

Abstract
Poly(ADP-ribose)-1 (PARP-1) is a key mediator of cell death in excitotoxicity, ischemia, and oxidative stress. PARP-1 activation leads to cytosolic NAD+ depletion and mitochondrial release of apoptosis-inducing factor (AIF), but the causal relationships between these two events have been difficult to resolve. Here, we examined this issue by using extracellular NAD+ to restore neuronal NAD+ levels after PARP-1 activation. Exogenous NAD+ was found to enter neurons through P2X7-gated channels. Restoration of cytosolic NAD+ by this means prevented the glycolytic inhibition, mitochondrial failure, AIF translocation, and neuron death that otherwise results from extensive PARP-1 activation. Bypassing the glycolytic inhibition with the metabolic substrates pyruvate, acetoacetate, or hydroxybutyrate also prevented mitochondrial failure and neuron death. Conversely, depletion of cytosolic NAD+ with NAD+ glycohydrolase produced a block in glycolysis inhibition, mitochondrial depolarization, AIF translocation, and neuron death, independent of PARP-1 activation. These results establish NAD+ depletion as a causal event in PARP-1-mediated cell death and place NAD+ depletion and glycolytic failure upstream of mitochondrial AIF release.
Introduction
The nuclear enzyme poly(ADP-ribose) polymerase-1 (PARP-1) is activated by DNA damage. PARP-1 cleaves the ADP-ribose moiety from NAD+ to generate poly(ADP-ribosyl)ation of specific nuclear acceptor proteins, including histones, DNA polymerases, and PARP-1 itself. This process is thought to facilitate DNA repair and prevent chromatid exchange (Schreiber et al., 2006); however, excessive PARP-1 activation can lead to cell death. PARP-1 has been shown to play a dominant role in neuronal death after excitotoxicity, ischemia–reperfusion, oxidative stress, and other conditions (Eliasson et al., 1997; Virág and Szabó, 2002). PARP family members other than PARP-1 may also be activated by DNA damage, but they do not significantly contribute to cell death in these settings (Schreiber et al., 2006).
How PARP-1 activation leads to neuronal death remains controversial. It was suggested >20 years ago that depletion of cytosolic NAD+ by PARP-1 causes cell death by energy failure (Berger, 1985). This energy failure could result from consumption of ATP for NAD+ resynthesis (Zhang et al., 1994) or from impairment of the NAD+-dependent steps of glycolysis (Ying et al., 2003; Zong et al., 2004). More recently, translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus was identified as a key step in the PARP-1 cell death process (Yu et al., 2002). AIF release can be triggered by the action of poly(ADP-ribose) (PAR) directly on mitochondria, suggesting that PARP-1-induced AIF release may be independent of NAD+ depletion (Andrabi et al., 2006; Yu et al., 2006). However, a causal link between NAD+ depletion and AIF release is suggested by studies showing that, in astrocytes, restoring NAD+ levels blocks PARP-1-induced mitochondrial depolarization, AIF release, and cell death (Alano et al., 2004). Thus, the question remains as to whether NAD+ depletion is a causative event or merely an epiphenomenon in PARP-1-mediated neuronal death.
A definitive way to address this issue is to manipulate intracellular NAD+ concentrations after PARP-1 activation. The cytosolic and mitochondrial NAD+ pools each account for ∼50% of total NAD+ in cultured neurons, but only the cytosolic pool is directly consumed by nuclear PARP-1 (Alano et al., 2007). Cytosolic NAD+ concentrations can be increased or maintained by the addition of exogenous NAD+ in the medium (Alano et al., 2004), and can be reduced by NAD+ glycohydrolase protein transfection. In this study, we used these approaches to examine the role of NAD+ in PARP-1-mediated neuronal death induced by DNA alkylation, peroxynitrite, chemical ischemia, and NMDA excitotoxicity. Results of these studies indicate that cytosolic NAD+ depletion is both necessary for PARP-1-mediated neuronal death, and sufficient, in the absence of PARP-1 activation, to induce glycolytic inhibition, mitochondrial depolarization, and AIF release.
Materials and Methods
Materials.
3,4-Dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone(DPQ) and N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-(N,N-dimethylamino)acetamide hydrochloride (PJ34) were obtained from Calbiochem. Tetramethylrhodamine methyl ester (TMRM) and 3-morpholinosydnonimine (SIN1) were obtained through Invitrogen. All other reagents were purchased from Sigma-Aldrich except where otherwise noted. The studies were approved by the San Francisco Veterans Affairs Medical Center animal studies committee.
Cell cultures.
Astrocyte monocultures were prepared from cerebral cortices of wild-type or PARP-1−/− mice, as previously described (Alano et al., 2004, 2007), and used when confluent, at 19–24 d in vitro. Cortical neuron monocultures were prepared from fetal (embryonic day 15) mice as previously described using wild-type and PARP-1−/− mice (Alano et al., 2004, 2007). Proliferation of non-neuronal cells was inhibited by the addition of 10 μm cytosine arabinoside 24 h after plating. After 24 h of cytosine arabinoside exposure, the plating medium was replaced with culture media consisting of the following: Neurobasal medium (Invitrogen) supplemented with B-27 supplement and 0.2 mm Glutamax. The medium was subsequently exchanged 50% with fresh culture medium every 3–4 d. Experiments were conducted when the neurons were 10–14 d in vitro. Immunostaining of the neuron monocultures with the astrocyte marker GFAP (glial fibrillary acidic protein) revealed <3% of the cells to be contaminating astrocytes after 12 d in vitro. Mixed neuron–astrocyte cultures were prepared by plating cortical neurons onto a preexisting confluent astrocyte layer. These cultures were subsequently maintained as described for the neuron monocultures. Mouse embryonic fibroblasts were prepared from 13.5-d-pregnant mice using a modified protocol (Nilausen and Green, 1965; Bryja et al., 2006). The cells were cultured in DMEM (Invitrogen) containing 10% FBS and 2 mm glutamine. Cells were cultured in 12-well (seeding density, 1.0 × 105 cells/well) or 24-well (seeding density, 5.0 × 104 cells/well) tissue culture plates and used at 90% confluence. These cultures were used for experiments within three passages.
Experimental procedures.
Experiments were initiated by placing the cultures in an artificial CSF (aCSF) composed of the following (in mm): 3.1 KCl, 134 NaCl, 1.2 CaCl2, 1.2 MgSO4, 0.25 KH2PO4, 15.7 NaHCO3, and 2 glucose (Alano et al., 2006). The pH was adjusted to 7.2 while the solution was equilibrated with 5% CO2 at 37°C. Osmolarity was verified at 280–310 mOsm with a vapor pressure osmometer. Concentrated drug stocks were prepared in aCSF and likewise adjusted to pH 7.2 and 290–310 mOsm. Excitotoxicity was induced by incubating the neurons with NMDA (100 μm) in aCSF. Chemical ischemia (CI) was induced by incubating the cultures in glucose-free aCSF containing 0.5 mm 2-deoxyglucose and 5 mm sodium azide in a 37°C, 5% CO2 incubator (Swanson and Benington, 1996; Garnier et al., 2003). Drug exposures were terminated by washing with aCSF.
Neuronal death.
Neuronal death was assessed by propidium iodide (PI) exclusion (Alano et al., 2006) or by calcein retention and propidium iodide exclusion as previously described (Alano et al., 2007). Live and dead neurons were counted, in a blinded manner, in four to six randomly picked optical fields within each experimental well. At least 200 neurons were counted in each well. Each experimental group was repeated in duplicate or triplicate wells per plate and averaged to give a single value per experiment.
Radiolabeled NAD+ uptake assay.
Pure neuronal cultures were washed twice with aCSF, and 0.01 μCi of 14C-labeled NAD+ (302 Ci/mol; GE Healthcare) was added into each well. The NAD+ is 14C-labeled at the adenine moiety. The cells were incubated at 37°C for 0–60 min, and then washed twice in aCSF and lysed in 0.01N NaOH/0.1% SDS. 14C was measured in the lysates by scintillation counting. To control for the possibility that 14C-labeled NAD+ binding to outer cell membranes could contribute to the NAD+ uptake measurements, control wells were exposed to 0.01 μCi of 14C-labeled NAD+ for only 30 s and otherwise treated identically.
Metabolite assays.
