

Abstract
Endothelin-1 (ET-1) is an important contributor to ventricular hypertrophy and failure, which are associated with arrhythmogenesis and sudden death. To elucidate the mechanism(s) underlying the arrhythmogenic effects of ET-1 we tested the hypothesis that long-term (24 hrs) exposure to ET-1 impairs impulse conduction in cultures of neonatal rat ventricular myocytes (NRVM). NRVM were seeded on micro-electrode-arrays (MEAs, Multi Channel Systems, Reutlingen, Germany) and exposed to 50 nM ET-1 for 24 hrs. Hypertrophy was assessed by morphological and molecular methods. Consecutive recordings of paced activation times from the same cultures were conducted at baseline and after 3, 6 and 24 hrs, and activation maps for each time period constructed. Gap junctional Cx43 expression was assessed using Western blot and confocal microscopy of immunofluorescence staining using anti-Cx43 antibodies. ET-1 caused hypertrophy as indicated by a 70% increase in mRNA for atrial natriuretic peptide (P < 0.05), and increased cell areas (P < 0.05) compared to control. ET-1 also caused a time-dependent decrease in conduction velocity that was evident after 3 hrs of exposure to ET-1, and was augmented at 24 hrs, compared to controls (P < 0.01). ET-1 increased total Cx43 protein by ∼40% (P < 0.05) without affecting non- phosphorylated Cx43 (NP-Cx43) protein expression. Quantitative confocal microscopy showed a ∼30% decrease in the Cx43 immunofluorescence per field in the ET-1 group (P < 0.05) and a reduced field stain intensity (P < 0.05), compared to controls. ET-1-induced hypertrophy was accompanied by reduction in conduction velocity and gap junctional remodelling. The reduction in conduction velocity may play a role in ET-1 induced susceptibility to arrhythmogenesis.
Keywords: Endothelin-1, connexin 43, conduction velocity, hypertrophy, neonatal rat ventricular myocytes
Introduction
Ventricular hypertrophy is associated with an increased incidence of spontaneous ventricular arrhythmias, cardiovascular events and arrhythmia-related deaths [1, 2] as well as greater vulnerability to the arrhythmogenic effects of acute ischaemia [3, 4]. Endothelin-1 (ET-1) is an important mediator of hypertrophy [5, 6] and has an acute pro-arrhythmic effect on its own and in the setting of ischaemia [7–10]. Several mechanisms have been proposed for the detrimental effects of ET-1: (i) ET-1 has direct toxic effects on cardiac myocytes [11, 12]; (ii) ET-1 causes hypertrophy of cardiac myocytes [5, 6] and increases myocardial fibrosis [13, 14]; (iii) ET-1 has a long-term positive inotropic effect in rats with congestive heart failure (CHF), and thus may contribute to the progression of CHF by increasing myocardial energy utilization [15]; (iv) ET-1 administration induces ventricular arrhythmias [16]. Complimenting these observations of the deleterious effects of ET-1, treatment with the ETA receptor antagonist BQ-123 greatly improved survival, ameliorated left ventricular dysfunction and prevented detrimental ventricular remodelling in rats with CHF [17]. Consistent with its hypertrophic effects in vivo, ET-1 causes hypertrophy of neonatal rat ventricular myocytes (NRVM) [6, 18]. Consequently, exposure of cultured ventricular myocytes to ET-1 has been widely used as a model for in vitro myocardial hypertrophy [19, 20].
Despite the established involvement of ET-1 in arrhythmogenesis in a variety of disease states, the mechanisms underlying this phenomenon are not entirely understood. We hypothesized that long-term (24 hrs) exposure to ET-1 impairs impulse conduction in cultures of NRVM concomitant with hypertrophy and gap junctional remodelling, thus forming a substrate for arrhythmogenesis. To test the hypothesis, control and ET-1-treated cultures were plated on micro-electrode-arrays (MEA), and extracellular electrical activity was recorded before and throughout the experimental intervention, such that each NRVM culture served as its own control. In this work we report that the hypertrophy induced by ET-1 was associated with decreased conduction velocity, gap junctional remodelling and alterations in the response of myocytes to electrical pacing.
Methods
The research conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no. 85–23; revised 1996).
Cultures of neonatal rat ventricular myocytes
NRVM cultures were prepared as previously described [21]. In brief, ventricles from 1–2-day-old Sprague-Dawley rats were dissociated enzymatically at room temperature, using the protease RDB (catalogue no. 300–0; IIBR, Israel). The myocytes were collected following 10–12 cycles of 10 min. digestion. The pooled cells were re-suspended in growth medium: Ham's F10 supplemented with 5% foetal calf serum, 5% horse serum, 100 U/ml penicillin, 100 mg/ml streptomycin (Biological Industries, Beit Haemek, Israel) and 1 mM CaCl2 (up to a total concentration of 1.3 mM). To reduce fibroblast content, cell suspensions were pre-plated on culture flasks for 1 hr in the incubator and 5 mg/100 ml bromodeoxyuridine (BrdU; Sigma, St. Louis, MO, USA) were added to the medium. Subsequently myocytes were plated on MEA plates pre-coated with collagen type I (Sigma C-8919) diluted 1:10 in acetic acid, at a density of 104 cell/mm2. The cultures were maintained in a humidified incubator, with 5% CO2+ 95% air at 37°C. Unsettled cells were washed out after 24 hrs and the medium was replaced. The medium was replaced again on alternating days. Cultures were maintained in the incubator 5 days prior to data recordings.
