickoloff JA, De Haro LP, Wray J, Hromas R. Mechanisms of leukemia translocations. Curr Opin Hematol. 2008;15:338–45. doi: 10.1097/MOH.0b013e328302f711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McClintock B. Chromosome organization and genic expression. Cold Spring Harb Symp Quant Biol. 1951;16:13–47. doi: 10.1101/sqb.1951.016.01.004. [DOI] [PubMed] [Google Scholar]
- 78.Tanaka H, Yao MC. Palindromic gene amplification--an evolutionarily conserved role for DNA inverted repeats in the genome. Nat Rev Cancer. 2009;9:216–224. doi: 10.1038/nrc2591. [DOI] [PubMed] [Google Scholar]
- 79.Tanaka H, Bergstrom DA, Yao MC, Tapscott SJ. Large DNA palindromes as a common form of structural chromosome aberrations in human cancers. Hum Cell. 2006;19:17–23. doi: 10.1111/j.1749-0774.2005.00003.x. [DOI] [PubMed] [Google Scholar]
- 80.Coquelle A, Pipiras E, Toledo F, Buttin G, Debatisse M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell. 1997;89:215–225. doi: 10.1016/s0092-8674(00)80201-9. [DOI] [PubMed] [Google Scholar]
- 81.Shaw CJ, Lupski JR. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet. 2005;116:1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- 82.Arlt MF, et al. Replication stress induces genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am J Hum Genet. 2009 doi: 10.1016/j.ajhg.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Durkin SG, et al. Replication stress induces tumor-like microdeletions in FHIT/FRA3B. Proc Natl Acad Sci U S A. 2008;105:246–251. doi: 10.1073/pnas.0708097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kuo MT, Vyas RC, Jiang LX, Hittelman WN. Chromosome breakage at a major fragile site associated with P-glycoprotein gene amplification in multidrug-resistant CHO cells. Mol Cell Biol. 1994;14:5202–5211. doi: 10.1128/mcb.14.8.5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Coquelle A, Rozier L, Dutrillaux B, Debatisse M. Induction of multiple double-strand breaks within an hsr by meganucleaseI-SceI expression or fragile site activation leads to formation of double minutes and other chromosomal rearrangements. Oncogene. 2002;21:7671–7679. doi: 10.1038/sj.onc.1205880. [DOI] [PubMed] [Google Scholar]
- 86.Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Albertini AM, Hofer M, Calos MP, Miller JH. On the formation of spontaneous deletions: the importance of short sequence homologies in the generation of large deletions. Cell. 1982;29:319–328. doi: 10.1016/0092-8674(82)90148-9. [DOI] [PubMed] [Google Scholar]
- 88.Farabaugh PJ, Schmeissner U, Hofer M, Miller JH. Genetic studies of the lac repressor. VII. On the molecular nature of spontaneous hotspots in the lacI gene of Escherichia coli. J Mol Biol. 1978;126:847–857. doi: 10.1016/0022-2836(78)90023-2. [DOI] [PubMed] [Google Scholar]
- 89.Ikeda H, Shimizu H, Ukita T, Kumagai M. A novel assay for illegitimate recombination in Escherichia coli: stimulation of lambda bio transducing phage formation by ultra-violet light and its independence from RecA function. Adv Biophys. 1995;31:197–208. doi: 10.1016/0065-227x(95)99392-3. [DOI] [PubMed] [Google Scholar]
- 90.Shimizu H, et al. Short-homology-independent illegitimate recombination in Escherichia coli: distinct mechanism from short-homology-dependent illegitimate recombination. J Mol Biol. 1997;266:297–305. doi: 10.1006/jmbi.1996.0794. [DOI] [PubMed] [Google Scholar]
- 91.Bi X, Liu LF. recA-independent and recA-dependent intramolecular plasmid recombination: differential homology and requirement and distance effect. J Mol Biol. 1994;235:414–423. doi: 10.1006/jmbi.1994.1002. [DOI] [PubMed] [Google Scholar]
- 92.Mazin AV, Kuzminov AV, Dianov GL, Salganik RI. Mechanisms of deletion formation in Escherichia coli plasmids. II. Deletions mediated by short direct repeats. Mol Gen Genet. 1991;228:209–214. doi: 10.1007/BF00282467. [DOI] [PubMed] [Google Scholar]