Metabolite samples from neuron cultures were extracted in perchloric acid (0.5N) and subsequently deproteinized and pH neutralized with potassium hydrogen phosphate. Dihydroacetone phosphate (DHAP), glyceraldehyde-3-phosphate (GAP), and fructose bisphosphate (FBP) were measured by enzyme-linked fluorometric methods (Lowry and Passonneau, 1972; Michal, 1988). DHAP was converted to glycerol-3-phosphate by glycerol-3-phosphate dehydrogenase, and the coupled oxidation of NADH to NAD+ was measured fluorometrically. GAP was subsequently converted to DHAP by TIM (triosephosphate isomerase), with the coupled oxidation of NADH to NAD+ measured fluorometrically. FBP was assayed by its conversion to DHAP plus GAP by aldolase, and these metabolites measured as above. Pyruvate was assayed by its conversion to lactate by LDH (lactate dehydrogenase), and the coupled production of NADH was followed colorimetrically (Lamprecht and Heinz, 1988). Standards of known concentrations (0–500 μm) were run in the same plates. Proteins were determined by bicinchoninic acid (BCA) method for standardization.
NAD+ and nicotinamide measurements.
NAD+ was measured in neuronal monocultures and isolated mitochondria as previously described (Alano et al., 2006, 2007) using the recycling assay described by Szabó et al. (1996). Samples were extracted in perchloric acid. Nicotinamide (NAM) was measured by HPLC as described previously (Gillmor et al., 1999), with minor modifications. Samples were injected using a Waters 717 plus Autosampler. HPLC was performed with a multiwavelength detector (Waters) using a Microsorb column (type C18 4.6 × 100 mm column; particle size, 3 μm; pore size, 100 Å; Rainin Instruments) using Waters 600E Multisolvent Delivery System at a flow rate of 1 ml/min. Both the NAD+ and nicotinamide measurements were calibrated against internal standards added to lysates before the deproteination step and normalized to protein content of the samples as determined by the BCA method.
ATP and AMP measurements.
ATP and AMP were assayed from perchloric acid extracts. Separations were performed on a Supelcosil LC-18 HPLC column (5 μm particle size; length by inner diameter, 25 cm × 4.6 mm; Phenomenex), using a Waters 600E Multisolvent Delivery System at a flow rate of 1 ml/min. ATP and AMP were detected by monitoring absorbance change at 254 nm using a Waters 2996 Photodiode Array Detector. The mobile phase and chromatographic conditions were as described previously (Crescentini and Stocchi, 1984), with minor modifications.
Mitochondrial isolation.
Mitochondrial isolation from cultured mouse neurons was performed as described previously (Alano et al., 2007). Mitochondrial enrichment was quantified by Western blots using an antibody targeted to mitochondrial protein voltage-dependent anion channel (VDAC).
Glycolysis assay.
Glucose utilization was measured as described previously (Ying et al., 2005), using a modification of the Sokoloff 2-deoxyglucose (2-DG) assay (Sokoloff et al., 1977). The medium was exchanged with aCSF containing 5 mm piperazine-1,4-bis(2-ethanesulfonic acid (PIPES) to maintain pH at 7.2 in a room air atmosphere. After a 30 min equilibration period, 2.5 μCi of [14C(U)]2-DG (340 Ci/mol; ARC) was added to each culture well. Incubations were continued for 30 min at 37°C, and then terminated by three washes in ice-cold aCSF. Cells were lysed in 0.01N NaOH/0.5% SDS, and the accumulated 14C was measured with a scintillation counter.
Western blots.
Western blots were prepared as described previously (Alano et al., 2006, 2007). Cell extracts were mixed with a 1:4 volume of SDS-PAGE loading buffer (10% β-mercaptoethanol, 10% glycerol, 4% SDS, 0.01% bromophenol blue, and 62.5 mm Tris-HCl, pH 6.8) and heated to 65°C for 15 min. Samples were loaded on a 10% resolving SDS-polyacrylamide gel and after electrophoresis were transferred to either polyvinylidene difluoride membranes or to nitrocellulose membranes (for PAR). The membranes were placed in blocking buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.1% Tween 20) containing 5% blotting grade nonfat dry milk for 60 min. Incubations with primary antibodies overnight at 4°C: anti-AIF Ab (1:2500; Sigma-Aldrich), anti-PAR (1:1000; Trevigen), anti-β-actin (1:5000; Sigma-Aldrich), or anti-VDAC (1:2000; Sigma-Aldrich), and subsequently exposed to peroxidase-labeled anti-IgG for 60 min. Bound antibody was detected via chemiluminescence using ECL+ WB Detection kit (GE Healthcare). Protein loading and transfer was evaluated by immunostaining for either anti-β-actin Ab (1:10,000) or anti-VDAC Ab (1:5000) and by Ponceau S staining (data not shown). Densitometry analysis was quantified with NIH ImageJ program. Values for each lane were normalized to protein loading. Immunoblots prepared in the absence of primary or secondary antibodies showed no signal (data not shown).
Immunostaining.
Cell fixation and immunostaining for AIF and PAR were performed as described previously (Alano et al., 2004, 2006), using anti-AIF polyclonal antibody (Sigma-Aldrich) at a 1:1500 dilution and anti-PAR polyclonal antibody (Trevigen) at a 1:2000 dilution. Immunostaining was visualized with fluorescence-tagged (Alexa 488) secondary antibody at 1:2500 dilution (Invitrogen). Nuclear counterstaining with propidium iodide was performed after removal of excess secondary antibody. Cell fluorescence was captured using a Zeiss LSM 510 META confocal laser scanning module mounted onto an inverted Axio Observer (Zeiss).
Mitochondrial membrane potential.
Mitochondrial membrane potential was evaluated by TMRM under nonquench conditions, as described previously (Alano et al., 2004). Stock TMRM was prepared with DMSO to a concentration of 100 μm, and this was diluted to 1–2 nm in recording buffer composed of the following (in mm): 3.1 KCl, 134 NaCl, 1.2 CaCl2, 1.2 MgSO4, 0.25 KH2PO4, 10 PIPES, 5.7 NaHCO3, 2 glucose, at pH 7.2. Fluorescence values were collected every minute as a field average for each excitation/emission pair, with 4–10 fields assessed per coverslip. These values were averaged for each “n” and normalized to the values obtained from control coverslips prepared in parallel. The nonquench loading condition (1–2 nm) was established by loading cultures with a range of TMRM concentrations and confirming a fluorescence decrease after incubation with 10 μm carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP), a mitochondrial uncoupler. In addition, FCCP was added at the end of each experimental recording to verify nonquench conditions (i.e., a decrease in TMRM fluorescence). Data were presented as fluorescence (F) over baseline fluorescence (Fo), or expressed as the percentage of fluorescence decrease calculated between the maximal fluorescence (baseline) and the minimal fluorescence (FCCP).
NAD+ glycohydrolase transfection.
NAD+ glycohydrolase from porcine brain (Sigma-Aldrich) was diluted in serum-free aCSF to 1–500 μg/ml and mixed with BioPORTER reagent (Gene Therapy Systems). Neuron cultures were incubated with the BioPORTER–protein complex for 3–4 h at 37°C and immediately used for experiments. Cell transfection rate was estimated using parallel transfections with fluorescein-labeled IgG or β-galactosidase.
Statistical analyses.
Data are presented as means ± SEM. Statistical significance was assessed using ANOVA and the Student–Newman–Keuls post hoc test to compare the indicated experimental groups. The “n” denotes the number of independent experiments, each performed with independently prepared cultures. Values from three to four culture wells were averaged to generate each data point within each experiment.
Results
PARP-1 activation leads to NAD+ depletion and neuronal death
Experiments were performed with cortical neuron cultures in aCSF medium, which contains glucose as the only metabolic substrate. PARP-1 activation was induced with the alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) to induce DNA strand breaks (Ha and Snyder, 1999), or with the peroxynitrite generator SIN1 to mimic peroxynitrite production in ischemic brain (Eliasson et al., 1997, 1999; Alano et al., 2006). Neuronal death resulting from these treatments was blocked in PARP-1−/− neurons and by the PARP inhibitors PJ34 and DPQ (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material), as previously reported (Szabó and Dawson, 1998; Yu et al., 2002; Alano and Swanson, 2006; Alano et al., 2007; Haddad et al., 2008). Cytosolic NAD+ depletion resulting from these treatments was also blocked by PARP inhibitors and in PARP-1−/− neurons (Alano et al., 2007) (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). Thus, both neuronal death and cytosolic NAD+ depletion are attributable to PARP-1 activation under these treatment conditions.
Extracellular NAD+ enters neurons throughP2X7-gated channels
We established a method of manipulating intracellular NAD+ concentrations to determine whether NAD+ depletion contributes to neuronal death during PARP-1 activation. Previous studies provide indirect evidence for uptake of extracellular NAD+ by neurons (Araki et al., 2004; Wang et al., 2005) and direct evidence for NAD+ uptake by other cell types (Bruzzone et al., 2001; Alano et al., 2004; Billington et al., 2008). Here, neurons incubated with [14C]NAD+ showed a time-dependent accumulation of the 14C label (Fig. 1A). This accumulation did not occur in the presence of NAD+ glycohydrolase, which cleaves the [14C]NAD+ to nicotinamide and [14C]ADP-ribose, thus excluding uptake ADP-ribose or other NAD+ metabolites containing only the 14C-labeled adenine moiety. Neurons incubated with millimolar concentrations of NAD+ in the medium exhibited a concentration-dependent increase in intracellular NAD+ content (Fig. 1B). This increase was blocked by brilliant blue and by preincubation with (ox-ATP), indicative of a P2X7-gated channel (Murgia et al., 1993; Jiang et al., 2000). To determine whether exogenous NAD+ can replenish cellular NAD+ after PARP-1 activation, we treated neurons with MNNG (75 μm) for 30 min and subsequently added NAD+ to the medium. The NAD+ depletion induced by MNNG was reversed with the addition of 5 mm NAD+ to the culture medium (Fig. 1C).