Recording of extracellular electrograms using the micro-electrode-array data acquisition system
We measured microscopic electrical propagation in the cultures by recording unipolar electrograms from NRVM plated on MEAs (Multi Channel Systems, Reutlingen, Germany) as previously described [21, 22]. The MEA is a 50 × 50 mm glass substrate, with 8 × 8 matrix of 60 titanium-nitride, 30 μm diameter electrodes, embedded in its centre, with an inter-electrode distance of 200 μm. For the electrophysiological measurements, MEAs were removed from the incubator, placed in the recording apparatus preheated to 37°C. The recording apparatus was connected to a PC-based data acquisition system. Electrical activity was recorded within 1–3 min. of placement. To ascertain that these measurements were performed within the stable period, in addition to the 1–3 min. time-points, conduction velocity was measured at 8 and 10 min. after removing the cultures from the incubator. As we previously reported [22] in control cultures conduction velocities normalized to the value measured at ∼2 min., were respectively: 1.02 ± 0.01 at 8 min. and 1.03 ± 0.01 at 10 min.
Cultures were paced via a pair of stimulation electrodes located 2 mm from the side of the electrode array at a basic cycle length (BCL) of 500 ms (2 Hz) using a stimulator (STG-series, Multi Channel Systems). The local activation time (LAT) at each electrode was defined as the time of occurrence of the maximal negative slope of the signal. Colour-coded activation maps were constructed by interpolating the LAT values for the areas between the electrodes. Conduction velocity was calculated using the LAT at each electrode and the inter-electrode distances. The value of conduction velocity presented for each measurement was taken as the mean value of local velocities of all 60 electrodes.
Incubating NRVM cultures with ET-1
Twenty-four hours before the actual experiment, the cultures were transferred to serum-free medium, 50%/50% DMEM/F-12 (Biological Industries) containing 2 mM L-glutamine, 0.1 mmol/l BrdU, insulin-trans-ferine-sodium selenite (ITS) media supplement (Sigma) and penicillin. On the day of the experiment, following a baseline recording, the culture medium was replaced with new medium alone (control) or with medium containing ET-1 (50 nM). Consecutive recordings were conducted at 3, 6 and 24 hrs after exposure to ET-1. Cell size and Cx43 expression were determined 24 hrs after exposure to ET-1.
Quantitative microscopy
(i) Measurements of cell surface area. Cell surface area (a common measure of hypertrophy) was determined from microscopic digital images of selected unfixed culture areas. Measuring the same cells precisely within the same areas at baseline and 24 hrs thereafter (in control or ET-1 treated cultures) enabled us to use myocytes as their own control. The area contained within the cell circumference was measured using Scion Image software for Windows based on the NIH Image for Macintosh, and expressed in arbitrary units determined by the number of pixels contained within the cell borders.
(ii) Immunohistochemical staining and analysis.
For immunostaining, cultures plated on glass cover slips were rinsed with phosphate buffered saline (PBS), fixed for 10 min. in 4% paraformalde-hyde in PBS at room temperature and permeabilized on ice with 0.2% Triton X-100 (Sigma) in PBS. The cultures were blocked with normal goat serum 10% (Biological Industries) for 1 hr at 37°C. The primary antibodies used in this study were: mouse monoclonal anti-α-actinin (Sarcomeric) (clone EA-53, Sigma), and mouse monoclonal anti-Cx43 antibody (MAB 3068; Chemicon International, Temecula, CA, USA). The secondary antibodies were FITC-conjugated goat antimouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for α-actinin, and CY5-conjugated donkey antimouse IgG (Chemicon International) for Cx43. After blocking, the preparations were incubated overnight at 4°C with the primary antibody. Subsequently, the cultures were rinsed extensively and incubated with the secondary antibody for 1 hr at room temperature.
Confocal microscopy was performed using a confocal scanning laser microscope (Radiance 2000 confocal, Bio-Rad, Hercules, CA, USA) connected to a Nikon e600 upright microscope and the Image-Pro® Plus version 5 software (MediaCybernetics, Silver Spring, MD, USA). Each recorded image was obtained using multi-channel scanning and consisted of 1024 × 1024 pixels. All cultures were immuno-labelled simultaneously using identical dilutions of primary and secondary antibodies, and scanned under identical scanning parameters. To determine cellular area, the positive α-actinin labelled area was defined as the number of pixels with α-actinin signal intensity exceeding the threshold of 15 on a 0–255 grey intensity scale. The α-actinin stained area was automatically identified by the Image-Pro Software, which measured the occupied stained area and intensity within the microscopic field. For Cx43 analysis, the threshold parameters (used throughout the analysis) were chosen and set in the Image-Pro so that only the densely fluorescent points representing junctional Cx43 immunostaining were selected. The same threshold parameters were used throughout the analysis. After selecting the points in a field, the number of points per field and the total fluorescence intensity (of all selected points in the field) were determined.
Protein expression analysis by Western blot
Western blot analysis of Cx43 protein expression was performed as described previously [22]. Monoclonal anti-Cx43 antibodies recognizing total-Cx43 (Chemicon International) or monoclonal anti-Cx43 antibodies recognizing non-phosphorylated-Cx43 (NP-Cx43) (Zymed Laboratories, San Francisco, CA, USA) were used. Cx43 band intensity was quantified by densitometry and normalized to actin (Chemicon International).
Determination of mRNA levels by reverse transcriptase-polymerase chain reaction (RT-PCR)
Total RNA was extracted from NRVM cultures using the EZ-RNA™ isolation kit (Biological Industries) according to the manufacture's protocol, and the RT reaction was conducted as described previously [23], using specific primers (Table 1). The PCR reaction was carried out under the following conditions. An initial denaturation step at 94°C for 2 min. for all primers, was followed by a final elongation step at 72°C for 10 min. The amplified products were analysed by 2% agarose gel and visualized using ultra violet fluorescence after staining with ethidium bromide. The relative levels of mRNA encoding the above products were quantified by densitometry and normalized to actin mRNA.
1.