- 93.Chedin F, Dervyn E, Dervyn R, Ehrlich SD, Noirot P. Frequency of deletion formation decreases exponentially with distance between short direct repeats. Mol Microbiol. 1994;12:561–569. doi: 10.1111/j.1365-2958.1994.tb01042.x. [DOI] [PubMed] [Google Scholar]
- 94.Lovett ST, Gluckman TJ, Simon PJ, Sutera VA, Jr, Drapkin PT. Recombination between repeats in Escherichia coli by a recA-independent, proximity-sensitive mechanism. Mol Gen Genet. 1994;254:294–300. doi: 10.1007/BF00290109. [DOI] [PubMed] [Google Scholar]
- 95.Bierne H, Vilette D, Ehrlich SD, Michel B. Isolation of a dnaE mutation which enhances RecA-independent homologous recombination in the Escherichia coli chromosome. Mol Microbiol. 1997;24:1225–1234. doi: 10.1046/j.1365-2958.1997.4381795.x. [DOI] [PubMed] [Google Scholar]
- 96.Saveson CJ, Lovett ST. Enhanced deletion formation by aberrant DNA replication in Escherichia coli. Genetics. 1997;146:457–470. doi: 10.1093/genetics/146.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bzymek M, Lovett ST. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci U S A. 2001;98:8319–8325. doi: 10.1073/pnas.111008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lovett ST, Feshenko VV. Stabilization of diverged tandem repeats by mismatch repair: evidence for deletion formation via a misaligned replication intermediate. Proc Natl Acad Sci USA. 1996;93:7120–7124. doi: 10.1073/pnas.93.14.7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Slack A, Thornton PC, Magner DB, Rosenberg SM, Hastings PJ. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genetics. 2006;2:e48. doi: 10.1371/journal.pgen.0020048. [DOI] [PMC free article] [PubMed] [Google Scholar]; Experimental evidence from Escherichia coli suggesting that chromosomal structural change occurs by a replicative mechanism, and also revealing that the characteristics of amplification in E, coli are similar to those of non-recurrent changes in human genomic disease.
- 101.Kugelberg E, Kofoid E, Reams AB, Andersson DI, Roth JR. Multiple pathways of selected gene amplification during adaptive mutation. Proc Natl Acad Sci U S A. 2006;103:17319–17324. doi: 10.1073/pnas.0608309103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Allgood ND, Silhavy TJ. Escherichia coli xonA (sbcB) mutants enhance illegitimate recombination. Genetics. 1991;127:671–680. doi: 10.1093/genetics/127.4.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bzymek M, Saveson CJ, Feschenko VV, Lovett ST. Slipped misalignment mechanisms of deletion formation: in vivo susceptibility to nucleases. J Bacteriol. 1999;181:477–482. doi: 10.1128/jb.181.2.477-482.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 105.VanHulle K, et al. Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol Cell Biol. 2007;27:2601–2614. doi: 10.1128/MCB.01740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Davis AP, Symington LS. RAD51-dependent break-induced replication in yeast. Mol Cell Biol. 2004;24:2344–2351. doi: 10.1128/MCB.24.6.2344-2351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McVey M, Adams M, Staeva-Vieira E, Sekelsky JJ. Evidence for multiple cycles of strand invasion during repair of double-strand gaps in Drosophila. Genetics. 2004;167:699–705. doi: 10.1534/genetics.103.025411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bindra RS, Crosby ME, Glazer PM. Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 2007;26:249–260. doi: 10.1007/s10555-007-9061-3. [DOI] [PubMed] [Google Scholar]
- 109.Huang LE, Bindra RS, Glazer PM, Harris AL. Hypoxia-induced genetic instability--a calculated mechanism underlying tumor progression. J Mol Med. 2007;85:139–148. doi: 10.1007/s00109-006-0133-6. [DOI] [PubMed] [Google Scholar]
- 110.Bindra RS, Glazer PM. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene. 2007;26:2048–2057. doi: 10.1038/sj.onc.1210001. [DOI] [PubMed] [Google Scholar]