Figure 1.
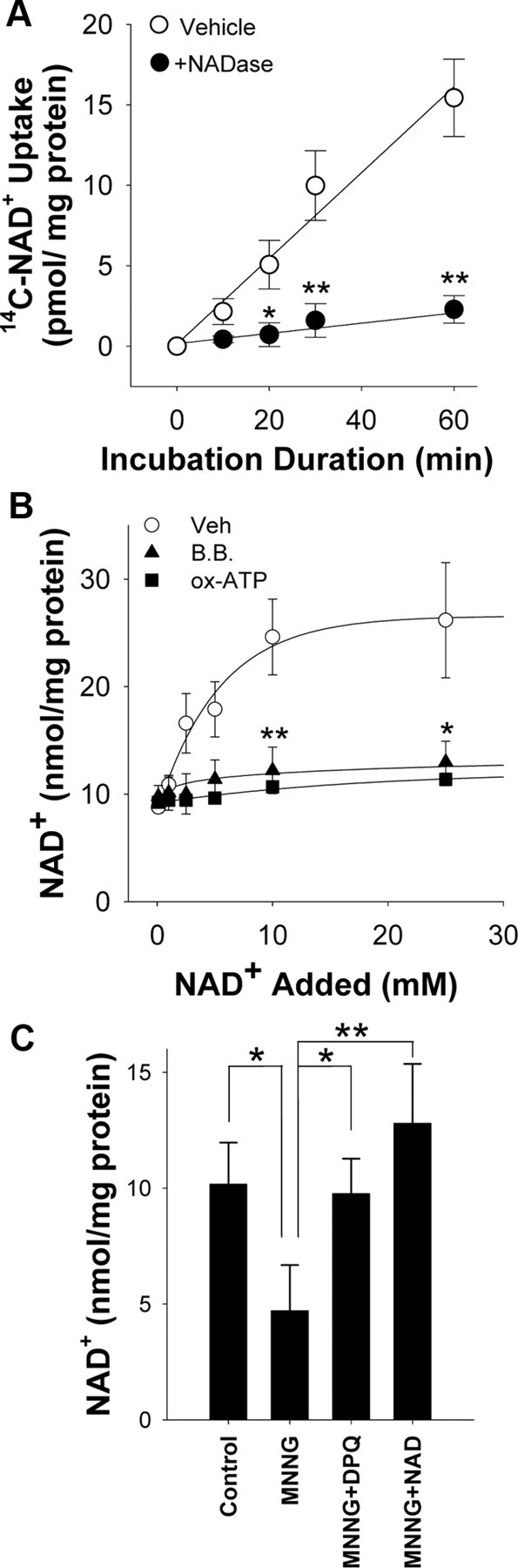
NAD+ enters neurons through P2X7-gated channels. A, Neurons incubated with [14C]NAD+, radiolabeled on the adenine moiety, exhibited a time-dependent accumulation of 14C that was abolished in the presence of 1 U/ml NADase. B, Thirty minute incubations with NAD+ produced a saturable increase in neuronal NAD+ content. The increase was blocked by preincubation with oxidized ATP (ox-ATP) (100 μm) or by coincubation with brilliant blue G (B.B.) (10 μm), both of which block P2X7 receptors. C, Neuronal NAD+ depletion induced by MNNG (75 μm; 30 min) was prevented by coincubation with the PARP inhibitor DPQ (25 μm) and reversed by the addition of 5 mm NAD+ to the medium after washout of the MNNG. NAD+ content was measured 30 min after washout of the MNNG. n = 3–4; *p < 0.01, **p < 0.001. Error bars indicate SEM.
NAD+ repletion prevents PARP-1-mediated neuronal death
We next determined whether reversal of the NAD+ depletion caused by PARP-1 can prevent PARP-1-induced neuronal death. Neurons were exposed to either MNNG or SIN1 for 30 min, these agents were washed out, and NAD+ was then added at varying concentrations. NAD+ reduced neuronal death induced by MNNG or SIN1, with a 75% reduction in cell death observed with 5 mm NAD+ (Fig. 2A,B). An end product of NAD+ metabolism by PARP-1 is nicotinamide, which is itself a weak PARP-1 inhibitor (Rankin et al., 1989). We therefore considered the possibility that the effects of NAD+ on neuronal survival might be attributable to inhibition of PARP-1 activity by augmented formation of nicotinamide. PAR formation, indicative of PARP-1 activity, was detectable in neuronal nuclei after 30 min of MNNG exposure. The PAR formation was blocked by the PARP inhibitor PJ34 but was not blocked by posttreatment with 5 mm NAD+ (Fig. 2C), arguing against an effect of NAD+ on PARP-1 activity. Western blot analysis of PAR formation under these same conditions confirmed that NAD+ did not inhibit PARP-1 activity but instead increased the amount of PAR polymer produced after MNNG exposure (Fig. 2D), presumably because of increased substrate availability for PARP-1. In addition, HPLC determinations of intracellular nicotinamide levels showed only modest elevations in neurons treated with NAD+ or NAD+ plus MNNG (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material), to levels well below the concentrations found necessary to reduce PARP-1-mediated death (supplemental Fig. 2B,C, available at www.jneurosci.org as supplemental material). Together, these data indicate that the neuroprotective effect of NAD+ is not attributable to inhibition of PARP-1 activity.
Figure 2.

NAD+ repletion prevents PARP-1-mediated neuron death. A, B, Neurons were incubated with the designated concentrations of either MNNG (A) for 30 min or SIN1 (B) for 60 min, and NAD+ (2.5–10 mm) was added after washout of the MNNG. Neuron death was assayed at 24 h. n = 3; *p < 0.05, **p < 0.01 versus no added NAD+. Error bars indicate SEM. C, Immunostaining for PAR at 60 min after MNNG washout showed no inhibition of PARP-1 activity by 5 mm NAD+. The PARP inhibitor PJ34 serves as a positive control. Images are representative of four independent experiments. Scale bar, 40 μm. D, Western blot analysis of PAR formation after MNNG exposure (75 μm for 30 min). NAD+ was used at 2 and 5 mm and PJ34 at 200 nm. Cells were lysed 30 min after MNNG washout.
We next evaluated the effect of NAD+ posttreatment on established the conditions resulting in PARP-1-induced neuronal death in neuron–astrocyte cocultures (Fig. 3A–C). Cocultures were used to complement the neuronal monocultures used for the NAD+ uptake and biochemical measurements because the cocultures more closely mimic the neuronal differentiation and astrocyte–neuron interactions present in vivo. As in the neuronal monocultures, neuronal death induced over a range of MNNG concentrations was reduced by >80% by the PARP inhibitor DPQ. Neuronal death induced by CI and NMDA was reduced ∼50% by DPQ, suggesting that this fraction of neuronal death is attributable to PARP-1 activation under these conditions. NAD+ had dose-dependent effects on neuronal survival in all three of these stressors, with a maximal effect in each condition achieved at 5 mm added NAD+ (Fig. 3D–F). NAD+ was found to significantly reduce neuronal death when given up to 2 h after washout of MNNG or CI, and up to 1 h after washout of NMDA (Fig. 3G–I). In contrast, DPQ had no effect when added at these delayed time points. The failure of DPQ to reduce neuronal death when added to the cultures at these delayed time points further confirms that the neuroprotective effect of NAD+ posttreatment is not attributable to PARP-1 inhibition.
Figure 3.

Effects of NAD+ on PARP-1-induced neuronal death. A–I, Neuronal death was induced by MNNG (A, D, G), NMDA (B, E, H), and chemical ischemia (C, F, I). Incubation times were 30 min for MNNG, 10 min for NMDA, and variable with CI. A–C, Dose–response curves are shown for each condition. NMDA-induced neuronal death was blocked by 10 μm MK801 as a positive control. Neuronal death in all three conditions was reduced by coincubation with the PARP inhibitor, DPQ (10 μm). **p < 0.01 versus control (wash only); n = 3–4. D–F show the effects of posttreatment with 1–10 mm NAD+ on neuronal death under each of the conditions shown in A–C. The NAD+ was added after washout of MNNG, CI, or NMDA. **p < 0.01 versus no added NAD+; n = 3–4. G–I show the effects of 5 mm NAD+ or 10 μm DPQ on neuronal survival when added to the culture medium at increasing time intervals after washout of MNNG, CI, or NMDA. **p < 0.01 versus no added NAD+; n = 3–4. Error bars indicate SEM.