PCR primers and protocols
| Gene | Primers | PCR protocol | ||
|---|---|---|---|---|
| ANP | Up: 5′- ATG GGC TCC TTC TCC ATC ACC -3′ | 94°C 30 sec., 58°C 30 sec., 72°C 1 min. | ||
| Down: 5′- GTA CCG GAA GCT GTT GCA GCC -3′ | 35 cycles | |||
Statistical analysis
The results are presented as mean ± S.E.M. Statistical analysis was performed using Sigmastat™ software. One-way repeated measures ANOVA, followed by Tukey post hoc testing, was used to compare effects of graded variables. Two-way repeated measures ANOVA, followed by Tukey post hoc testing, was used to compare a graded variable between two groups. ‘Student's t-test’ was used to compare control versus treatment groups in non-graded variables. P < 0.05 was considered statistically significant.
Results
ET-1 induced hypertrophy of NRVM
NRVM were treated with ET-1 50 nM, a concentration consistent with previous reports employing 10–100 nM ET-1 [18–20, 25]. To validate the hypertrophic response to ET-1, we determined its effects on the cell dimensions and on the mRNA levels of atrial natriuretic peptide (ANP) – a common marker of hypertrophy [23]. Figure 1A depicts representative light microscopy images of NRVM at baseline and 24 hrs later, in the absence and presence of ET-1. Importantly, the same cells were monitored throughout the experiment via a special marking device (‘Object Marker’, Nikon, MBW10010, circular diameter 1.8 mm, Nikon, Tokyo, Japan) interchangeable with an objective lens. The device contains an ink stamp pad that when comes in contact with the glass slide it marks a 1.8-mm-diameter circle around the focus point.
1.
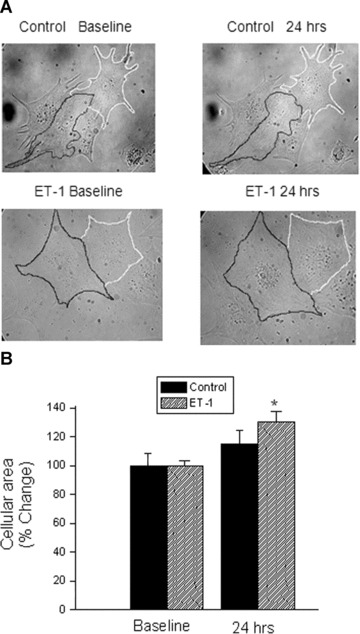
Treatment of NRVM with ET-1 for 24 hrs causes hypertrophy. (A) Representative myocytes photographed at baseline (left) and at 24 hrs (right), from control (top panel) and ET-1 treated (bottom panel) cultures. To illustrate the changes in myocyte size, the perimeters of the cells were traced manually. (B) Morphometric analysis of the same myocytes followed throughout a 24 hrs period, using light microscopy. The figure shows a summary of the changes in myocytes size. The results are expressed as percent change from control. Control, n= 16 cell groups; ET-1, n= 24 cell groups, *P < 0.05 versus ET-1 cultures.
As seen in the representative pictures in which two myocytes in each group were traced manually (Fig. 1A) and by the summary of these experiments (Fig. 1B), after 24 hrs the increase in size in the ET-1 group was twice that observed in the control group (30%versus 15%, P < 0.01). Next, we assessed the hypertrophy by measuring the stained intensity of sarcomeric α-actinin. Figure 2A shows examples of fluorescent immunostaining with anti-α-actinin in control and in ET-1 treated (24 hrs) cultures. As shown in Fig. 2B, the fluorescence intensity of the α-actinin area was significantly larger in the ET-1 group than in the control group (ET-1 group n = 8 cultures; control group n = 9 cultures, P < 0.05). A complementary (non-morphological) measure of hypertrophy was ANP mRNA levels (Fig. 2C). As depicted by the representative gel and the quantitative densitometry analysis, at 24 hrs ET-1 increased ANP mRNA compared to the control group (P < 0.05), supporting the hypertrophic effect of ET-1.
2.
Treatment of NRVM with ET-1 for 24 hrs causes hypertrophy: increase in cellular area and ANP mRNA levels. (A) Immunofluorescence staining for α-actinin from control (left panel) and ET-1 (right panel) cultures. (B) Summary of field α-actinin staining intensity. Control, n= 9 fields; ET-1, n= 8 fields, P < 0.05 versus Control. (C) mRNA levels of ANP. The upper panel depicts representative gels of ANP mRNA (top) and actin (bottom). The lower panel shows quantitative densitometric analysis of control and ET-1 cultures (24 hrs). Each value was divided by its corresponding actin value, and normalized to control. Control, n= 3 cultures; ET-1, n= 3 cultures. *P < 0.05.
The effects of ET-1 on activation and conduction velocity
On the day of the experiment baseline electrical activity was recorded from NRVM cultures, initially during spontaneous activity and then during pacing at a BCL of 500 ms (0.5 Hz). The cultures were randomly assigned to two treatment groups: control group in which the medium was replaced with the regular culture medium and an ET-1 group with medium containing ET-1 (50 nM). Consecutive recordings were conducted at 3, 6 and 24 hrs after exposure to ET-1. In some cultures we were able to maintain pacing throughout; in others pacing failed to capture and spontaneous activation patterns were recorded. At each time-point, both the spontaneous activity and, when possible, the paced activity initiated by the same pair of pacing electrodes as those used at baseline, were recorded. Activation maps were generated by calculating the LAT at each electrode of the array. The mean conduction velocity within the array area of three consecutive action potentials was calculated using the activation time difference and inter-electrode distances. As depicted in Fig. 3, 90% of the control cultures (11/12) could be paced throughout the 24-hr period, whereas only 17% of the ET-1 group captured.
3.
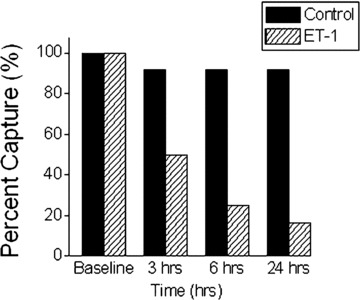
Percent capture of control and ET-1 treated cultures at baseline, 3, 6 and 24 hrs. The histograms depict the percent of cultures that were captured by pacing at both time-points. Control, n= 12 cultures; ET-1, n= 18 cultures.