- 111.Coquelle A, Toledo F, Stern S, Bieth A, Debatisse M. A new role for hypoxia in tumor progression: induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol Cell. 1998;2:259–265. doi: 10.1016/s1097-2765(00)80137-9. [DOI] [PubMed] [Google Scholar]
- 112.Hastings PJ, Bull HJ, Klump JR, Rosenberg SM. Adaptive amplification: an inducible chromosomal instability mechanism. Cell. 2000;103:723–731. doi: 10.1016/s0092-8674(00)00176-8. [DOI] [PubMed] [Google Scholar]
- 113.Lombardo MJ, Aponyi I, Rosenberg SM. General stress response regulator RpoS in adaptive mutation and amplification in Escherichia coli. Genetics. 2004;166:669–680. doi: 10.1534/genetics.166.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ponder RG, Fonville NC, Rosenberg SM. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell. 2005;19:791–804. doi: 10.1016/j.molcel.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 115.Mullighan CG, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–764. doi: 10.1038/nature05690. [DOI] [PubMed] [Google Scholar]
- 116.Shao L, et al. Identification of chromosome abnormalities in subtelomeric regions by microarray analysis: a study of 5,380 cases. Am J Med Genet A. 2008;146A:2242–2251. doi: 10.1002/ajmg.a.32399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yatsenko SA, et al. Molecular mechanisms for subtelomeric rearrangements associated with the 9q34.3 microdeletion syndrome. Hum Mol Genet. 2009 doi: 10.1093/hmg/ddp114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang L, Lu HH, Chung WY, Yang J, Li WH. Patterns of segmental duplication in the human genome. Mol Biol Evol. 2005;22:135–141. doi: 10.1093/molbev/msh262. [DOI] [PubMed] [Google Scholar]
- 119.She X, et al. The structure and evolution of centromeric transition regions within the human genome. Nature. 2004;430:857–864. doi: 10.1038/nature02806. [DOI] [PubMed] [Google Scholar]
- 120.Nguyen DQ, Webber C, Ponting CP. Bias of selection on human copy-number variants. PLoS Genet. 2006;2:e20. doi: 10.1371/journal.pgen.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Visser R, et al. Identification of a 3.0-kb major recombination hotspot in patients with Sotos syndrome who carry a common 1.9-Mb microdeletion. Am J Hum Genet. 2005;76:52–67. doi: 10.1086/426950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.de Smith AJ, et al. Small deletion variants have stable breakpoints commonly associated with alu elements. PLoS ONE. 2008;3:e3104. doi: 10.1371/journal.pone.0003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bacolla A, et al. Breakpoints of gross deletions coincide with non-B DNA conformations. Proc Natl Acad Sci U S A. 2004;101:14162–14167. doi: 10.1073/pnas.0405974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bacolla A, Wojciechowska M, Kosmider B, Larson JE, Wells RD. The involvement of non-B DNA structures in gross chromosomal rearrangements. DNA Repair (Amst) 2006;5:1161–1170. doi: 10.1016/j.dnarep.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 125.Inagaki H, et al. Chromosomal instability mediated by non-B DNA: Cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res. 2009;19:191–8. doi: 10.1101/gr.079244.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Myers S, Freeman C, Auton A, Donnelly P, McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat Genet. 2008;40:1124–9. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 127.Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet. 2004;13(Spec No 1):R57–64. doi: 10.1093/hmg/ddh073. [DOI] [PubMed] [Google Scholar]
- 128.Bi W, et al. Increased LIS1 expression affects human and mouse brain development. Nat Genet. 2009;41:168–77. doi: 10.1038/ng.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Galhardo RS, Hastings PJ, Rosenberg SM. Mutation as a stress response and the regulation of evolvability. Crit Rev Biochem Mol Biol. 2007;42:399–435. doi: 10.1080/10409230701648502. [DOI] [PMC free article] [PubMed] [Google Scholar]; A broad review of stress-induced mutation and its meaning for evolution.