PARP-1 activation blocks glycolysis
NAD+ is an essential cofactor at the glyceradehyde-3-phosphate dehydrogenase (GAPDH) step of glycolysis, and the depletion of NAD+ by PARP-1 has been shown to block glucose utilization and lead to cell death in non-neuronal cell types (Ying et al., 2002; Zong et al., 2004). Here, neurons treated with MNNG or SIN1 showed >75% reductions in glucose utilization as measured by the 2-deoxyglucose method (Sokoloff et al., 1977), and these reductions were blocked by the PARP inhibitor PJ34 and in PARP-1−/− neurons (Fig. 4A). Measures of neuronal pyruvate concentration corroborated these results; both MNNG and SIN1 reduced neuronal pyruvate concentrations, and these reductions were also prevented by PJ34 and in PARP-1−/− neurons (Fig. 4B). Importantly, the reductions in both glycolytic rate and pyruvate content were reversed in neurons treated with 5 mm NAD+ added to the medium after PARP-1 activation (Fig. 4C,D), whereas neurons treated with 0.1 mm NAD+, which is insufficient to raise intracellular NAD+ concentrations, did not show recovery of glycolysis or pyruvate levels (data not shown). Neuronal energy status measured under these conditions showed parallel changes: MNNG produced ATP depletion and a rise in AMP, both of which were blocked by cotreatment with the PARP inhibitors DPQ or PJ34 and by posttreatment with NAD+ or pyruvate (Fig. 4E,F). A block at the GAPDH step of glycolysis should also increase levels of glycolytic intermediates upstream of this step. As shown in Table 1, neurons exposed to MNNG had markedly elevated levels of dihydroxyacetone phosphate, glyceraldehyde-3-phosphate, and fructose biphosphate, and these elevations were prevented both by PARP-1 inhibitors and exogenous NAD+ treatment.
Figure 4.

PARP-1 activation causes a block in neuronal glycolysis that is reversed by NAD+. Neurons were treated with 75 μm MNNG for 30 min or 2.5 mm SIN1 for 60 min. Where used, PJ34 (200 nm) and DPQ (10 μm) were added with MNNG or SIN1. A, B, Glycolytic rate and pyruvate content were measured 1 h after MNNG or SIN1 washout. n = 3; **p < 0.001 versus control. C, D, Five millimolar NAD+ or 2.5 mm pyruvate was added to the after washout of MNNG or SIN1. Glycolytic rate was restored by NAD+, but not pyruvate, whereas intracellular pyruvate concentrations were restored by both NAD+, but not pyruvate. PJ34 or DPQ were given simultaneously with MNNG or SIN1. n = 3; **p < 0.001 versus no posttreatment (white bar). E, F, ATP and AMP were measured 1 h after MNNG or SIN1 washout. The effects of MNNG on ATP and AMP levels were blocked by cotreatment with PJ34 or DPQ, and also blocked by posttreatment with NAD+ or pyruvate. Error bars indicate SEM.
Table 1.
Effects of PARP-1 activation on glycolytic intermediates upstream of GAPDH
| Condition | DHAP (pmol/mg) | GAP (pmol/mg) | FBP (pmol/mg) |
|---|---|---|---|
| Control | 2.24 ± 0.74 | 0.93 ± 0.26 | 1.64 ± 0.97 |
| MNNG | 11.18 ± 2.57# | 2.37 ± 0.62# | 8.52 ± 1.69# |
| MNNG + PJ34 | 2.28 ± 0.96** | 1.06 ± 0.23* | 2.22 ± 1.29** |
| MNNG + DPQ | 2.58 ± 0.84** | 1.20 ± 0.34 | 3.19 ± 1.15** |
| MNNG + NAD | 1.52 ± 0.90** | 1.13 ± 0.22* | 3.39 ± 1.29** |
| IA | 9.83 ± 2.06# | 3.10 ± 1.14# | 6.93 ± 0.73# |
Mouse cortical neurons were exposed to 75 μm MNNG for 30 min. PJ34 (200 nm) and DPQ (25 μm) were added with MNNG, and NAD+ (5 mm) was added after MNNG washout. Levels of DHAP, GAP, and FBP were measured at 30 min after MNNG washout. Iodoacetate (IA) (250 μm) was used as a positive control for GAPDH inhibition. Data are means ± SEM (n = 3).
#p < 0.01 versus control; *p < 0.05, **p < 0.01 versus MNNG.
PARP-1-induced neuronal death is prevented by nonglycolytic substrates
If PARP-1-induced neuronal death stems from a block in glycolysis, then nonglucose substrates that do not require cytosolic NAD+ for energy metabolism should preserve neuronal viability after PARP-1 activation. Accordingly, cultures treated with pyruvate or α-ketoglutarate after washout of MNNG or SIN1 showed reductions in cell death that were comparable with that obtained by coincubation with the PARP inhibitors, DPQ and PJ34 (Fig. 5A–C). Near-complete reductions in neuronal death were also observed using acetoacetate or hydroxybutyrate as nonglycolytic substrates (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). The aCSF culture medium used for these studies contains only glucose as a metabolic substrate, to mimic conditions present in situ. However, most commercially available culture media also contain additional components, notably glutamine and pyruvate, that can serve as energy substrates in the absence of glycolysis and that could thereby mask the effects of PARP-1-induced glycolytic blockade. We therefore compared MNNG toxicity in neurons placed in either aCSF or standard culture medium (Neurobasal medium with B27 supplement) (see Materials and Methods). As expected, neurons were much less sensitive to MNNG in the standard culture medium (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material). This patterns was also observed with mouse cortical astrocytes and mouse embryonic fibroblasts (supplemental Fig. 3D,E, available at www.jneurosci.org as supplemental material).
Figure 5.

PARP-1-induced neuronal death is prevented by nonglycolytic substrates. A, B, Neurons were provided with pyruvate (Pyr) (2.5 mm) or α-ketoglutarate (αKG) (2.5 mm) after washout of MNNG or SIN1. The reduction in cell death was comparable with that achieved by coincubation with the PARP inhibitors, DPQ (10 μm) and PJ34 (200 nm). n = 3; **p < 0.001 versus MNNG or SIN1 alone. Error bars indicate SEM. C, Representative images of neurons 24 h after the designated treatments. Cell fields were photographed with phase contrast optics and with calcein fluorescence to identify live cells and PI fluorescence to identify dead cells.
Mitochondrial depolarization induced by PARP-1 activation is blocked by NAD+ and pyruvate
Depolarization of the mitochondrial membrane potential (Δψm) occurs after PARP-1 activation in astrocytes, neurons, and other cell types (Alano et al., 2004; Yu et al., 2006; Formentini et al., 2009). Cytosolic NAD+ has an essential role in providing the mitochondria with glucose carbon in the form of pyruvate, and reducing equivalents in the form of NADH (via the malate–aspartate shuttle). Cytosolic NAD+ depletion should disrupt this substrate flux and thereby lead to mitochondrial depolarization unless other energy substrates are present. Here, we followed changes in Δψm with TMRM (1 nm) under nonquench conditions (Vesce et al., 2005; Tao et al., 2007). Neurons exposed to either MNNG or SIN1 exhibited a decrease in TMRM signal, indicating Δψm depolarization (Fig. 6). This decrease was blocked by the PARP inhibitor PJ34 and by posttreatment with either NAD+ or pyruvate (Fig. 6B,C). These findings suggest that PARP-1-induced Δψm depolarization results from glycolytic inhibition and that either reversing this block with NAD+ or bypassing this block with pyruvate can preserve Δψm. The effects of nonglucose substrates on neuron viability and mitochondrial membrane potential after PARP-1 activation also provide a functional confirmation that the mitochondrial NAD+ pool is not depleted within the first few hours of PARP-1 activation (Di Lisa et al., 2001; Alano et al., 2007).
Figure 6.