Cultures that were paced throughout
Figure 4A depicts representative activation maps and their corresponding conduction velocities generated from two experiments from control and ET-1-treated cultures, both paced at a BCL of 500 ms. Whereas over the 24-hr period the control conduction velocity was only slightly reduced (from 19.7 to 18.1 cm/sec), in the ET-1 culture conduction velocity decreased from 21.5 to 13.5 cm/sec, as demonstrated by the denser isochrones in the activation map. As shown by the summary of these experiments (Fig. 4B), at 24 hrs conduction velocity in the control group decreased by ∼15% compared to baseline, while the reduction in the ET-1 group was more pronounced (∼40%, P <0.05), and was evident within 3 hrs (P < 0.05).
4.
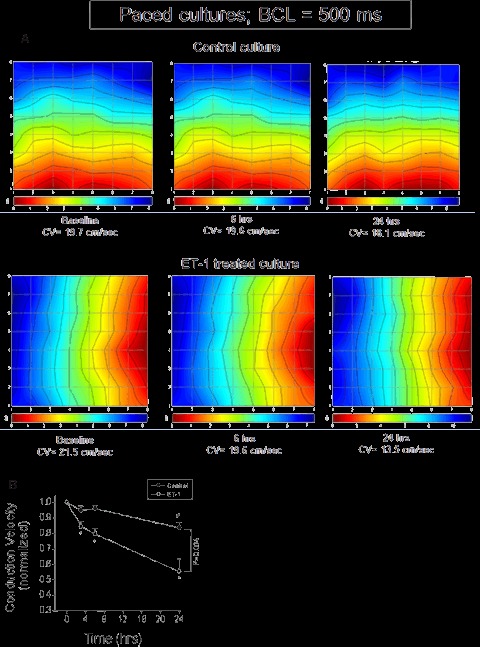
Effects of ET-1 on activation and conduction velocity in paced NRVM cultures, determined by the micro-electrode-array (MEA) data acquisition system. (A) Representative activation maps serving as a visual representation of the activation sequence, recorded at baseline, 6 and 24 hrs in paced Control and ET-1-treated cultures. The intersections of the straight lines mark the location of the recording electrodes. The map activation time (the time duration between first and last activations) is represented by the lower scale at the bottom of the map. The colour strip below the map represents the colour spectrum and its scaling according to time. Colour-coding: red – early; blue -late. Isochronal lines are overlaid on the maps and are spaced 1 ms apart. Notice the changes in the inter-isochronal spacing as an indication of conduction velocity changes. The calculated conduction velocities are shown below the activation maps. (B) The effect of ET-1 on conduction velocity. Mean conduction velocities in Control and in ET-1 exposed cultures paced at a BCL of 500 ms. The values shown are normalized to baseline conduction velocity which was set as 1. Control, n= 15 cultures; ET-1, n= 17 cultures. P= 0.004 for the trend differences between Control and ET-1. *P < 0.05 compared to baseline in the Control group. #P < 0.05 compared to baseline in the ET-1 group.
Cultures that could not be paced throughout
A representative control experiment in which pacing could not be maintained throughout the study period is shown in Figure 5A. In this experiment in a culture permitted to beat spontaneously for 24 hrs, conduction velocity remained unchanged throughout the 24-hr recording session. In contrast, in the presence of ET-1, conduction slowed markedly (Fig. 5B). Because pacing had failed at 24 hrs in 84% of cultures exposed to ET-1 (Fig. 3), we used a previously described method [22] to determine the rate-dependency relationships of conduction velocity in these cultures. To generate these relationships which enabled us to compare conduction velocities of pacing-unresponsive cultures beating at different rates, a subgroup of 23 spontaneously beating cultures were paced at different BCLs (1000 to 250 ms) and activation maps constructed (Fig. 6A). The rate-dependencies of the activation properties are depicted in Figs. 6A-C, illustrating that as BCL was increased, activation became faster. This is evident by: (1) the map activation times (Fig. 6A). At 250, 400 and 1000 ms, the activation times were approximately 10, 9 and 8 ms, respectively. While the activation time decreased, the activation path (i.e. the electrode activation sequence) remained unchanged; (2) the time delays between the stimulus artefact and the activation spikes of the same electrode became shorter as BCL was increased (Fig. 6B). Figure 6C summarizes the relationships between conduction velocity and BCL; as BCL increased from 250 to 1000 ms, conduction velocity increased by ∼12% (n= 23, P < 0.001). The curve was fitted (R2= 0.99) by a single exponential:
5.
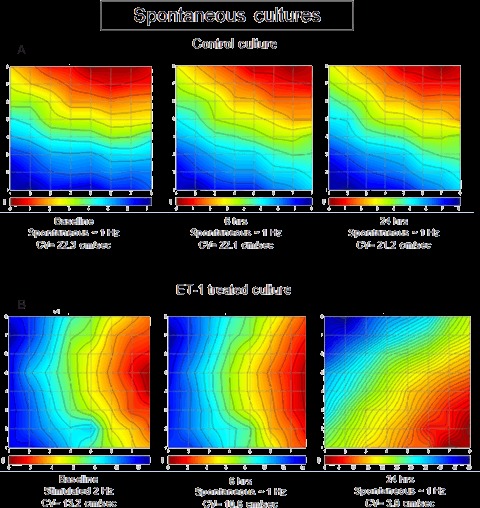
Effects of ET-1 on activation and conduction velocity in NRVM cultures that could not be paced. Representative activation maps serving as a visual representation of the activation sequence, recorded at baseline, 6 and 24 hrs in spontaneously beating Control (A) and ET-1 treated (B) cultures. The intersections of the straight lines mark the location of the recording electrodes. The map activation time (the time duration between first and last activations) is represented by the lower scale at the bottom of the map. The colour strip below the map represents the colour spectrum and its scaling according to time. Colour coding: red – early; blue – late. Isochronal lines are overlaid on the maps and are spaced 1 ms apart. Notice the changes in the inter-isochronal spacing as an indication of conduction velocity changes. The calculated conduction velocities are shown below the activation maps.