- 130.Rosenberg SM. Evolving responsively: adaptive mutation. Nature Rev Genet. 2001;2:504–515. doi: 10.1038/35080556. [DOI] [PubMed] [Google Scholar]
- 131.Cirz RT, et al. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005;3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Riesenfeld C, Everett M, Piddock LJ, Hall BG. Adaptive mutations produce resistance to ciprofloxacin. Antimicrob Agents Chemother. 1997;41:2059–60. doi: 10.1128/aac.41.9.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bindra RSS, et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol Cell Biol. 2004;24:8504–8518. doi: 10.1128/MCB.24.19.8504-8518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that homologous recombination enzymes are down-regulated by stress in human cells, and that this is accompanied by a reduction in HR.
- 134.Mihaylova VT, et al. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol Cell Biol. 2003;23:3265–3273. doi: 10.1128/MCB.23.9.3265-3273.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kolodner RD, et al. Germ-line msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–5074. [PubMed] [Google Scholar]
- 136.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–3079. [PubMed] [Google Scholar]
- 137.Modrich P. Mismatch repair, genetic stability and tumour avoidance. Phil Trans R Soc London B. 1995;347:89–95. doi: 10.1098/rstb.1995.0014. [DOI] [PubMed] [Google Scholar]
- 138.Nguyen DQ, et al. Reduced purifying selection prevails over positive selection in human copy number variant evolution. Genome Res. 2008;18:1711–1723. doi: 10.1101/gr.077289.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Perry GH, et al. Diet and the evolution of human amylase gene copy number variation. Nat Genet. 2007;39:1256–1260. doi: 10.1038/ng2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gonzalez E, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–1440. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 141.Higgs DR, et al. A review of the molecular genetics of the human alpha-globin gene cluster. Blood. 1989;73:1081–1104. [PubMed] [Google Scholar]
- 142.Nozawa M, Kawahara Y, Nei M. Genomic drift and copy number variation of sensory receptor genes in humans. Proc Natl Acad Sci U S A. 2007;104:20421–20426. doi: 10.1073/pnas.0709956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jakobsson M, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. doi: 10.1038/nature06742. [DOI] [PubMed] [Google Scholar]
- 144.Locke DP, et al. Linkage disequilibrium and heritability of copy-number polymorphisms within duplicated regions of the human genome. Am J Hum Genet. 2006;79:275–290. doi: 10.1086/505653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sharp AJ, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fortna A, et al. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2004;2:E207. doi: 10.1371/journal.pbio.0020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.McCarroll SA, et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet. 2008;40:1166–1174. doi: 10.1038/ng.238. [DOI] [PubMed] [Google Scholar]
- 148.Turner DJ, et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lam KW, Jeffreys AJ. Processes of copy-number change in human DNA: the dynamics of {alpha}-globin gene deletion. Proc Natl Acad Sci U S A. 2006;103:8921–8927. doi: 10.1073/pnas.0602690103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lam KW, Jeffreys AJ. Processes of de novo duplication of human alpha-globin genes. Proc Natl Acad Sci U S A. 2007;104:10950–10955. doi: 10.1073/pnas.0703856104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Flores M, et al. Recurrent DNA inversion rearrangements in the human genome. Proc Natl Acad Sci U S A. 2007;104:6099–6106. doi: 10.1073/pnas.0701631104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liang Q, Conte N, Skarnes WC, Bradley A. Extensive genomic copy number variation in embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105:17453–17456. doi: 10.1073/pnas.0805638105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lupski JR. Genomic rearrangements and sporadic disease. Nat Genet. 2007;39:S43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 154.van Ommen GJ. Frequency of new copy number variation in humans. Nature Genet. 2005;37:333–334. doi: 10.1038/ng0405-333. [DOI] [PubMed] [Google Scholar]
- 155.Tuzun E, Bailey JA, Eichler EE. Recent segmental duplications in the working draft assembly of the brown Norway rat. Genome Res. 2004;14:493–506. doi: 10.1101/gr.1907504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Graubert TA, et al. A high-resolution map of segmental DNA copy number variation in the mouse genome. PLoS Genetics. 2007;3:e3. doi: 10.1371/journal.pgen.0030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lovett ST. Encoded errors: mutations and rearrangements mediated by misalignment at repetitive DNA sequences. Mol Microbiol. 2004;52:1243–1253. doi: 10.1111/j.1365-2958.2004.04076.x. [DOI] [PubMed] [Google Scholar]