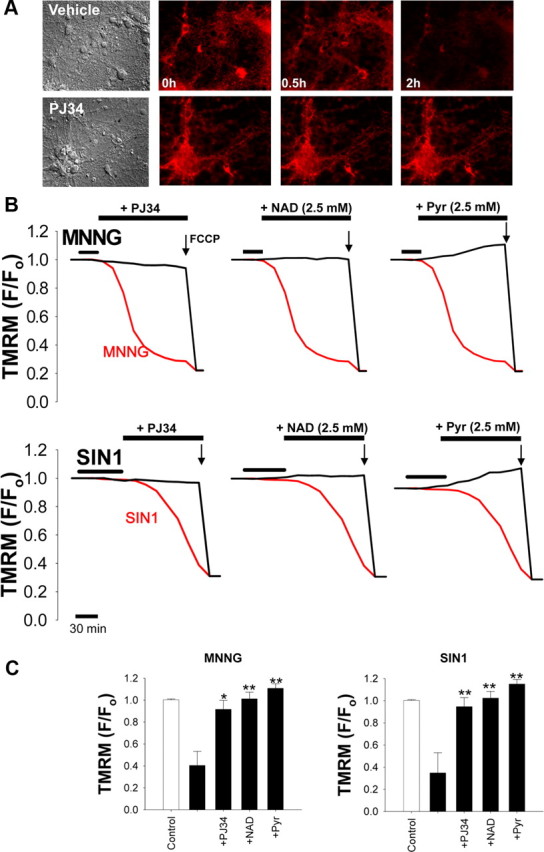
PARP-1-induced mitochondrial depolarization is prevented by NAD+ and pyruvate. A, Representative images of TMRM fluorescence in neurons after 30 min incubation with 2 mm SIN1. The loss of TMRM fluorescence, indicating loss of mitochondrial membrane potential, was prevented by coincubation with 200 nm PJ34. B, Mitochondrial depolarization over time after PARP-1 activation (red traces) with either MNNG (75 μm) or SIN1 (2 mm). The decrease in TMRM fluorescence was prevented by coincubation with the PARP inhibitor PJ34 (200 nm) or by postincubation with either NAD+ or pyruvate (2.5 mm). Neurons were exposed to FCCP (1 μm) at the end of each experiment to calibrate complete mitochondrial depolarization. C, Quantified TMRM fluorescence 2 h after MNNG or SIN1 treatment. n = 3; *p < 0.01, **p < 0.001 versus SIN1 or MNNG alone. Error bars indicate SEM.
Depletion of cytosolic NAD+ mimics PARP-1 activation
We next sought to determine whether NAD+ depletion alone was sufficient to induce the neuronal death that results from PARP-1 activation. The BioPORTER protein transfection system (Zelphati et al., 2001) was used to introduce NAD+ glycohydrolase (NADase) into the neuronal cytosol. Neurons transfected with NADase showed a significant decrease in NAD+ content within 3 h (Fig. 7A). NAD+ content in mitochondria isolated from these cells was unchanged (data not shown), confirming that NADase transfection depleted only the cytosolic NAD+ pool. The maximal degree of NAD+ depletion resulting from NADase transfection was ∼50%, consistent with our previous report finding that cytosolic NAD+ accounts for ∼50% of the total neuronal NAD+ (Alano et al., 2007). The decrease in NAD+ was prevented in cells cocultured with 0.2 mm NAM, an NADase inhibitor. Cultures treated with NADase also exhibited a dose-dependent loss of neuronal viability that was prevented by NAM (Fig. 7B) and by NAD+ repletion with exogenous NAD+ (Fig. 7C). These effects of NADase on NAD+ content and neuronal viability were comparable in wild-type and PARP-1−/− neurons (Fig. 6A), suggesting that the decrease in cytosolic NAD+ alone, independent of PARP-1 activation, is sufficient to cause neuronal death. NADase, like PARP-1 activation, also blocked neuronal glucose utilization and reduced pyruvate concentrations (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Also like PARP-1 activation, the neuronal death resulting from NADase transfection was prevented by exogenous pyruvate, whereas the glycolytic block was not (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Figure 7.
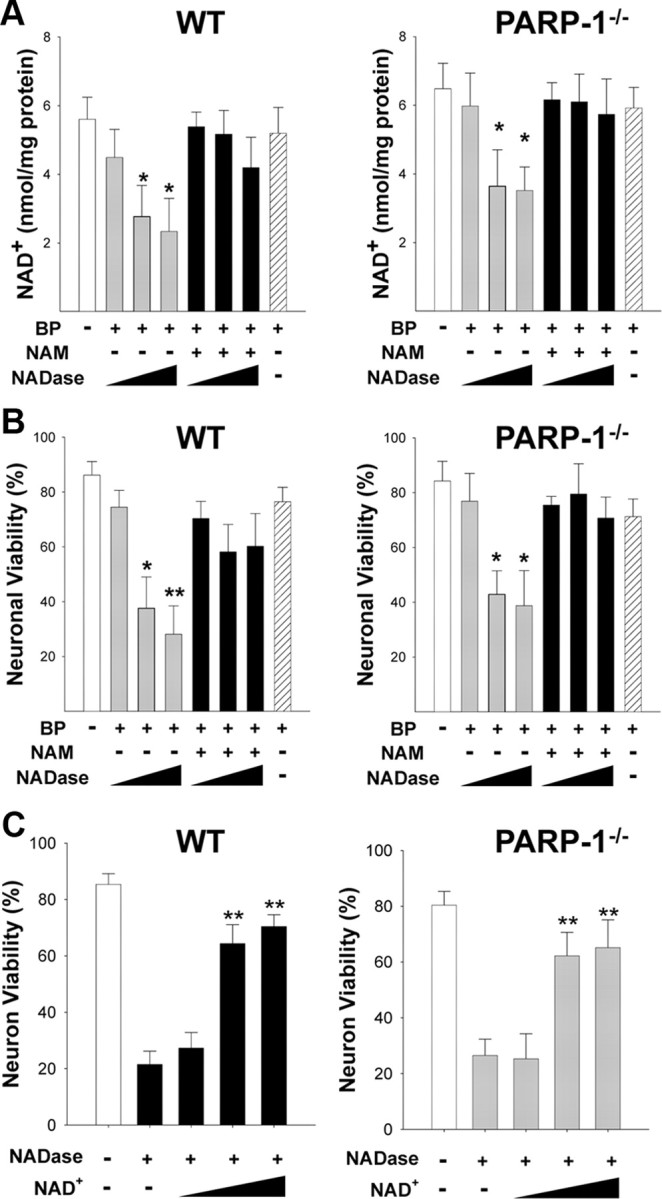
Depletion of cytosolic NAD+ with NADase kills neurons independent of PARP-1 activation. A, NADase transfection (10, 30, 100 μg/ml) produced a dose-dependent decrease in neuronal NAD+ content in both wild-type and PARP-1−/− neurons, as measured 3 h after BioPORTER transfection (BP). This decrease was blocked by the NADase inhibitor NAM (200 μm) and not observed in cultures treated with the BioPORTER vehicle alone (far right bar). B, NADase transfection also produced a dose-dependent neuronal death in both wild-type and PARP-1−/− neurons, evaluated 24 h later. This decrease was blocked by the NADase inhibitor NAM (200 μm) and not observed in cultures treated with the BioPORTER vehicle alone. C, Neuron death caused by NADase transfection (30 μg/ml) was significantly prevented by exogenous NAD+ treatment (2.5, 5, and 10 mm) in both wild-type and PARP-1−/− neurons. Error bars indicate SEM. *p < 0.01, **p < 0.001 (in A and B, versus control; in C, versus NADase alone).
AIF translocation induced by PARP-1 or NADase is prevented by pyruvate
Translocation of AIF from mitochondria to nucleus is considered a necessary step in the PARP-1 cell death pathway. Here, AIF immunoreactivity was reproducibly observed in the nucleus beginning 3–4 h after 30 min incubations with 75 μm MNNG (Fig. 8A). The AIF translocation was blocked by PJ34, confirming the role of PARP-1 activation in this process. The AIF translocation was also blocked by the addition of 5 mm NAD+ after MNNG washout, suggesting that cytosolic NAD+ depletion is necessary for PARP-1-induced AIF translocation. We then used NADase to determine whether NAD+ depletion is sufficient to induce AIF translocation. NADase transfection induced nuclear AIF translocation in both wild-type and PARP-1−/− neurons (Fig. 8B,C), and the AIF translocation induced by either PARP-1 activation or NADase was blocked by pyruvate (Fig. 8A–C). These results obtained by immunostaining were confirmed by Western blots for AIF in mitochondria isolated from neurons at designated time points after NADase transfection. The Western blots showed significant loss of AIF from the mitochondria 4 h after NADase treatment, and this was blocked by the addition of pyruvate (Fig. 8D,E). Effectiveness of the subcellular fractionation was verified by the enrichment of the mitochondrial marker VDAC and absence of the nuclear marker histone H2B (data not shown). For technical reasons, it was not possible to isolate a nuclear fraction that was completely devoid of mitochondrial contamination (data not shown).
Figure 8.