6.
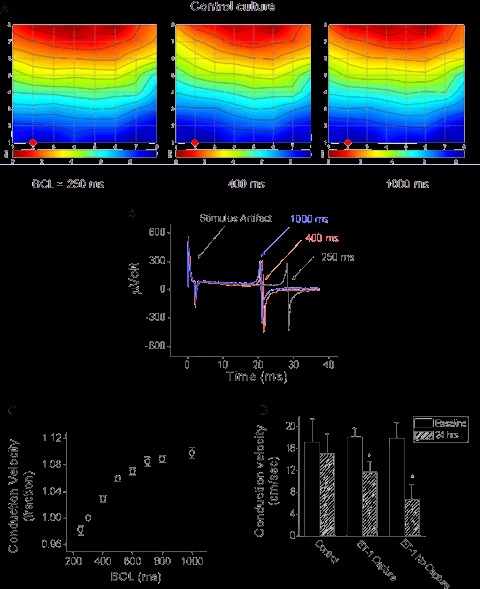
The effects of pacing on conduction velocity in Control, and ET-1-treated cultures. (A) Activation maps from a culture stimulated at BCLs of 250, 400 and 1000 ms. Each activation map represents propagation of one action potential. The map activation time (the time duration between first and last activations) is represented by the lower scale at the bottom of the map. The colour strip below the map represents the colour spectrum and its scaling according to time. Colour coding: red – early; blue – late. (B) Three spikes recorded from electrode #12 (denoted by the red circles in A) at BCL = 1000 ms (blue trace), 400 ms (red trace) and 250 ms (black trace). The spikes are overlaid and synchronized relative to the stimulus artefact occurrence. (C) The relationships between conduction velocity and BCL. The conduction velocity values were normalized to the values at BCL = 300 ms. n= 23 cultures, P < 0.001, one-way ANOVA analysis. (D) Conduction velocity values at baseline and at 24 hrs, from Control (n= 5), ET-1 treated cultures that captured pacing (n= 4) and ET-1 cultures unresponsive to pacing (n= 6). *P < 0.005 for the interaction between treatment versus time. In the non-capturing cultures beating spontaneously, the conduction velocity values were rate-corrected according to the rate correction equation discussed in the text.
Equation 1: The relationships between conduction velocity and BCL.
Next, we used this equation to determine whether the non-capturing ET-1 cultures have different conduction velocities than the ET-1 responsive cultures. Figure 6D depicts the conduction velocities of 3 groups of cultures: control (paced at BCL = 500 ms), ET-1 pacing-responsive (paced at BCL = 500 ms) and ET-1 pacing-unresponsive cultures (rate corrected to BCL = 500 ms). The unresponsive cultures (n= 6) had a significantly (P < 0.001) lower conduction velocity than both the control group (n= 5) and the responsive ET-1 group (n= 4).
Gap junctional remodelling by ET-1
Western blot and immunofluorescence analyses of Cx43
Since gap junctions are important determinants of conduction velocity, we investigated the effects of ET-1 on Cx43 protein levels and expression, using Western blot analysis and quantitative confocal microscopy of Cx43 immunofluorescence staining, respectively. Figure 7A depicts different representative Western blots for total-Cx43 (upper blot), NP-Cx43 (middle blot) and actin (lower blot) in control and ET-1 (24 hrs) treated cultures. Please note that the 41 kD band appears in two different blots; the upper lane generated by the total-Cx43 antibody and the middle lane by the NP-Cx43 antibody. The total-Cx43 antibody recognized two bands (top panel): one major dense band at 44–46 kD that is the summation of the two phosphorylated isoforms, and another band at 41 kD that comprises the NP-Cx43 (middle panel). Figures 7B and C show the summary of densitometry analysis for total Cx43 and NP-Cx43, respectively, illustrating that exposure to ET-1 for 24 hrs increased total Cx43 by 40% (Control, n= 16; ET-1, n= 5, P < 0.05) without affecting the NP-Cx43 level.
7.
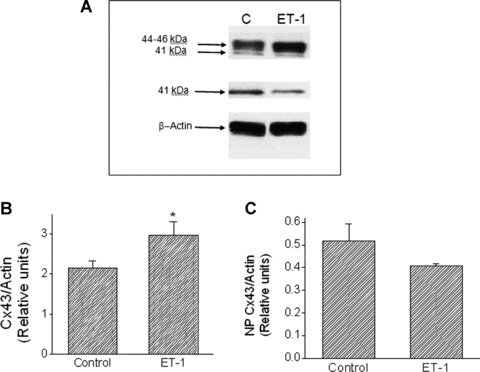
The effect of ET-1 on Cx43 protein expression. (A) Representative Western blots from Control and ET-1 cultures. Culture lysates were probed for total-Cx43 (upper panel) and NP-Cx43 (middle panel). Upper and lower arrows indicate the positions of the 44–46 and 41-kD bands, respectively. (B) Quantitative densitometric analysis of total-Cx43 in Control (n= 16 cultures) and ET-1 (n= 5 cultures). *P < 0.05 versus Control. (C) Quantitative densitometric analysis of NP-Cx43 in control (n= 16 cultures) and ET-1 (n= 5 samples). Equivalency of loading was verified with an antibody against actin (lower panel). Each value was normalized to its corresponding actin value (A, lower panel). Each sample is a pooling of two to three cultures.
Next, we determined the effect of ET-1 on Cx43 immunofluorescence expression as illustrated by the representative confocal microscopy images from control (left) and ET-1 treated (right) cultures (Fig. 8A); Cx43-containing gap junctions are represented by an intense immunofluorescence signal at discrete spots along the cell perimeter. It appears that ET-1 decreased the number of apparent gap junctions on the perimeter. As shown in Fig. 8B, the number of Cx43 gap junctions visualized in the membrane per microscopic field was reduced after 24 hrs of ET-1 (Control, n= 8; ET-1, n= 7, P < 0.05). Further, as depicted in Fig. 8C, the mean Cx43 fluorescence intensity was significantly decreased by ET-1 (Control, n= 8; ET-1; n= 7, P < 0.05).