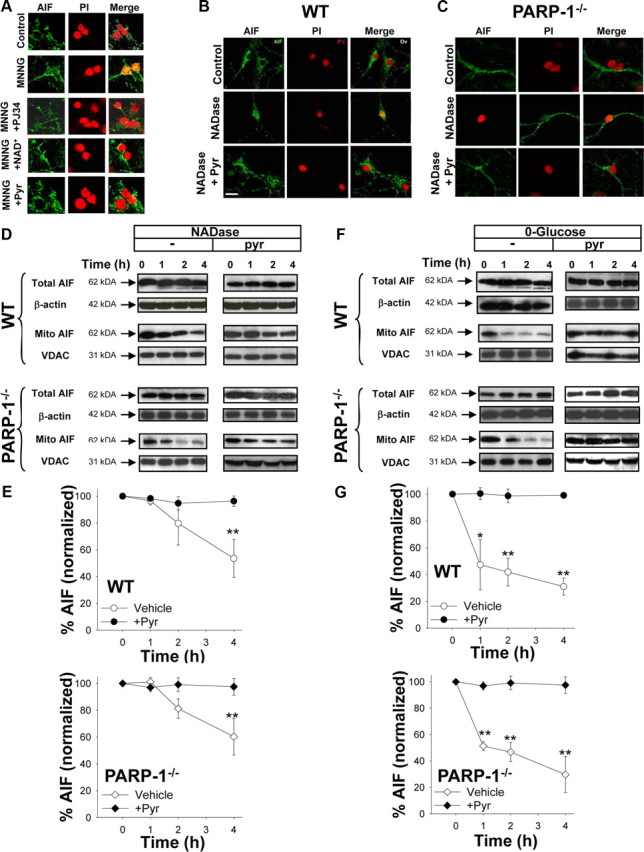
AIF translocation induced by PARP-1 or NADase is prevented by pyruvate. A, Immunostaining for AIF (green) in cultures fixed 3 h after incubation with MNNG alone (75 μm for 30 min), MNNG with 200 nm PJ34, MNNG followed by 5 mm NAD+, MNNG followed by 2.5 mm pyruvate, or medium exchanges only (control). Nuclei are counterstained with PI (red). Merged images show that DPQ, NAD+, and pyruvate block AIF translocation to the nucleus. B, C, Neurons transfected with NADase showed AIF translocation to the nucleus in both wild-type and PARP-1−/− neurons fixed 4 h after BioPORTER transfection. The AIF translocation was blocked by 2.5 mm pyruvate. Images are representative of four independent experiments. Scale bar, 40 μm. D, E, Western blots show both total and mitochondrial AIF content at designated time points (hours) after transfection with NADase, with or without pyruvate. The Western blots were quantified after normalizing to either β-actin for total AIF, or the mitochondrial protein VDAC for mitochondrial AIF. Mitochondrial AIF release occurred in both wild-type and PARP-1−/− neurons and was blocked by 2.5 mm pyruvate. F, G, Western blots show both total and mitochondrial AIF content at designated time points (hours) after placement in glucose-free medium, with or without pyruvate. Total AIF was quantified after normalizing to β-actin, and mitochondrial AIF was quantified after normalizing to the mitochondrial marker, VDAC. Mitochondrial AIF release occurred in both wild-type and PARP-1−/− neurons and was blocked by 2.5 mm pyruvate. n = 3; *p < 0.01, **p < 0.001. Error bars indicate SEM.
The ability of pyruvate to block AIF translocation induced by either PARP-1 or NADase suggests that AIF release results from the glycolytic block and mitochondrial depolarization induced by NAD+ depletion. To confirm that glycolytic failure can lead to AIF release, we assayed mitochondrial AIF after placing neurons in glucose-free medium. This condition replicated the effects of PARP-1 activation and NADase; glucose-free media led to a decrease in mitochondrial AIF in both wild-type and PARP-1−/− neurons, and this decrease was prevented by the addition of pyruvate (Fig. 8F,G).
Mitochondrial permeability transition inhibitors block AIF release
Mitochondrial permeability transition (MPT) can be triggered by mitochondrial depolarization, and previous studies have shown that MPT can trigger AIF release from mitochondria (Petit et al., 1998; Alano et al., 2004; Polster et al., 2005). MPT induced by mitochondrial depolarization could thus provide a mechanism linking cytosolic NAD+ depletion to mitochondrial release of AIF. We evaluated this possibility using both the classical MPT inhibitor cyclosporine A (CsA) (250 nm) and an alternative MPT inhibitor sanglifehrin A (SfA) (250 nm), which is a potent inhibitor of the mitochondrial permeability transition (Clarke et al., 2002), and which does not inhibit calcineurin activity (Zenke et al., 2001; Sànchez-Tilló et al., 2006). Both agents prevented the mitochondria-to-nucleus translocation of AIF that otherwise resulted from neuronal PARP-1 activation (Fig. 9A–C). The MPT inhibitors, unlike pyruvate, did not prevent the mitochondrial membrane depolarization (Δψm) that occurred during the initial 30 min after PARP-1 activation, thus arguing against the possibility that MPT was a cause rather than a result of Δψm (Fig. 9D). However, treatment with either CsA or SfA at the 30 min time point did prevent the total collapse of Δψm otherwise seen at the 4 h time point, consistent with an effect on MPT (Fig. 9D).
Figure 9.
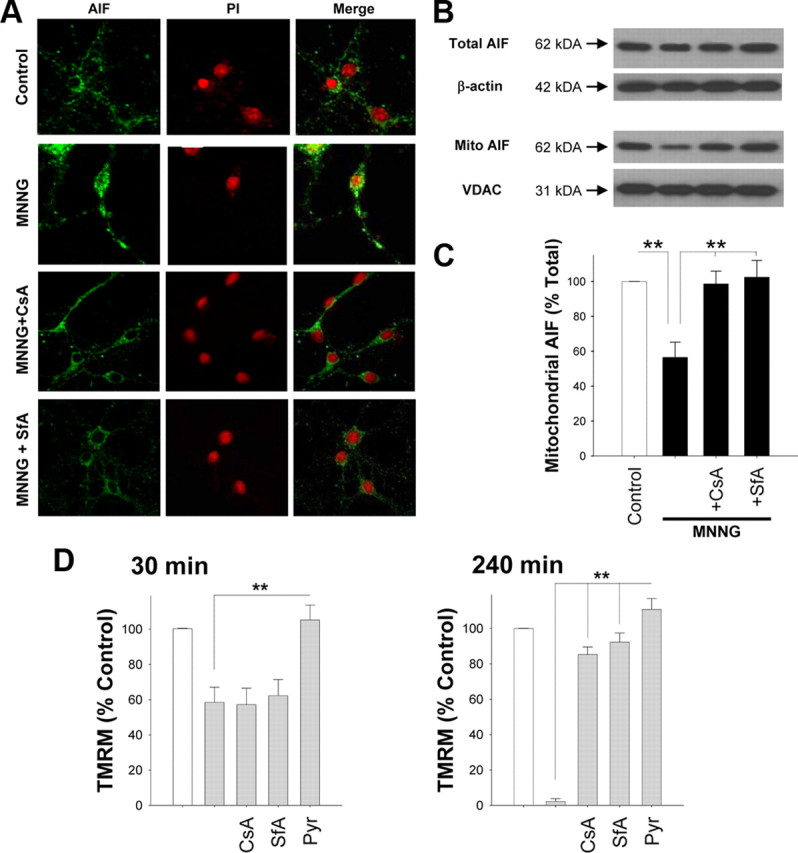
MPT inhibitors block mitochondrial AIF release. A, Immunostaining for AIF (green) in cultures fixed 3 h after incubation with MNNG alone (75 μm for 30 min), MNNG with CsA (250 nm), or MNNG with SfA (250 nm). CsA and SfA were added 1 h before PARP-1 activation. Nuclei are counterstained with PI (red). Merged images show that CsA and SfA prevent AIF translocation to the nucleus. B, C, Western blot analysis (B) of experiment as in A, showing total AIF and mitochondrial AIF. n = 3; **p < 0.001 versus MNNG alone. D, Effects of MPT inhibitors on mitochondrial membrane potential, as quantified by TMRM fluorescence, 30 min and 4 h after MNNG treatment. CsA (250 nm) and SfA (250 nm) added 1 h before MNNG, and pyruvate (2.5 mm) was given immediately after MNNG washout. Neurons were exposed to FCCP (1 μm) at the end of each experiment to calibrate complete mitochondrial depolarization (data not shown). n = 3; **p < 0.001. Error bars indicate SEM.
Discussion
PARP-1 activation is a major cause of neuronal death in brain ischemia, trauma, and other settings, but the role of NAD+ depletion in this cell death pathway has been unresolved. The present findings show that PARP-1-mediated neuronal death is blocked when NAD+ depletion is prevented with exogenous NAD+. NAD+ repletion also prevents the intermediary steps in this cell death pathway that otherwise result from PARP-1 activation: glycolytic inhibition, mitochondrial depolarization, and mitochondrial AIF release. Conversely, depletion of cytosolic NAD+ with NAD+ glycohydrolase causes glycolytic inhibition and mitochondrial AIF release, independent of PARP-1 activation. These findings establish cytosolic NAD+ depletion as a necessary and sufficient event in the PARP-1 cell death pathway.