8.
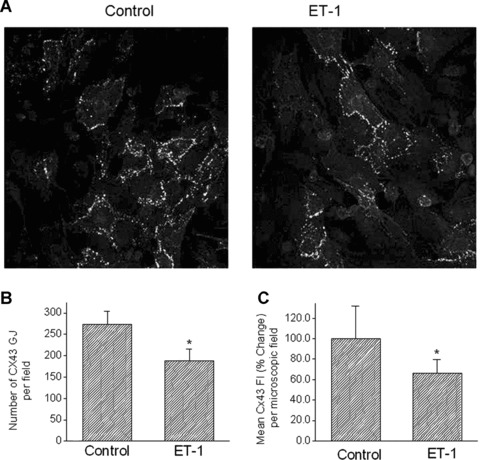
The effect of ET-1 on the morphological arrangement of Cx43 in NRVM cultures: quantitative confocal microscopy analysis of immunostained preparations. (A) Represen tative confocal images of a control culture and ET-1 treated culture. (B) The number of Cx43 gap junctions (GJ) per microscopic field. (C) Mean fluorescence intensity (FI) of Cx43 (expressed as percent change from control) per microscopic field. In (B) and (C): Control, n= 8 fields; ET-1, n= 7 fields, *P < 0.05 compared to control cultures.
Discussion
In this study we tested the hypothesis that long-term (24 hrs) exposure to ET-1 impairs impulse conduction in NRVM cultures concomitant with hypertrophy and gap junctional remodelling, thus forming a substrate for arrhythmogenesis. The motivation for this study is the association of hypertrophy with increased risk for arrhythmias and cardiac mortality. Specifically, we demonstrated for the first time that ET-1-induced hypertrophy is accompanied by decrease in conduction velocity and gap junctional remodelling. The latter is apparent from a reduction in the number of Cx43 gap junctions per field and mean Cx43 gap junction fluorescence intensity. However, these changes were unaccounted for by the quantitative changes in total Cx43 protein expression which was increased by ET-1.
The hypertrophic response to ET-1, gap junctional remodelling and their relationship to changes in conduction velocity
In the first stage of this study, we confirmed that in agreement with previous reports [6, 18] ET-1 causes hypertrophy of NRVM, as demonstrated by increased (at 24 hrs) cell area, α-actinin stain intensity, ANP mRNA level and Cx43 protein expression. To determine the effect of ET-1 on activation and propagation, electrograms were recorded non-invasively throughout the entire experimental protocol, such that each culture served as its own control. As seen in Fig. 4B, exposure to ET-1 for 3–24 hrs decreased conduction velocity (∼40% at 24 hrs) without altering the propagation pathway (see Fig. 2B). The decrease in conduction velocity observed here (both in the pacing-responsive and unresponsive cultures) differs from reports of an increase in conduction velocity following 24 hrs exposure to hypertrophic agents such as dibutyryl cAMP [30] concomitantly with an increase in Cx43 protein content and Cx43 immunofluorescence staining.
Our experiments on gap junctional remodelling showed that exposure to ET-1 was associated with increased total Cx43 protein by ∼40% (and a small statistically insignificant decrease in NP-Cx43), and a ∼30% decrease in the number of Cx43 gap junctions per field and in the mean Cx43 fluorescence intensity. This increase in Cx43 protein expression was in the same direction as, albeit smaller than the effect (130%) reported by Polontchouk et al.[25] in a similar experimental system. The seeming contradiction between the Cx43 protein and immunofluorescence data can be explained as follows: while Western blot results (which showed that the Cx43 protein level was increased by ET-1) were obtained via analysis of protein levels in whole cell lysates, the confocal analysis was based on detecting Cx43 immunofluorescence message at the membrane. This would suggest that even though the overall protein is increased, there is a redistribution of connexins from the membrane to cytosol, which would be concordant with a decrease in conduction velocity. These findings show that the increased total Cx43 protein levels is not associated with increased conduction velocity, a dichotomy which emphasizes the need to measure not only the effect of an intervention on total quantitative protein level, but also on morphological gap junctional remodelling, as was done here.
The relationships between quantitative changes in Cx43 expression and conduction velocity have been studied by means of empirical and theoretical studies. For example, a decrease of ∼30% in conduction velocity in both neonatal and adult heterozygous Cx43+∼ mice expressing ∼50% of the wild type Cx43 level [26–28] was reported, and a similar relationship appears to hold for increased Cx43 expression [29]. Nevertheless, some studies showed no change in conduction velocity in the Cx43 heterozygous hearts [30–32]. As shown by Bursac et al. [33], the level of Cx43 expression in cultured NRVM is only ∼11–20% of Cx43 expression in the neonatal rat heart. The lower basal coupling level of the cultured NRVM used in our study as compared to that in whole heart would be expected to render conduction velocity in the NRVM more sensitive to changes in intercellular coupling.
Finally, an additional mechanism that may have played a role in the changes observed in conduction velocity is the increase in cellular size (hypertrophy). In this regard, Spach et al. [34] showed that the distribution pattern of gap junctions has a relatively small effect on the average cell-to-cell delay of the electrical impulse (as an indirect measure of conduction velocity), while cell size has a major effect. Whereas they showed a positive relationship between cell size and conduction velocity, we found a decrease in conduction velocity while cell size increased. One possible explanation for this seemingly contradiction is the culture settings within the MEA plate; in the present study we used high seeding density (104 cells/mm2) to attain cell confluence on the MEA. Consequently, myocytes in the MEA are smaller and are packed densely, frequently overriding one another. Therefore, in the MEA plates, hypertrophy can only be achieved by an increase in the axis perpendicular to the MEA plane, namely – cell ‘thickness’. However, Spach et al. have described their findings in a two-dimensional model, and the effect of an isolated increase in cell thickness was not determined.