The cytosolic and mitochondrial NAD+ pools are physically separated by the mitochondrial membrane, and only the cytosolic pool is accessible to nuclear PARP-1 unless mitochondrial disruption or mitochondrial permeability transition occur (Di Lisa et al., 2001; Alano et al., 2007). Cytosolic NAD+ is required for the GAPDH step of glycolysis, and studies using other cell types have shown that PARP-1 activation can cause a block in glycolysis (Zong et al., 2004; Verrax et al., 2007) that is reversible with NAD+ repletion (Alano et al., 2004; Zong et al., 2004). The glycolytic inhibition that results from NAD+ depletion blocks the flux of glucose-derived pyruvate and NADH reducing equivalents to the mitochondria. This block provides a biochemical link between cytosolic NAD+ depletion and mitochondrial failure, because the mitochondrial membrane potential cannot be maintained in the absence of energy substrates. Moreover, the rise in AMP that results from this block can inhibit the mitochondrial adenine transporter (Formentini et al., 2009), an effect that could further contribute to mitochondrial depolarization. Mitochondrial depolarization promotes mitochondrial membrane permeability transition (MPT), and MPT can in turn trigger mitochondrial AIF release (Petit et al., 1998; Alano et al., 2004; Polster et al., 2005). Here, a role for MPT in PARP-1-induced AIF release was supported by the suppression of AIF release in cells treated with the MPT inhibitors CsA and SfA. The use of CsA is highly criticized because, in addition to binding cyclophilin D to inhibit MPT pore opening, it can also bind to other cyclophilins, such as cyclophilin A, which in turn inhibits calcineurin, a calcium-dependent protein phosphatase demonstrated to have many intracellular roles. Although SfA has been shown to bind cyclophilin A, it does not inhibit calcineurin activity (Zenke et al., 2001; Sànchez-Tilló et al., 2006).
Previous studies using PAR neutralizing antibody and exogenous delivery of PAR to neurons have suggested an essential role for PAR itself in AIF release and PARP-1-mediated neuronal death (Andrabi et al., 2006). In addition, AMP generated by PAR degradation can contribute to mitochondrial depolarization (Formentini et al., 2009). However, results of the present studies suggest that PARP-1-induced mitochondrial failure and cell death can occur independent of these effects, because (1) AIF translocation was blocked by NAD+ repletion and by pyruvate treatment, despite abundant PAR formation, and (2) AIF translocation and cell death occurred after NAD+ depletion even in PARP-1−/− neurons, in which PAR production is minimal (Schreiber et al., 2006). We propose that the effects of NAD+ depletion and PAR on AIF translocation may be context dependent. The present studies were performed using a medium containing glucose as the only energy substrate, to approximate the composition of brain extracellular fluid. The flux of carbon and reducing equivalents to the mitochondria is almost entirely dependent on glycolysis under these conditions. Standard culture medium, unlike brain extracellular fluid, contains substantial concentrations of amino acids and other substrates that do not require glycolysis or NAD+ for entry into oxidative metabolism and that have been shown to rescue neurons and other cell types from PARP-1-induced cell death (Ying et al., 2002; Zong et al., 2004). Consequently, studies performed in standard culture medium may not replicate this aspect of PARP-1 activation in brain.
MNNG and SIN1 were used in these studies to induce nuclear PARP-1 activity and NAD+ depletion, but these compounds can also have direct effects on mitochondria (Dodoni et al., 2004). The lack of any detectable effect on mitochondria membrane potential or cell viability when these agents were used in the presence of PARP inhibitors or in PARP-1−/− cells argues against a significant direct effect on mitochondria under the conditions used here. It is nevertheless possible that these agents could have a more subtle “sensitizing” effect on mitochondria, whereby they increase the likelihood of MPT when mitochondria are depolarized (Dodoni et al., 2004). This possibility cannot be excluded, but the results obtained with NADase transfection indicate that cytosolic NAD+ depletion alone is sufficient to induce mitochondrial AIF release and neuronal death.
Although previous studies have shown that exogenous NAD+ can replenish neuronal NAD+ levels (Araki et al., 2004; Wang et al., 2008), the mechanism of uptake into neurons has not previously been established. Results presented here indicate that NAD+ uptake occurs through P2X7 receptor-gated channels. P2X7 receptors are activated by ATP and other signals (Faria et al., 2005) and exhibit a basal state of conductance in some cell types (Nagasawa et al., 2009). P2X7 receptors are expressed on many cell types, including neurons (Sperlágh et al., 2006). Recent evidence suggests that the pore itself is formed by a separate protein, pannexin-1, rather than by the P2X7 receptor alone (Pelegrin and Surprenant, 2006). Brilliant blue and oxidized ATP are potent inhibitors of the P2X7 receptor, and in the present studies these inhibitors completely blocked NAD+ uptake into neurons (Anderson and Nedergaard, 2006), but we cannot entirely exclude a role for other purinergic receptors that are also associated with pore formation.
Measures of brain NAD+ changes in vivo are influenced by both mitochondrial and cytosolic NAD+ pools, and by the presence of both neuronal and non-neuronal cells. Nevertheless, an early fall in total NAD+ has been reported after brain ischemia and brain trauma in most studies in which this has been evaluated (Endres et al., 1997; Paschen et al., 2000; Yang et al., 2002; Clark et al., 2007; Liu et al., 2009). PARP-1 inhibitors prevent this drop and provide robust neuroprotection in these settings but must be administered within a very short period of time after these insults to be effective (Clark et al., 2007; Moroni, 2008). A notable aspect of the studies presented here is that NAD+ repletion can prevent PARP-1-induced neuronal death when added hours after the PARP-1-inducing injury stimulus has ceased and PARP-1 inhibitors no longer have a neuroprotective effect. The effect of NAD+ at these delayed time points suggests that it may be possible to similarly target downstream events in the PARP-1 cell death pathway in brain ischemia, brain trauma, or other settings to extend the window of opportunity for intervening in these disorders.
Footnotes
This work was supported by National Institutes of Health Grant NS14543 (R.A.S.), the Department of Veterans Affairs (C.C.A., R.A.S., W.Y.), and the American Heart Association (C.C.A.).