In summary, this study demonstrates that the hypertrophy caused by ET-1 is associated with reduction in conduction velocity and in the responsiveness to electrical pacing. The mechanisms underlying these effects are complex, and appear to be explained, at least in part, by morphological changes in gap junction organization despite the hypertrophy-related increase in Cx43 protein content.
Acknowledgments
This work was supported by the Rappaport Family Institute for Research in the Medical Sciences, the German-Israel Foundation (GIF), the US-Israel Binational Science Foundation (BFS), the Johns Hopkins University – Technion Foundation, and USPHS-NHLBI grants HL-28958 and HL-67101.
References
- 1.Kannel WB. Left ventricular hypertrophy as a risk factor: the Framingham experience. J Hypertens Suppl. 1991;9:S3–8. doi: 10.1097/00004872-199112002-00002. [DOI] [PubMed] [Google Scholar]
- 2.Messerli FH, Soria F. Hypertension, left ventricular hypertrophy, ventricular ectopy, and sudden death. Am J Med. 1992;93:21S–26S. doi: 10.1016/0002-9343(92)90291-i. [DOI] [PubMed] [Google Scholar]
- 3.Bril A, Forest MC, Gout B. Ischemia and reperfusion-induced arrhythmias in rabbits with chronic heart failure. Am J Physiol. 1991;261:301–7. doi: 10.1152/ajpheart.1991.261.2.H301. [DOI] [PubMed] [Google Scholar]
- 4.Winterton SJ, Turner MA, O'Gorman DJ, Flores NA, Sheridan DJ. Hypertrophy causes delayed conduction in human and guinea pig myocardium: accentuation during ischaemic perfusion. Cardiovasc Res. 1994;28:47–54. doi: 10.1093/cvr/28.1.47. [DOI] [PubMed] [Google Scholar]
- 5.Ito H, Hiroe M, Hirata Y, Fujisaki H, Adachi S, Akimoto H, Ohta Y, Marumo F. Endothelin ETA receptor antagonist blocks cardiac hypertrophy provoked by hemodynamic overload. Circulation. 1994;89:2198–203. doi: 10.1161/01.cir.89.5.2198. [DOI] [PubMed] [Google Scholar]
- 6.Shubeita HE, McDonough PM, Harris AN, Knowlton KU, Glembotski CC, Brown JH, Chien KR. Endothelin induction of inositol phospholipid hydrolysis, sarcomere assembly, and cardiac gene expression in ventricular myocytes. A paracrine mechanism for myocardial cell hypertrophy. J Biol Chem. 1990;265:20555–62. [PubMed] [Google Scholar]
- 7.Geller L, Merkely B, Szokodi I, Szabo T, Vecsey T, Juhasz-Nagy A, Toth M, Horkay F. Electrophysiological effects of intrapericardial infusion of endothelin-1. Pacing Clin Electrophysiol. 1998;21:151–6. doi: 10.1111/j.1540-8159.1998.tb01079.x. [DOI] [PubMed] [Google Scholar]
- 8.Merkely B, Geller L, Toth M, Kiss O, Kekesi V, Solti F, Vecsey T, Horkay F, Tenczer J, Juhasz-Nagy A. Mechanism of endothelin-induced malignant ventricular arrhythmias in dogs. J Cardiovasc Pharmacol. 1998;31:S437–S9. doi: 10.1097/00005344-199800001-00125. [DOI] [PubMed] [Google Scholar]
- 9.Salvati P, Chierchia S, Dho L, Ferrario RG, Parenti P, Vicedomini G, Patrono C. Proarrhythmic activity of intracoronary endothelin in dogs: relation to the site of administration and to changes in regional flow. J Cardiovasc Pharmacol. 1991;17:1007–14. doi: 10.1097/00005344-199106000-00022. [DOI] [PubMed] [Google Scholar]
- 10.Yorikane R, Shiga H, Miyake S, Koike H. Evidence for direct arrhythmogenic action of endothelin. Biochem Biophys Res Commun. 1990;173:457–62. doi: 10.1016/s0006-291x(05)81080-0. [DOI] [PubMed] [Google Scholar]
- 11.Prasad MR. Endothelin stimulates degradation of phospholipids in isolated rat hearts. Biochem Biophys Res Commun. 1991;174:952–7. doi: 10.1016/0006-291x(91)91511-a. [DOI] [PubMed] [Google Scholar]
- 12.Stawski G, Olsen UB, Grande P. Cytotoxic effect of endothelin-1 during ‘stimulated’ ischaemia in cultured myocytes. Eur J Pharmacol. 1991;201:123–4. doi: 10.1016/0014-2999(91)90334-m. [DOI] [PubMed] [Google Scholar]
- 13.Fujisaki H, Ito H, Hirata Y, Tanaka M, Hata M, Lin M, Adachi S, Akimoto H, Marumo F, Hiroe M. Natriuretic peptides inhibit angiotensin II-induced proliferation of rat cardiac fibroblasts by blocking endothelin-1 gene expression. J Clin Invest. 1995;96:1059–65. doi: 10.1172/JCI118092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramires FJ, Nunes VL, Fernandes F, Mady C, Ramires JA. Endothelins and myocardial fibrosis. J Card Fail. 2003;9:232–7. doi: 10.1054/jcaf.2003.26. [DOI] [PubMed] [Google Scholar]
- 15.Sakai S, Miyauchi T, Sakurai T, Kasuya Y, Ihara M, Yamaguchi I, Goto K, Sugishita Y. Endogenous endothelin-1 participates in the maintenance of cardiac function in rats with congestive heart failure. Marked increase in endothelin-1 production in the failing heart. Circulation. 1996;93:1214–22. doi: 10.1161/01.cir.93.6.1214. [DOI] [PubMed] [Google Scholar]
- 16.Yorikane R, Koike H. The arrhythmogenic action of endothelin in rats. Jpn J Pharmacol. 1990;53:259–63. doi: 10.1254/jjp.53.259. [DOI] [PubMed] [Google Scholar]