References
- Alano CC, Swanson RA. Players in the PARP-1 cell-death pathway: JNK1 joins the cast. Trends Biochem Sci. 2006;31:309–311. doi: 10.1016/j.tibs.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem. 2004;279:18895–18902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- Alano CC, Kauppinen TM, Valls AV, Swanson RA. Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc Natl Acad Sci U S A. 2006;103:9685–9690. doi: 10.1073/pnas.0600554103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano CC, Tran A, Tao R, Ying W, Karliner JS, Swanson RA. Differences among cell types in NAD+ compartmentalization: a comparison of neurons, astrocytes, and cardiac myocytes. J Neurosci Res. 2007;85:3378–3385. doi: 10.1002/jnr.21479. [DOI] [PubMed] [Google Scholar]
- Anderson CM, Nedergaard M. Emerging challenges of assigning P2X7 receptor function and immunoreactivity in neurons. Trends Neurosci. 2006;29:257–262. doi: 10.1016/j.tins.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T, Zhang Z, Koehler RC, Hurn PD, Poirier GG, Dawson VL, Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- Billington RA, Travelli C, Ercolano E, Galli U, Roman CB, Grolla AA, Canonico PL, Condorelli F, Genazzani AA. Characterization of NAD uptake in mammalian cells. J Biol Chem. 2008;283:6367–6374. doi: 10.1074/jbc.M706204200. [DOI] [PubMed] [Google Scholar]
- Bruzzone S, Franco L, Guida L, Zocchi E, Contini P, Bisso A, Usai C, De Flora A. A self-restricted CD38-connexin 43 cross-talk affects NAD+ and cyclic ADP-ribose metabolism and regulates intracellular calcium in 3T3 fibroblasts. J Biol Chem. 2001;276:48300–48308. doi: 10.1074/jbc.M107308200. [DOI] [PubMed] [Google Scholar]
- Bryja V, Bonilla S, Arenas E. Derivation of mouse embryonic stem cells. Nat Protoc. 2006;1:2082–2087. doi: 10.1038/nprot.2006.355. [DOI] [PubMed] [Google Scholar]
- Clark RS, Vagni VA, Nathaniel PD, Jenkins LW, Dixon CE, Szabó C. Local administration of the poly(ADP-ribose) polymerase inhibitor INO-1001 prevents NAD+ depletion and improves water maze performance after traumatic brain injury in mice. J Neurotrauma. 2007;24:1399–1405. doi: 10.1089/neu.2007.0305. [DOI] [PubMed] [Google Scholar]
- Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J Biol Chem. 2002;277:34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- Crescentini G, Stocchi V. Fast reversed-phase high-performance liquid chromatographic determination of nucleotides in red blood cells. J Chromatogr. 1984;290:393–399. doi: 10.1016/s0021-9673(01)93592-7. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Menabò R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- Dodoni G, Canton M, Petronilli V, Bernardi P, Di Lisa F. Induction of the mitochondrial permeability transition by the DNA alkylating agent N-methyl-N′-nitro-N-nitrosoguanidine. Sorting cause and consequence of mitochondrial dysfunction. Biochim Biophys Acta. 2004;1658:58–63. doi: 10.1016/j.bbabio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, Moskowitz MA. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;19:5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Faria RX, Defarias FP, Alves LA. Are second messengers crucial for opening the pore associated with P2X7 receptor? Am J Physiol Cell Physiol. 2005;288:C260–C271. doi: 10.1152/ajpcell.00215.2004. [DOI] [PubMed] [Google Scholar]
- Formentini L, Macchiarulo A, Cipriani G, Camaioni E, Rapizzi E, Pellicciari R, Moroni F, Chiarugi A. Poly(ADP-ribose) catabolism triggers AMP-dependent mitochondrial energy failure. J Biol Chem. 2009;284:17668–17676. doi: 10.1074/jbc.M109.002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier P, Ying W, Swanson RA. Ischemic preconditioning by caspase cleavage of poly(ADP-ribose) polymerase-1. J Neurosci. 2003;23:7967–7973. doi: 10.1523/JNEUROSCI.23-22-07967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillmor HA, Bolton CH, Hopton M, Moore WP, Perrett D, Bingley PJ, Gale EA. Measurement of nicotinamide and N-methyl-2-pyridone-5-carboxamide in plasma by high performance liquid chromatography. Biomed Chromatogr. 1999;13:360–362. doi: 10.1002/(SICI)1099-0801(199908)13:5<360::AID-BMC893>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A. 1999;96:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad M, Beray-Berthat V, Coqueran B, Palmier B, Szabó C, Plotkine M, Margaill I. Reduction of hemorrhagic transformation by PJ34, a poly(ADP-ribose)polymerase inhibitor, after permanent focal cerebral ischemia in mice. Eur J Pharmacol. 2008;588:52–57. doi: 10.1016/j.ejphar.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Jiang LH, Mackenzie AB, North RA, Surprenant A. Brilliant blue G selectively blocks ATP-gated rat P2X7 receptors. Mol Pharmacol. 2000;58:82–88. [PubMed] [Google Scholar]
- Lamprecht W, Heinz F. Ed 3. Weinheim, Germany: VCH Publishers; 1988. Methods of enzymatic analysis. [Google Scholar]
- Liu D, Gharavi R, Pitta M, Gleichmann M, Mattson MP. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromolecular Med. 2009;11:28–42. doi: 10.1007/s12017-009-8058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Passonneau JV. New York: Academic; 1972. A flexible system of enzymatic analysis. [Google Scholar]
- Michal G. d-Fructose 1,6-bisphosphate, dihydroxyacetone phosphate and d-glyceraldehyde 3-phosphate. In: Bergmeyer HU, editor. Methods of enzymatic analysis. Ed 3. New York: VCH; 1988. pp. 342–350. [Google Scholar]
- Moroni F. Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol. 2008;8:96–103. doi: 10.1016/j.coph.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Murgia M, Hanau S, Pizzo P, Rippa M, Di Virgilio F. Oxidized ATP. An irreversible inhibitor of the macrophage purinergic P2Z receptor. J Biol Chem. 1993;268:8199–8203. [PubMed] [Google Scholar]
- Nagasawa K, Escartin C, Swanson RA. Astrocyte cultures exhibit P2X7 receptor channel opening in the absence of exogenous ligands. Glia. 2009;57:622–633. doi: 10.1002/glia.20791. [DOI] [PubMed] [Google Scholar]
- Nilausen K, Green H. Reversible arrest of growth in G1 of an established fibroblast line (3T3) Exp Cell Res. 1965;40:166–168. doi: 10.1016/0014-4827(65)90306-x. [DOI] [PubMed] [Google Scholar]
- Paschen W, Oláh L, Mies G. Effect of transient focal ischemia of mouse brain on energy state and NAD levels: no evidence that NAD depletion plays a major role in secondary disturbances of energy metabolism. J Neurochem. 2000;75:1675–1680. doi: 10.1046/j.1471-4159.2000.0751675.x. [DOI] [PubMed] [Google Scholar]
- Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit PX, Goubern M, Diolez P, Susin SA, Zamzami N, Kroemer G. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett. 1998;426:111–116. doi: 10.1016/s0014-5793(98)00318-4. [DOI] [PubMed] [Google Scholar]
- Polster BM, Basañez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- Rankin PW, Jacobson EL, Benjamin RC, Moss J, Jacobson MK. Quantitative studies of inhibitors of ADP-ribosylation in vitro and in vivo. J Biol Chem. 1989;264:4312–4317. [PubMed] [Google Scholar]
- Sànchez-Tilló E, Wojciechowska M, Comalada M, Farrera C, Lloberas J, Celada A. Cyclophilin A is required for M-CSF-dependent macrophage proliferation. Eur J Immunol. 2006;36:2515–2524. doi: 10.1002/eji.200535270. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- Sperlágh B, Vizi ES, Wirkner K, Illes P. P2X7 receptors in the nervous system. Prog Neurobiol. 2006;78:327–346. doi: 10.1016/j.pneurobio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Benington JH. Astrocyte glucose metabolism under normal and pathological conditions in vitro. Dev Neurosci. 1996;18:515–521. doi: 10.1159/000111448. [DOI] [PubMed] [Google Scholar]
- Szabó C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- Szabó C, Zingarelli B, O'Connor M, Salzman AL. DNA strand breakage, activation of poly(ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Karliner JS, Simonis U, Zheng J, Zhang J, Honbo N, Alano CC. Pyrroloquinoline quinone preserves mitochondrial function and prevents oxidative injury in adult rat cardiac myocytes. Biochem Biophys Res Commun. 2007;363:257–262. doi: 10.1016/j.bbrc.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrax J, Vanbever S, Stockis J, Taper H, Calderon PB. Role of glycolysis inhibition and poly(ADP-ribose) polymerase activation in necrotic-like cell death caused by ascorbate/menadione-induced oxidative stress in K562 human chronic myelogenous leukemic cells. Int J Cancer. 2007;120:1192–1197. doi: 10.1002/ijc.22439. [DOI] [PubMed] [Google Scholar]
- Vesce S, Jekabsons MB, Johnson-Cadwell LI, Nicholls DG. Acute glutathione depletion restricts mitochondrial ATP export in cerebellar granule neurons. J Biol Chem. 2005;280:38720–38728. doi: 10.1074/jbc.M506575200. [DOI] [PubMed] [Google Scholar]
- Virág L, Szabó C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xing Z, Vosler PS, Yin H, Li W, Zhang F, Signore AP, Stetler RA, Gao Y, Chen J. Cellular NAD replenishment confers marked neuroprotection against ischemic cell death: role of enhanced DNA repair. Stroke. 2008;39:2587–2595. doi: 10.1161/STROKEAHA.107.509158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Klaidman LK, Nalbandian A, Oliver J, Chang ML, Chan PH, Adams JD., Jr The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in Wistar rats. Neurosci Lett. 2002;333:91–94. doi: 10.1016/s0304-3940(02)01005-4. [DOI] [PubMed] [Google Scholar]
- Ying W, Chen Y, Alano CC, Swanson RA. Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J Cereb Blood Flow Metab. 2002;22:774–779. doi: 10.1097/00004647-200207000-00002. [DOI] [PubMed] [Google Scholar]
- Ying W, Garnier P, Swanson RA. NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem Biophys Res Commun. 2003;308:809–813. doi: 10.1016/s0006-291x(03)01483-9. [DOI] [PubMed] [Google Scholar]
- Ying W, Alano CC, Garnier P, Swanson RA. NAD+ as a metabolic link between DNA damage and cell death. J Neurosci Res. 2005;79:216–223. doi: 10.1002/jnr.20289. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelphati O, Wang Y, Kitada S, Reed JC, Felgner PL, Corbeil J. Intracellular delivery of proteins with a new lipid-mediated delivery system. J Biol Chem. 2001;276:35103–35110. doi: 10.1074/jbc.M104920200. [DOI] [PubMed] [Google Scholar]
- Zenke G, Strittmatter U, Fuchs S, Quesniaux VF, Brinkmann V, Schuler W, Zurini M, Enz A, Billich A, Sanglier JJ, Fehr T. Sanglifehrin A, a novel cyclophilin-binding compound showing immunosuppressive activity with a new mechanism of action. J Immunol. 2001;166:7165–7171. doi: 10.4049/jimmunol.166.12.7165. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]