- 17.Sakai S, Miyauchi T, Kobayashi M, Yamaguchi I, Goto K, Sugishita Y. Inhibition of myocardial endothelin pathway improves long-term survival in heart failure. Nature. 1996;384:353–5. doi: 10.1038/384353a0. [DOI] [PubMed] [Google Scholar]
- 18.Ito H, Hirata Y, Hiroe M, Tsujino M, Adachi S, Takamoto T, Nitta M, Taniguchi K, Marumo F. Endothelin-1 induces hypertrophy with enhanced expression of muscle-specific genes in cultured neonatal rat car diomyocytes. Circ Res. 1991;69:209–15. doi: 10.1161/01.res.69.1.209. [DOI] [PubMed] [Google Scholar]
- 19.Choukroun G, Hajjar R, Kyriakis JM, Bonventre JV, Rosenzweig A, Force T. Role of the stress-activated protein kinases in endothelin-induced cardiomyocyte hypertrophy. J Clin Invest. 1998;102:1311–20. doi: 10.1172/JCI3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luodonpaa M, Vuolteenaho O, Eskelinen S, Ruskoaho H. Effects of adrenomedullin on hypertrophic responses induced by angiotensin II, endothelin-1 and phenyle-phrine. Peptides. 2001;22:1859–66. doi: 10.1016/s0196-9781(01)00505-8. [DOI] [PubMed] [Google Scholar]
- 21.Meiry G, Reisner Y, Feld Y, Goldberg S, Rosen M, Ziv N, Binah O. Evolution of action potential propagation and repolarization in cultured neonatal rat ventricular myocytes. J Cardiovasc Electrophysiol. 2001;12:1269–77. doi: 10.1046/j.1540-8167.2001.01269.x. [DOI] [PubMed] [Google Scholar]
- 22.Zeevi-Levin N, Barac YD, Reisner Y, Reiter I, Yaniv G, Meiry G, Abassi Z, Kostin S, Schaper J, Rosen MR, Resnick N, Binah O. Gap junctional remodeling by hypoxia in cultured neonatal rat ventricular myocytes. Cardiovasc Res. 2005;66:64–73. doi: 10.1016/j.cardiores.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 23.Barac YD, Zeevi-Levin N, Yaniv G, Reiter I, Milman F, Shilkrut M, Coleman R, Abassi Z, Binah O. The 1,4,5-inositol trisphosphate pathway is a key component in Fas-mediated hypertrophy in neonatal rat ventricular myocytes. Cardiovasc Res. 2005;68:75–86. doi: 10.1016/j.cardiores.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 24.Darrow BJ, Fast UG, Beyer EC, Saffitz JE. Functional and structural assessment of intercellular communication. Increased conduction velocity and enhanced connexin expression in dibutyryl cAMP-treated cultured cardiac myocytes. Circ Res. 1996;79:174–83. doi: 10.1161/01.res.79.2.174. [DOI] [PubMed] [Google Scholar]
- 25.Polontchouk L, Ebelt B, Jackels M, Dhein S. Chronic effects of endothelin 1 and angiotensin II on gap junctions and intercellular communication in cardiac cells. FASEB J. 2002;16:87–9. doi: 10.1096/fj.01-0381fje. [DOI] [PubMed] [Google Scholar]
- 26.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA, Saffitz JE. Slow ventricular conduction in mice heterozygous for a connexin43 null mutation. J Clin Invest. 1997;99:1991–8. doi: 10.1172/JCI119367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas SA, Schuessler RB, Berul CI, Beardslee MA, Beyer EC, Mendelsohn ME, Saffitz JE. Disparate effects of deficient expression of connexin43 on atrial and ventricular conduction – evidence for chamber-specific molecular determinants of conduction. Circulation. 1998;97:686–91. doi: 10.1161/01.cir.97.7.686. [DOI] [PubMed] [Google Scholar]
- 28.Eloff BC, Lerner DL, Yamada KA, Schuessler RB, Saffitz JE, Rosenbaum DS. High resolution optical mapping reveals conduction slowing in connexin43 deficient mice. Cardiovasc Res. 2001;51:681–90. doi: 10.1016/s0008-6363(01)00341-8. [DOI] [PubMed] [Google Scholar]
- 29.Zhuang J, Yamada KA, Saffitz JE, Kleber AG. Pulsatile stretch remodels cell-to-cell communication in cultured myocytes. Circ Res. 2000;87:316–22. doi: 10.1161/01.res.87.4.316. [DOI] [PubMed] [Google Scholar]
- 30.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361–75. doi: 10.1111/j.1540-8167.1999.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 31.Thomas SP, Kucera JP, Bircher-Lehmann L, Rudy Y, Saffitz JE, Kleber AG. Impulse propagation in synthetic strands of neonatal cardiac myocytes with genetically reduced levels of connexin43. Circ Res. 2003;92:1209–16. doi: 10.1161/01.RES.0000074916.41221.EA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaidya D, Tamaddon HS, Lo CW, Taffet SM, Delmar M, Morley GE, Jalife J. Null mutation of connexin43 causes slow propagation of ventricular activation in the late stages of mouse embryonic development. Circ Res. 2001;88:1196–202. doi: 10.1161/hh1101.091107. [DOI] [PubMed] [Google Scholar]
- 33.Bursac N, Papadaki M, White JA, Eisenberg SR, Vunjak-Novakovic G, Freed LE. Cultivation in rotating bioreactors promotes maintenance of cardiac myocyte electrophysiology and molecular properties. Tissue Eng. 2003;9:1243–53. doi: 10.1089/10763270360728152. [DOI] [PubMed] [Google Scholar]
- 34.Spach MS, Heidlage JF, Dolber PC, Barr RC. Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth. Circ Res. 2000;86:302–11. doi: 10.1161/01.res.86.3.302. [DOI] [PubMed] [Google Scholar]